
Synthesis and Vibrational Circular Dichroism Analysis of N-Heterocyclic Carbene Precursors Containing Remote Chirality Centers

 , and
, and 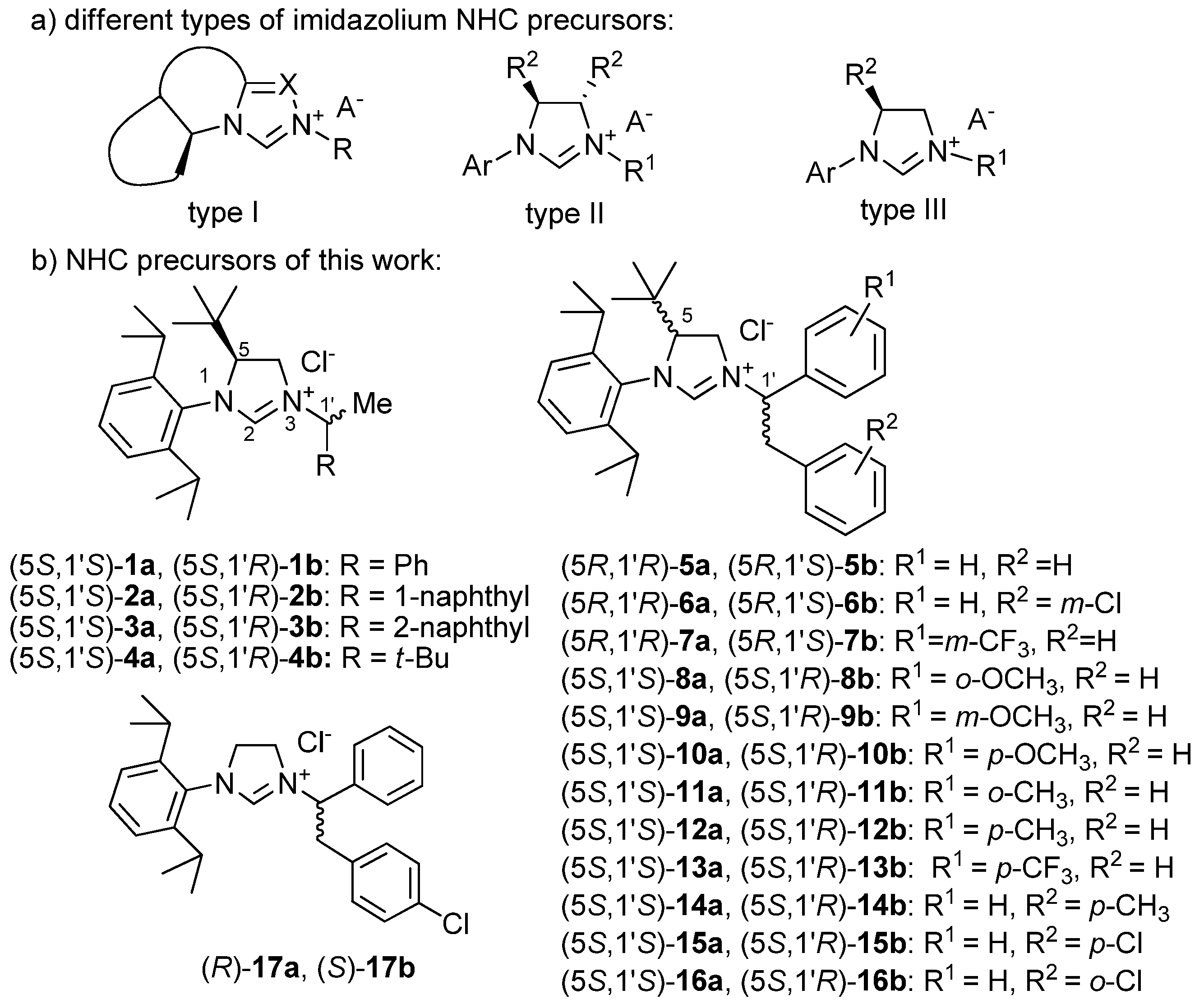{kind=link}
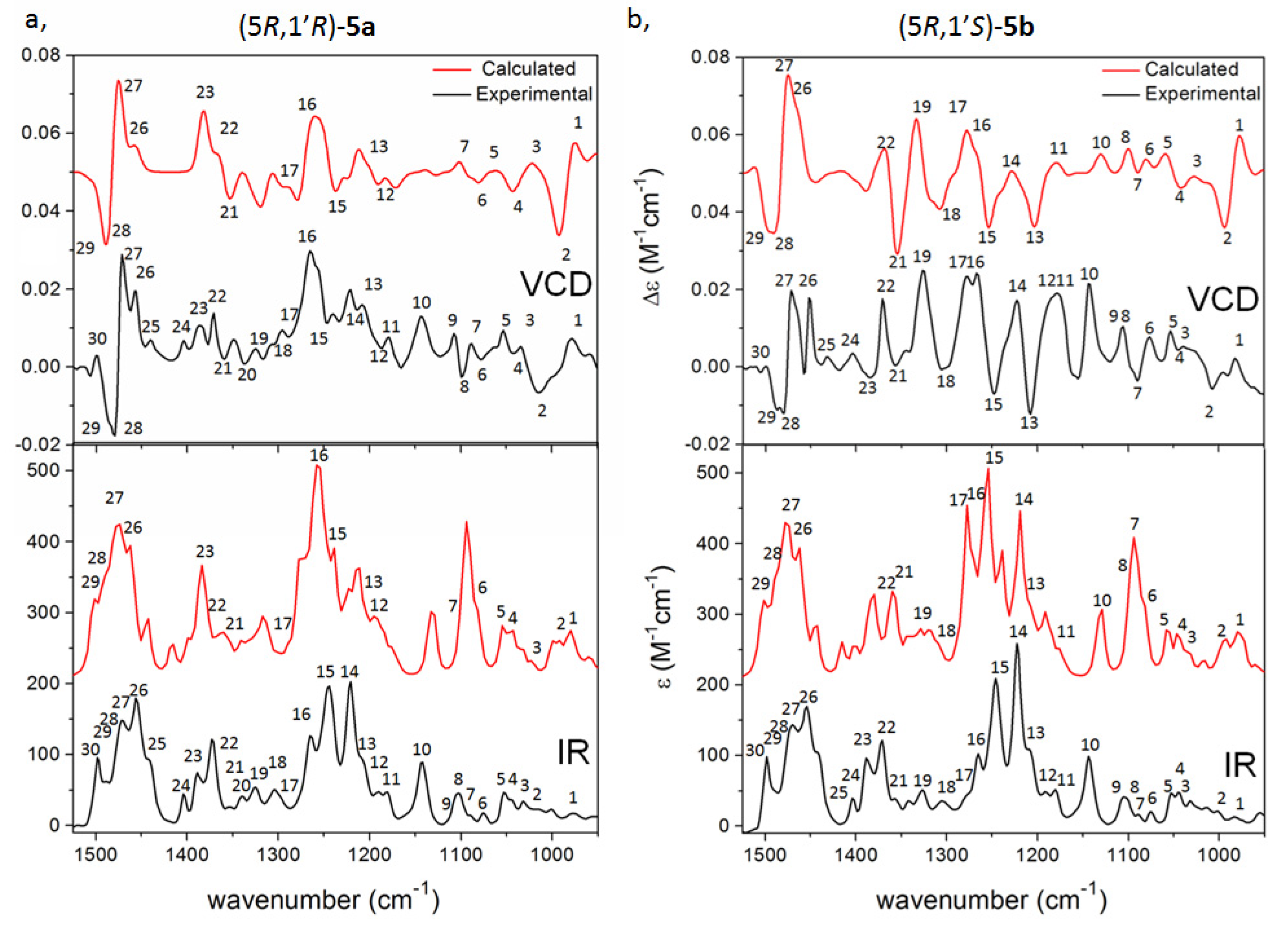{kind=link}
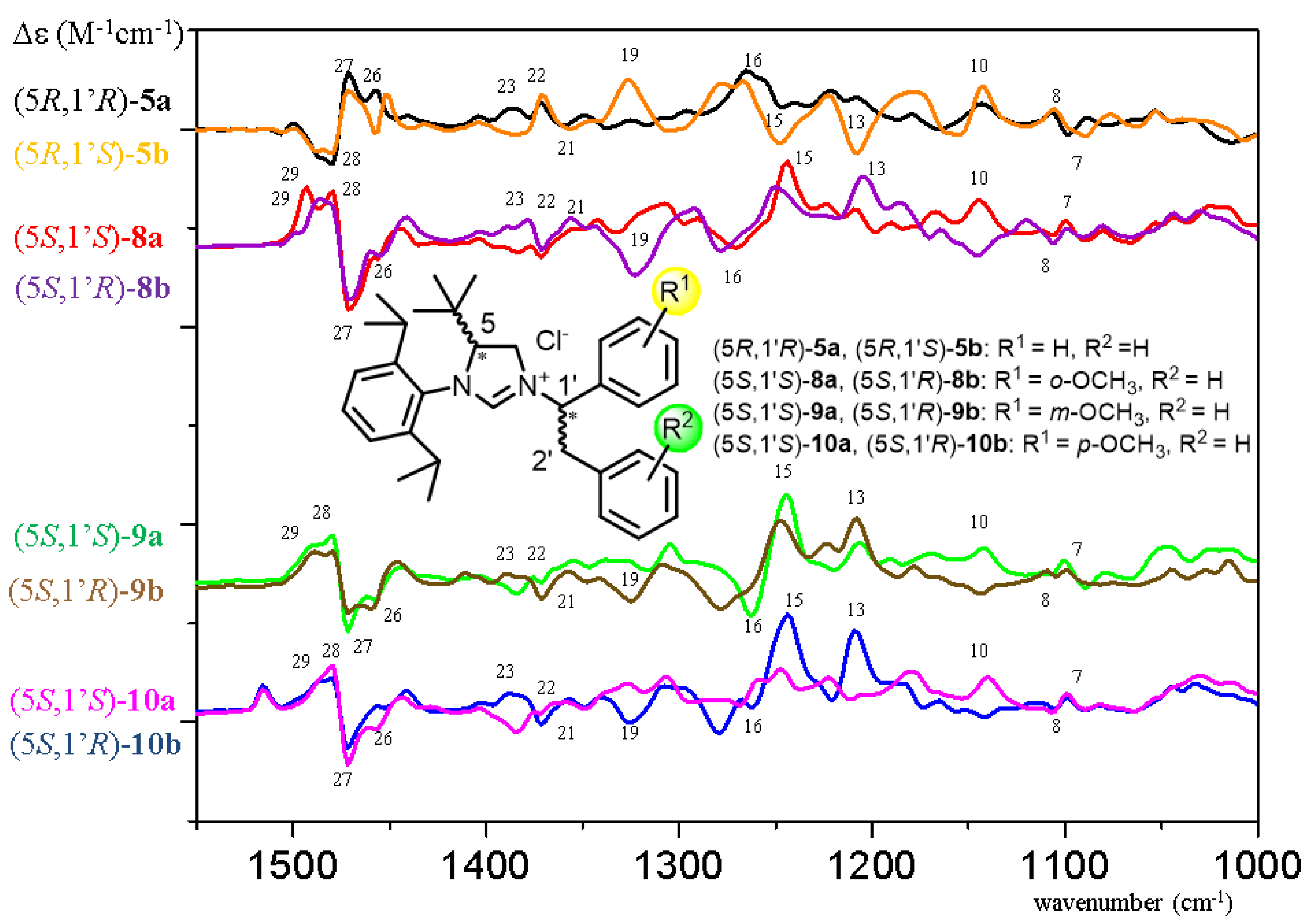{kind=link}
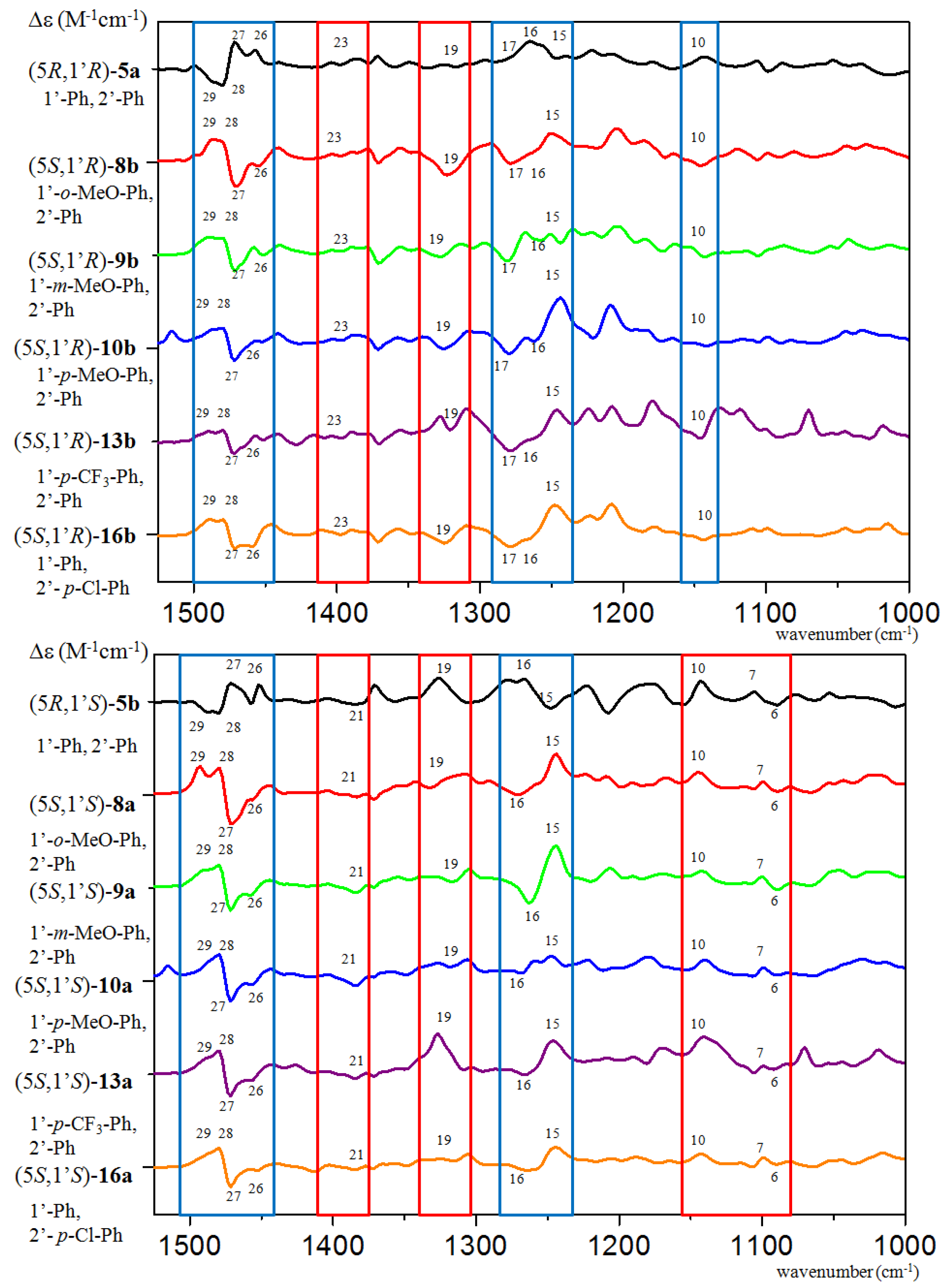{kind=link}
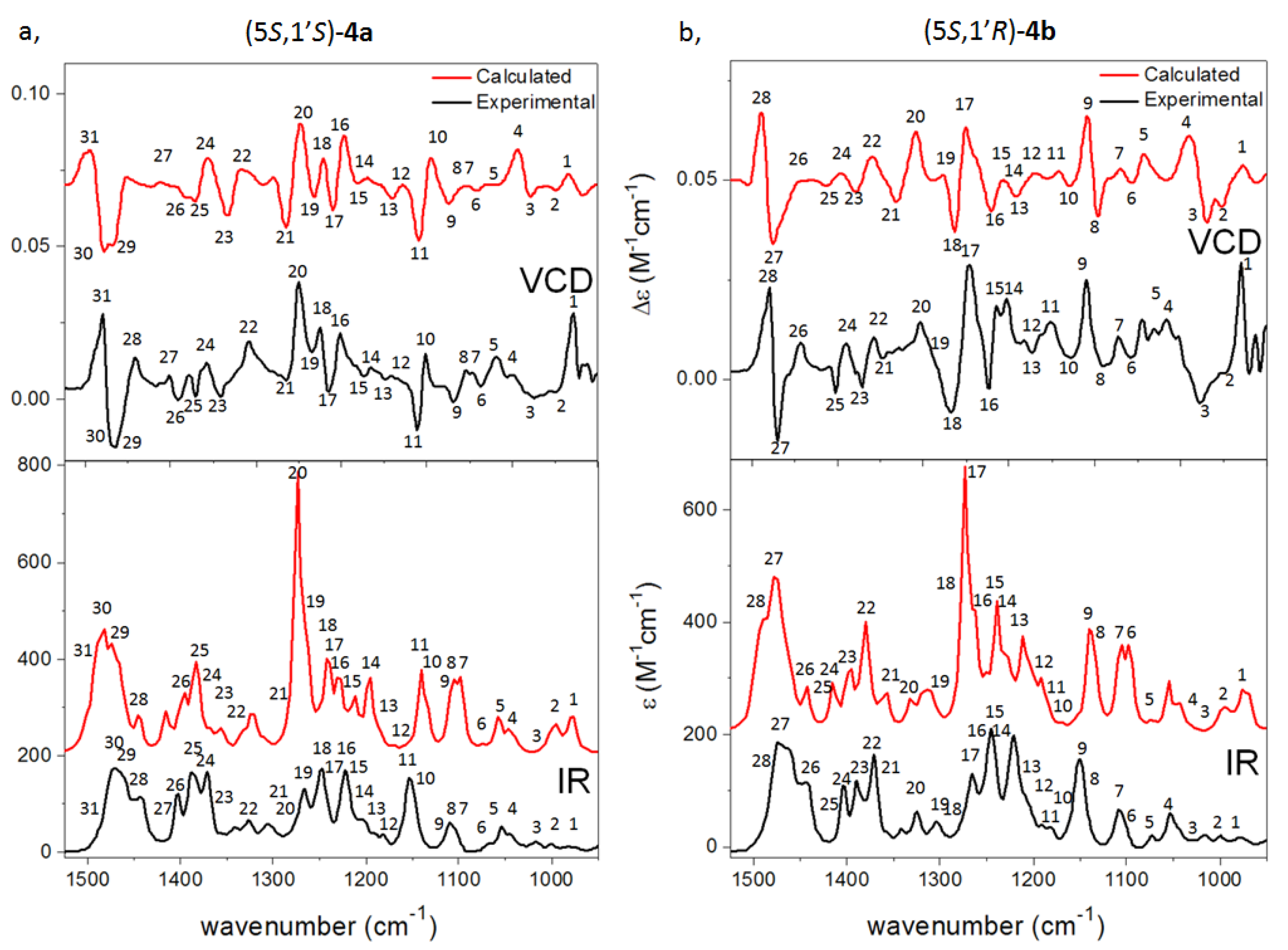{kind=link}
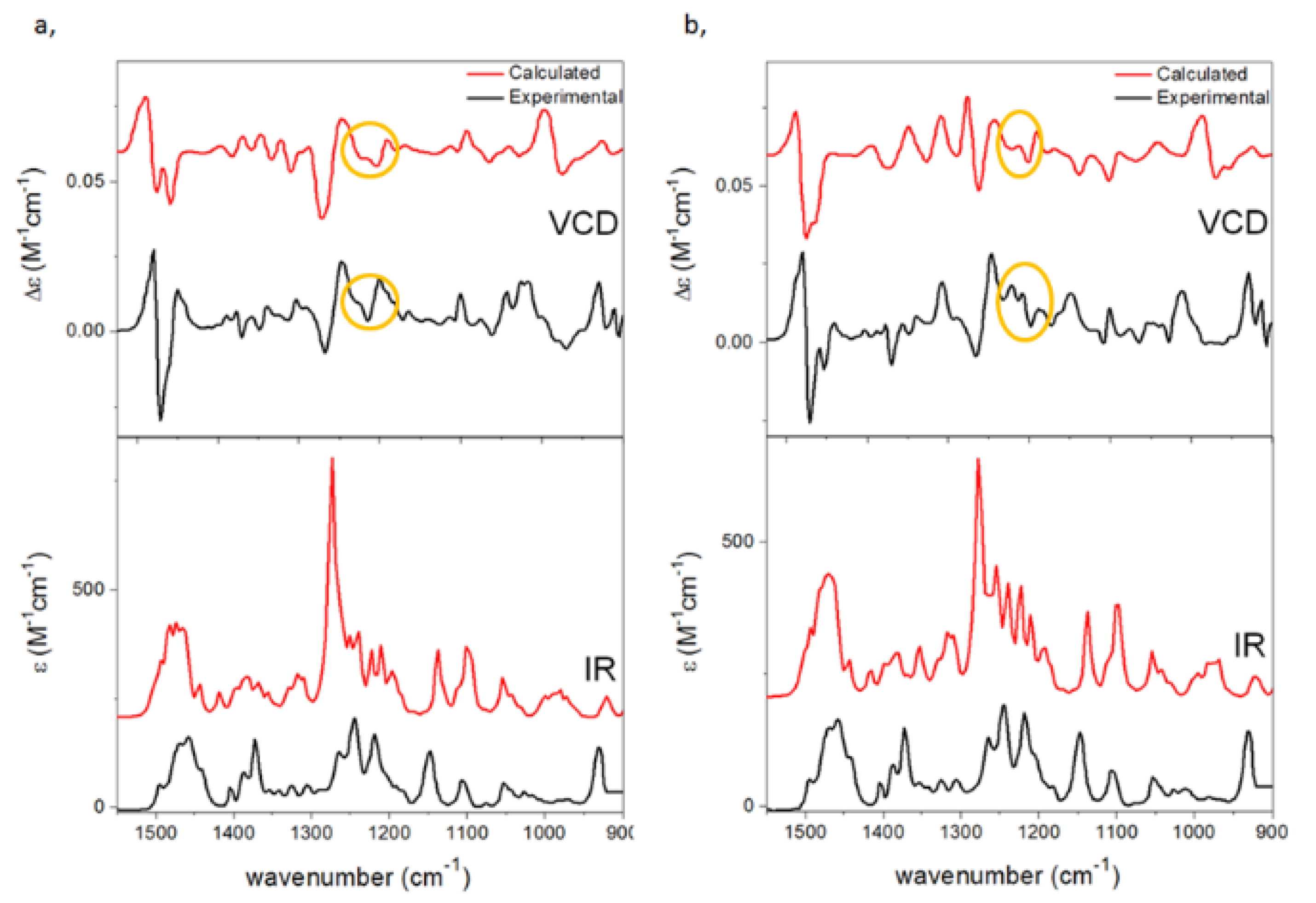{kind=link}
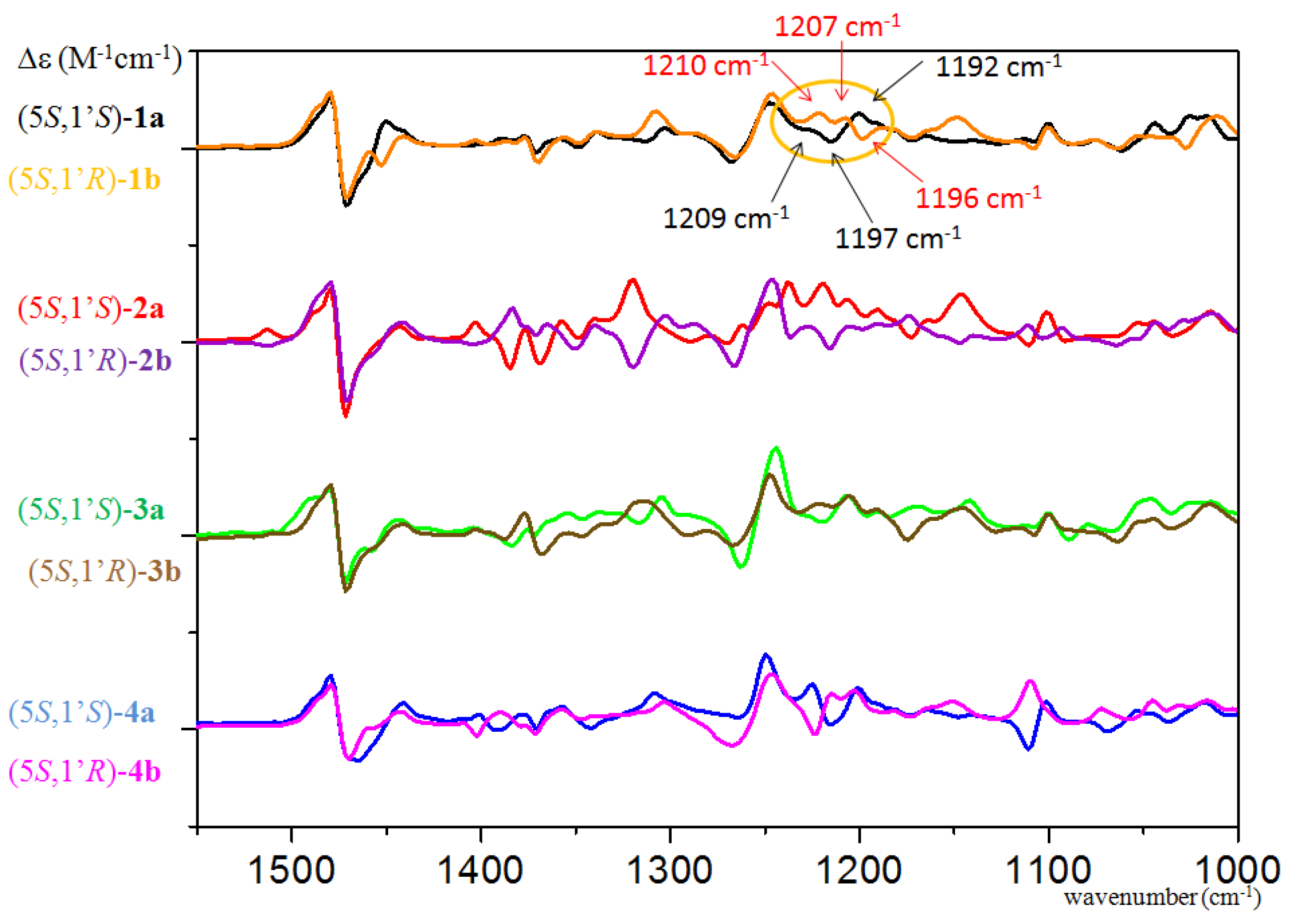{kind=link}
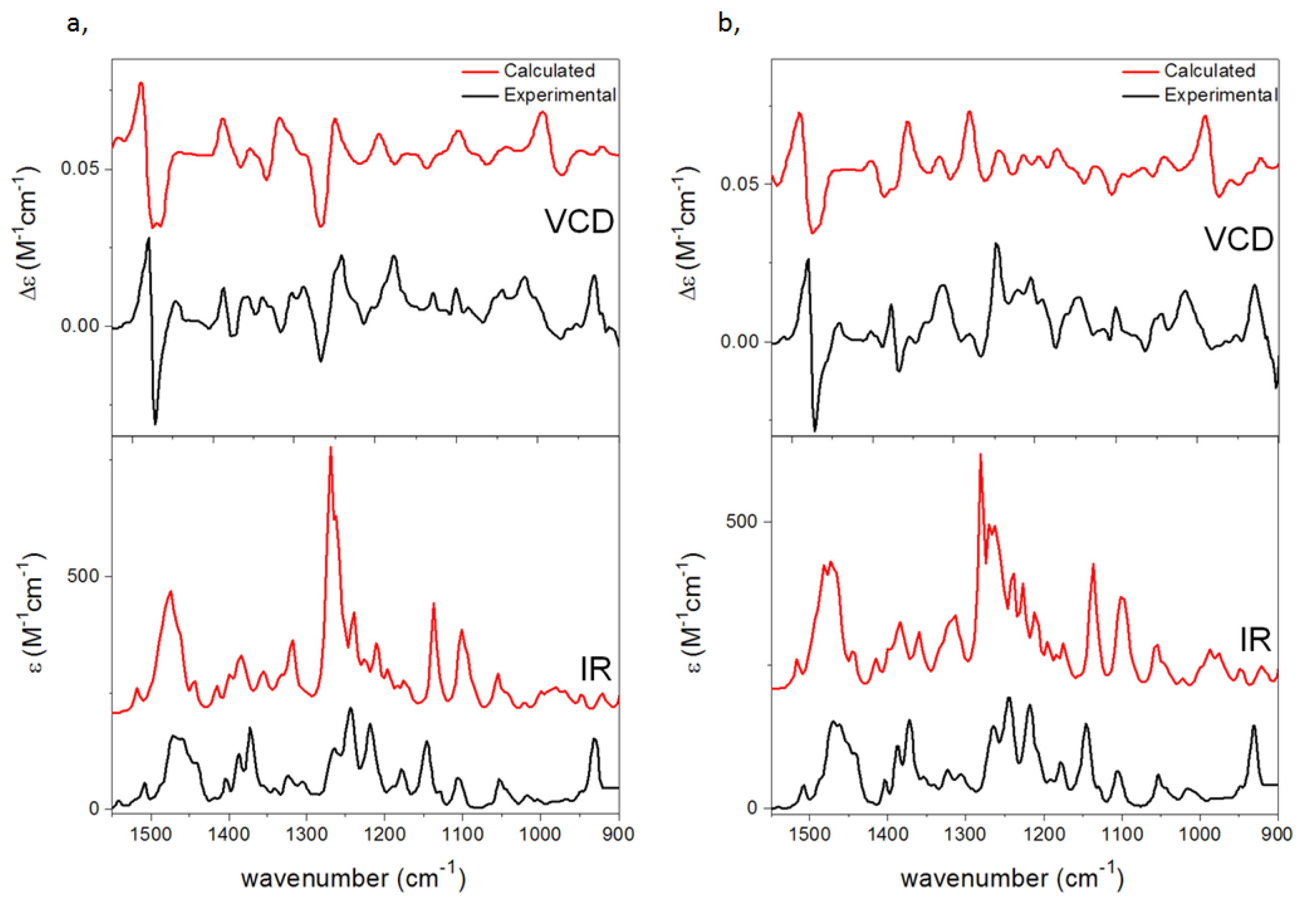{kind=link}
{kind=link}
Abstract
:1. Introduction
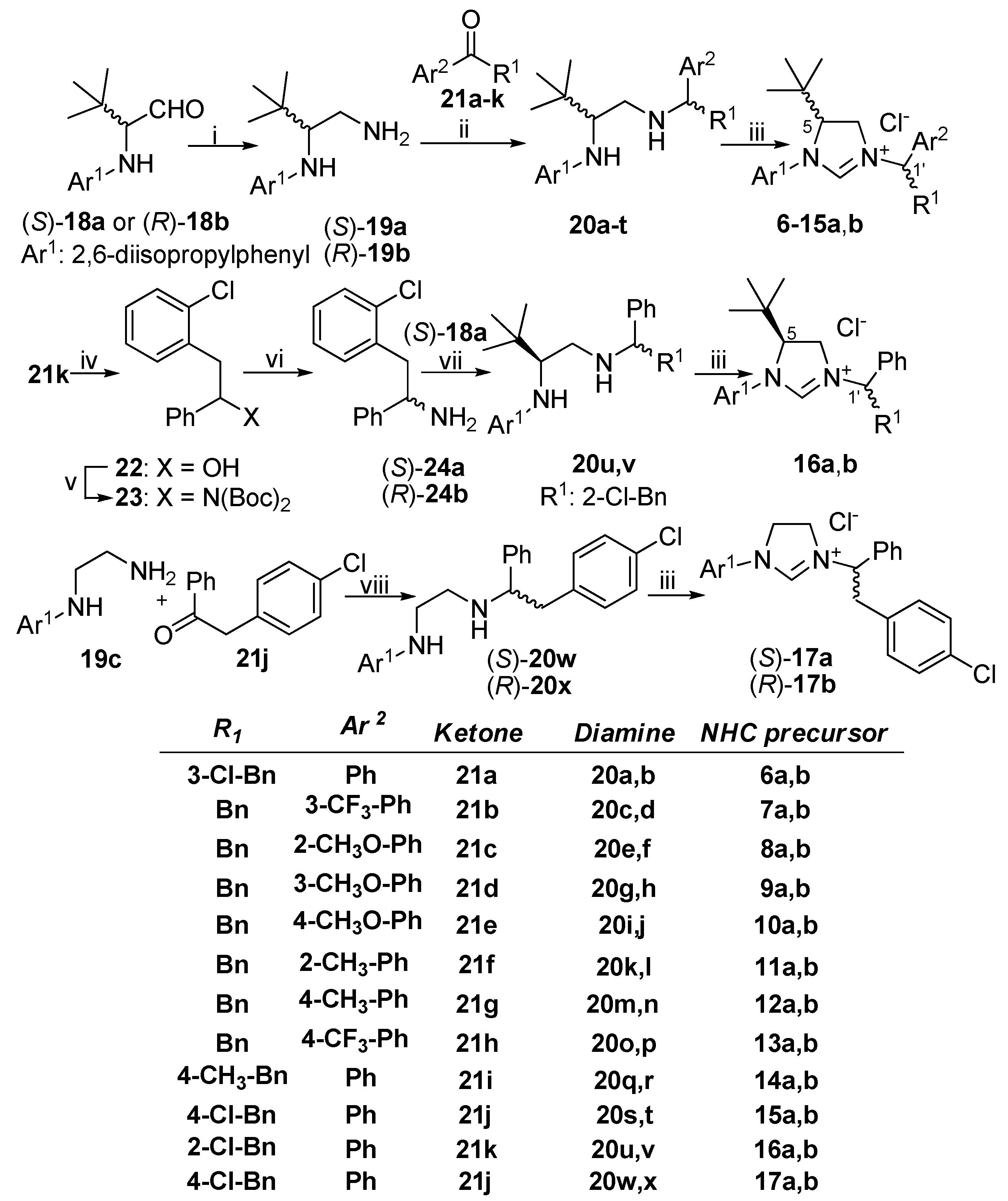
2. Results and Discussion
3. Materials and Methods
3.1. General Procedures
3.2. Computational Section
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janssen-Müller, D.; Schlepphorst, C.; Glorius, F. Privileged chiral N-heterocyclic carbene ligands for asymmetric transition-metal catalysis. Chem. Soc. Rev. 2017, 46, 4845–4854. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Candish, L.; Paul, D.; Glorius, F. N-Heterocyclic Carbenes in Asymmetric Hydrogenation. ACS Catal. 2016, 6, 5978–5988. [Google Scholar] [CrossRef]
- Paradiso, V.; Costabile, C.; Grisi, F. Ruthenium-based olefin metathesis catalysts with monodentate unsymmetrical NHC ligands. Beilstein J. Org. Chem. 2018, 14, 3122–3149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czerwiński, P.J.; Michalak, M. Synthetic Approaches to Chiral Non-C2-symmetric N-Heterocyclic Carbene Precursors. Synthesis 2019, 51, 1689–1714. [Google Scholar]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [Green Version]
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Arduengo, A.J. Looking for Stable Carbenes: The Difficulty in Starting Anew. Acc. Chem. Res. 1999, 32, 913–921. [Google Scholar] [CrossRef]
- Herrmann, W.A. N-Heterocyclic Carbenes: A New Concept in Organometallic Catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Perry, M.C.; Burgess, K. Chiral N-heterocyclic carbene-transition metal complexes in asymmetric catalysis. Tetrahedron Asymm. 2003, 14, 951–961. [Google Scholar] [CrossRef]
- Crudden, C.M.; Allen, D.P. Stability and reactivity of N-heterocyclic carbene complexes. Coord. Chem. Rev. 2004, 248, 2247–2273. [Google Scholar] [CrossRef]
- César, V.; Bellemin-Laponnaz, S.; Gade, L.H. Chiral N-heterocyclic carbenes as stereodirecting ligands in asymmetric catalysis. Chem. Soc. Rev. 2004, 33, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Díez-González, S.; Marion, N.; Nolan, S.P. N-Heterocyclic Carbenes in Late Transition Metal Catalysis. Chem. Rev. 2009, 109, 3612–3676. [Google Scholar] [CrossRef] [PubMed]
- Díez-González, S. N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools; RSC Publishing: London, UK, 2011. [Google Scholar]
- Wang, F.; Liu, L.; Wang, W.; Li, S.; Shi, M. Chiral NHC–metal-based asymmetric catalysis. Coord. Chem. Rev. 2012, 256, 804–853. [Google Scholar] [CrossRef]
- Nolan, S.P. N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Zhang, D.; Zi, G. N-heterocyclic carbene (NHC) complexes of group 4 transition metals. Chem. Soc. Rev. 2015, 44, 1898–1921. [Google Scholar] [CrossRef] [Green Version]
- Charra, V.; de Frémont, P.; Braunstein, P. Multidentate N-heterocyclic carbene complexes of the 3d metals: Synthesis, structure, reactivity and catalysis. Coord. Chem. Rev. 2017, 341, 53–176. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef]
- Fürstner, A.; Alcarazo, M.; César, V.; Lehmann, C.W. Convenient, scalable and flexible method for the preparation of imidazolium salts with previously inaccessible substitution patterns. Chem. Commun. 2006, 37, 2176–2178. [Google Scholar] [CrossRef] [Green Version]
- Hirano, K.; Urban, S.; Wang, C.; Glorius, F. A Modular Synthesis of Highly Substituted Imidazolium Salts. Org. Lett. 2009, 11, 1019–1022. [Google Scholar] [CrossRef]
- Sheehan, J.C.; Hara, T. Asymmetric thiazolium salt catalysis of the benzoin condensation. J. Org. Chem. 1974, 39, 1196–1199. [Google Scholar] [CrossRef]
- Enders, D.; Gielen, H.; Breuer, K. Immobilized Triazolium Salts as Precursors to Chiral Carbenes-Rhodium-Catalyzed Asymmetric Hydrosilylation as a First Test Reaction. Mol. Online 1998, 2, 105–109. [Google Scholar] [CrossRef]
- Bao, Y.; Kumagai, N.; Shibasaki, M. Design and synthesis of a bis(hydroxyphenyl)diamide bearing a pendant thiazolium unit; application to the catalytic asymmetric intramolecular Stetter reaction. Tetrahedron Asymm. 2014, 25, 1401–1408. [Google Scholar] [CrossRef] [Green Version]
- Fournier, P.-A.; Collins, S.K. A Highly Active Chiral Ruthenium-Based Catalyst for Enantioselective Olefin Metathesis. Organometallics 2007, 26, 2945–2949. [Google Scholar] [CrossRef]
- Savoie, J.; Stenne, B.; Collins, S.K. Improved Chiral Olefin Metathesis Catalysts: Increasing the Thermal and Solution Stability via Modification of a C1-Symmetrical N-Heterocyclic Carbene Ligand. Adv. Synth. Catal. 2009, 351, 1826–1832. [Google Scholar] [CrossRef]
- Paradiso, V.; Bertolasi, V.; Grisi, F. Novel Olefin Metathesis Ruthenium Catalysts Bearing Backbone-Substituted Unsymmetrical NHC Ligands. Organometallics 2014, 33, 5932–5935. [Google Scholar] [CrossRef]
- Paradiso, V.; Bertolasi, V.; Costabile, C.; Caruso, T.; Dąbrowski, M.; Grela, K.; Grisi, F. Expanding the Family of Hoveyda–Grubbs Catalysts Containing Unsymmetrical NHC Ligands. Organometallics 2017, 36, 3692–3708. [Google Scholar] [CrossRef]
- Seiders, T.J.; Ward, D.W.; Grubbs, R.H. Enantioselective Ruthenium-Catalyzed Ring-Closing Metathesis. Org. Lett. 2001, 3, 3225–3228. [Google Scholar] [CrossRef] [Green Version]
- Rix, D.; Labat, S.; Toupet, L.; Crévisy, C.; Mauduit, M. Design of Chiral Hydroxyalkyl- and Hydroxyarylazolinium Salts as New Chelating Diaminocarbene Ligand Precursors Devoted to Asymmetric Copper-Catalyzed Conjugate Addition. Eur. J. Inorg. Chem. 2009, 2009, 1989–1999. [Google Scholar] [CrossRef]
- Tiede, S.; Berger, A.; Schlesiger, D.; Rost, D.; Lühl, A.; Blechert, S. Highly Active Chiral Ruthenium-Based Metathesis Catalysts through a Monosubstitution in the N-Heterocyclic Carbene. Angew. Chem. Int. Ed. 2010, 49, 3972–3975. [Google Scholar] [CrossRef]
- Latham, C.M.; Blake, A.J.; Lewis, W.; Lawrence, M.; Woodward, S. Short Synthesis of Chiral 4-Substituted (S)-Imidazolinium Salts Bearing Sulfonates and Their Use in γ-Selective Reactions of Allylic Halides with Grignard Reagents. Eur. J. Org. Chem. 2012, 2012, 699–707. [Google Scholar] [CrossRef]
- Kannenberg, A.; Rost, D.; Eibauer, S.; Tiede, S.; Blechert, S. A Novel Ligand for the Enantioselective Ruthenium-Catalyzed Olefin Metathesis. Angew. Chem. Int. Ed. 2011, 50, 3299–3302. [Google Scholar] [CrossRef]
- Bonnet, L.G.; Douthwaite, R.E.; Kariuki, B.M. Synthesis of New Chiral N-Heterocyclic Carbene−Imine Ligands and Their Application to an Asymmetric Allylic Alkylation Reaction. Organometallics 2003, 22, 4187–4189. [Google Scholar] [CrossRef]
- Szabó, Z.; Timári, M.; Kassai, R.; Szokol, B.; Bényei, A.C.; Gáti, T.; Paczal, A.; Kotschy, A. Modular Synthesis of Chiral NHC Precursors and Their Silver and Gold Complexes. Organometallics 2020, 39, 3572–3589. [Google Scholar] [CrossRef]
- Yang, Q.; Fusè, M.; Bloino, J.; Barone, V. Interplay of stereo-electronic, vibronic and environmental effects in tuning the chiroptical properties of an Ir(III) cyclometalated N-heterocyclic carbene. Spectrochim. Acta A 2021, 254, 119631. [Google Scholar] [CrossRef] [PubMed]
- Reimers, T.; Haberhauer, G.; Benkhäuser, C.; Schmidtchen, F.P.; Lützen, A.; Lüning, U. Enantiopure Chiral Concave N-Heterocyclic Carbene Precursors. Eur. J. Org. Chem. 2013, 2013, 7556–7566. [Google Scholar] [CrossRef]
- Yi, H.; Osten, K.M.; Levchenko, T.I.; Veinot, A.J.; Aramaki, Y.; Ooi, T.; Nambo, M.; Crudden, C.M. Synthesis and enantioseparation of chiral Au13 nanoclusters protected by bis-N-heterocyclic carbene ligands. Chem. Sci. 2021, 12, 10436–10440. [Google Scholar] [CrossRef]
- Avello, M.G.; de la Torre, M.C.; Sierra, M.A.; Gornitzka, H.; Hemmert, C. Central (S) to Central (M = Ir, Rh) to Planar (Metallocene, M = Fe, Ru) Chirality Transfer Using Sulfoxide-Substituted Mesoionic Carbene Ligands: Synthesis of Bimetallic Planar Chiral Metallocenes. Chem. Eur. J. 2019, 25, 13344–13353. [Google Scholar] [CrossRef]
- Ding, W.-Y.; Yan, Y.-M.; Meng, X.-H.; Nafie, A.L.; Xu, T.; Dukor, R.K.; Qin, H.-B.; Cheng, Y.-X. Isolation, Total Synthesis, and Absolute Configuration Determination of Renoprotective Dimeric N-Acetyldopamine–Adenine Hybrids from the Insect Aspongopus chinensis. Org. Lett. 2020, 22, 5726–5730. [Google Scholar] [CrossRef]
- Hopmann, K.H.; Šebestík, J.; Novotná, J.; Stensen, W.; Urbanová, M.; Svenson, J.; Svendsen, J.S.; Bouř, P.; Ruud, K. Determining the Absolute Configuration of Two Marine Compounds Using Vibrational Chiroptical Spectroscopy. J. Org. Chem. 2012, 77, 858–869. [Google Scholar] [CrossRef]
- Polavarapu, P.L.; Santoro, E. Vibrational optical activity for structural characterization of natural products. Nat. Prod. Rep. 2020, 37, 1661–1699. [Google Scholar] [CrossRef]
- Mándi, A.; Kurtán, T. Applications of OR/ECD/VCD to the structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 889–918. [Google Scholar] [CrossRef]
- Qiu, S.; de Gussem, E.; Tehrani, K.A.; Sergeyev, S.; Bultinck, P.; Herrebout, W. Stereochemistry of the Tadalafil Diastereoisomers: A Critical Assessment of Vibrational Circular Dichroism, Electronic Circular Dichroism, and Optical Rotatory Dispersion. J. Med. Chem. 2013, 56, 8903–8914. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Hammam, M.A.S.; Taniguchi, T.; Suga, Y.; Monde, K. What Is the True Structure of D609, a Widely Used Lipid Related Enzyme Inhibitor? Org. Lett. 2016, 18, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hopmann, K.H.; Hudecová, J.; Isaksson, J.; Novotná, J.; Stensen, W.; Andrushchenko, V.; Urbanová, M.; Svendsen, J.-S.; Bouř, P.; et al. Determination of Absolute Configuration and Conformation of a Cyclic Dipeptide by NMR and Chiral Spectroscopic Methods. J. Phys. Chem. A 2013, 117, 1721–1736. [Google Scholar] [CrossRef] [PubMed]
- Koenis, M.A.J.; Chibueze, C.S.; Jinks, M.A.; Nicu, V.P.; Visscher, L.; Goldup, S.M.; Buma, W.J. Vibrational circular dichroism spectroscopy for probing the expression of chirality in mechanically planar chiral rotaxanes. Chem. Sci. 2020, 11, 8469–8475. [Google Scholar] [CrossRef]
- Nakahashi, A.; Taniguchi, T.; Miura, N.; Monde, K. Stereochemical Studies of Sialic Acid Derivatives by Vibrational Circular Dichroism. Org. Lett. 2007, 9, 4741–4744. [Google Scholar] [CrossRef]
- Nakahashi, A.; Siddegowda, A.K.C.; Hammam, M.A.S.; Gowda, S.G.B.; Murai, Y.; Monde, K. Stereochemical Study of Sphingosine by Vibrational Circular Dichroism. Org. Lett. 2016, 18, 2327–2330. [Google Scholar] [CrossRef]
- Demarque, D.P.; Pinho, D.R.; Lopes, N.P.; Merten, C. Revisiting empirical rules for the determination of the absolute configuration of cascarosides and other (ox-)anthrones. Chirality 2018, 30, 432–438. [Google Scholar] [CrossRef]
- Demarque, D.P.; Heinrich, S.; Schulz, F.; Merten, C. Sensitivity of VCD spectroscopy for small structural and stereochemical changes of macrolide antibiotics. Chem. Commun. 2020, 56, 10926–10929. [Google Scholar] [CrossRef]
- Kurtán, T.; Antus, S.; Pescitelli, G. Electronic CD of benzene and other aromatic chromophores for determination of absolute configuration. In Comprehensive Chiroptical Spectroscopy: Applications in Stereochemical Analysis of Synthetic Compounds, Natural Products, And Biomolecules; Berova, N., Polavarapu, P.L., Nakanishi, K., Woody, R.W., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; Volume 2, pp. 73–114. [Google Scholar]
- Covington, C.L.; Polavarapu, P.L. CDSpecTech: A single software suite for multiple chiroptical spectroscopic analyses. Chirality 2017, 29, 178–192. [Google Scholar] [CrossRef]
- Debie, E.; de Gussem, E.; Dukor, R.K.; Herrebout, W.; Nafie, L.A.; Bultinck, P. A confidence level algorithm for the determination of absolute configuration using vibrational circular dichroism or raman optical activity. ChemPhysChem 2011, 12, 1542–1549. [Google Scholar] [CrossRef] [Green Version]
- MacroModel; Schrödinger LLC.: New York, NY, USA, 2015. Available online: https://www.schrodinger.com/products/macromodel (accessed on 23 February 2022).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Chai, J.D.; Head-Gordon, M.J. Systematic optimization of long-range corrected hybrid density functionals. Chem. Phys. 2008, 128, 84106. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Harada, N. ECD cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef] [PubMed]
- El-Kashef, D.H.; Daletos, G.; Plenker, M.; Hartmann, R.; Mándi, A.; Kurtán, T.; Weber, H.; Lin, W.; Ancheeva, E.; Proksch, P. Polyketides and a Dihydroquinolone Alkaloid from a Marine-Derived Strain of the Fungus Metarhizium marquandii. J. Nat. Prod. 2019, 82, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Covington, C.L.; Polavarapu, P.L. CDspecTech: Computer Programs for Calculating Similarity Measures of Comparison between Experimental and Calculated Dissymmetry Factors and Circular Intensity Differentials. Version 22.0. 2017. Available online: https://sites.google.com/site/cdspectech1/ (accessed on 23 February 2022).
- Varetto, U. MOLEKEL 5.4; Swiss National Supercomputing Centre: Manno, Switzerland, 2009. [Google Scholar]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szabó, Z.; Paczal, A.; Kovács, T.; Mándi, A.; Kotschy, A.; Kurtán, T. Synthesis and Vibrational Circular Dichroism Analysis of N-Heterocyclic Carbene Precursors Containing Remote Chirality Centers. Int. J. Mol. Sci. 2022, 23, 3471. https://doi.org/10.3390/ijms23073471
Szabó Z, Paczal A, Kovács T, Mándi A, Kotschy A, Kurtán T. Synthesis and Vibrational Circular Dichroism Analysis of N-Heterocyclic Carbene Precursors Containing Remote Chirality Centers. International Journal of Molecular Sciences. 2022; 23(7):3471. https://doi.org/10.3390/ijms23073471
Chicago/Turabian StyleSzabó, Zita, Attila Paczal, Tibor Kovács, Attila Mándi, Andras Kotschy, and Tibor Kurtán. 2022. "Synthesis and Vibrational Circular Dichroism Analysis of N-Heterocyclic Carbene Precursors Containing Remote Chirality Centers" International Journal of Molecular Sciences 23, no. 7: 3471. https://doi.org/10.3390/ijms23073471
APA StyleSzabó, Z., Paczal, A., Kovács, T., Mándi, A., Kotschy, A., & Kurtán, T. (2022). Synthesis and Vibrational Circular Dichroism Analysis of N-Heterocyclic Carbene Precursors Containing Remote Chirality Centers. International Journal of Molecular Sciences, 23(7), 3471. https://doi.org/10.3390/ijms23073471