
Immunotherapy for Cervical Cancer: Are We Ready for Prime Time?



 and
and 
Abstract
:1. Introduction
2. The Rationale for Immunotherapy in Cervical Cancer
3. Immune Checkpoint Inhibitors (ICI): Mechanism of Action
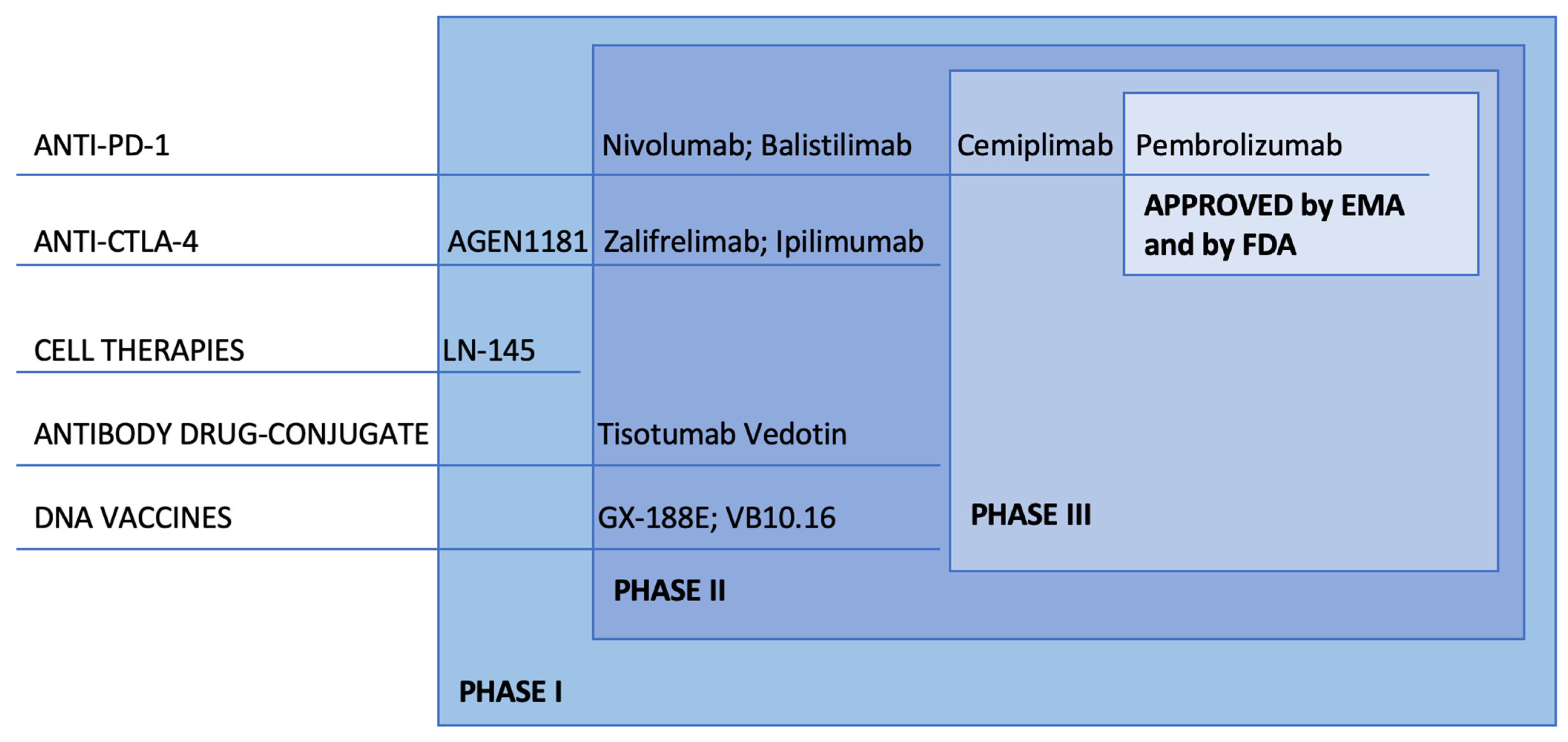
4. ICI: Clinical Development
4.1. Combination of PD-L1 Inhibition and CTLA-4 Inhibition
4.2. Combination of Immunotherapy and Antiangiogenic Agents
4.3. Combination of Immunotherapy and Radiation: The Abscopal Effect
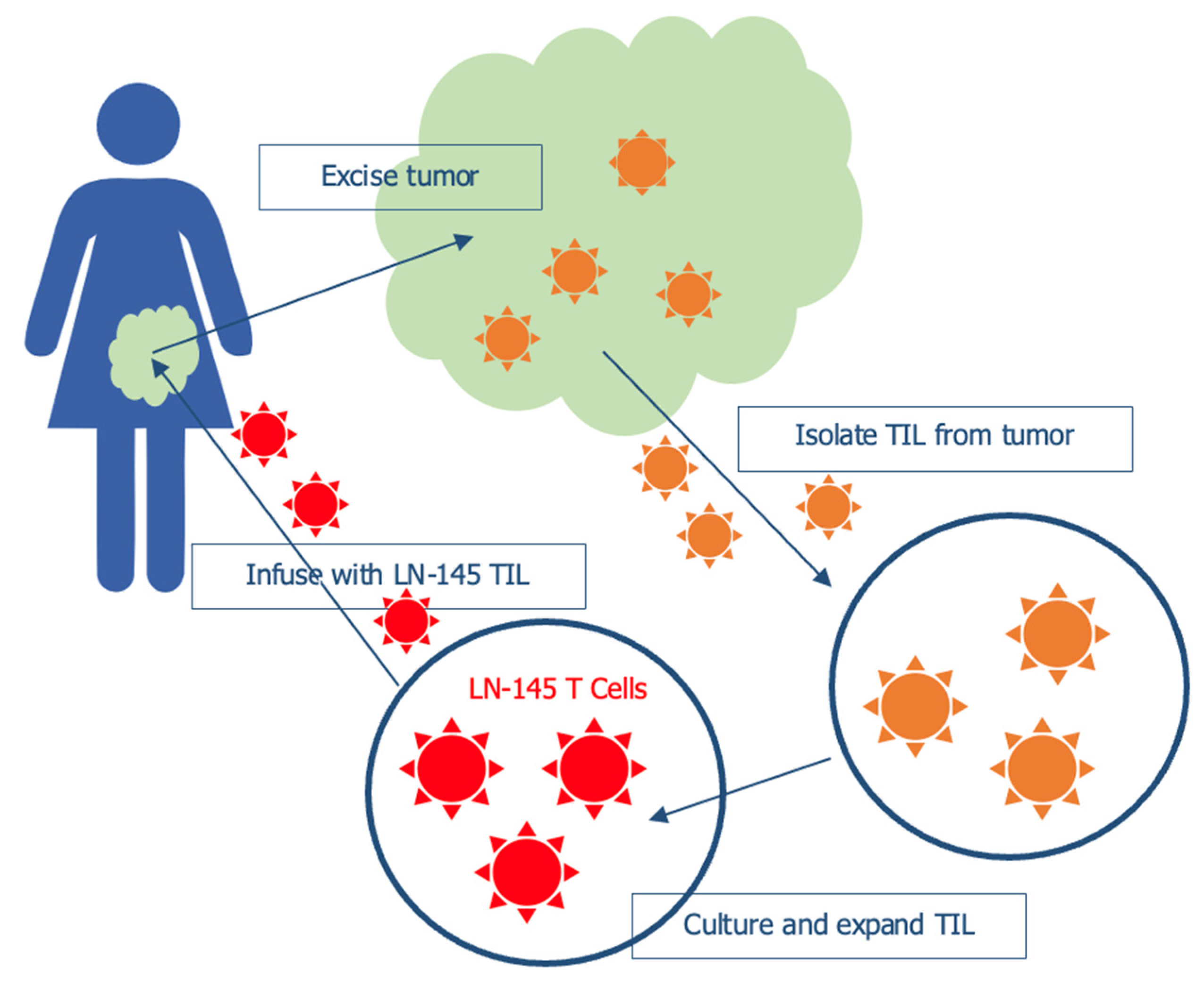
5. Tumor Infiltrating Lymphocytes (TILs)
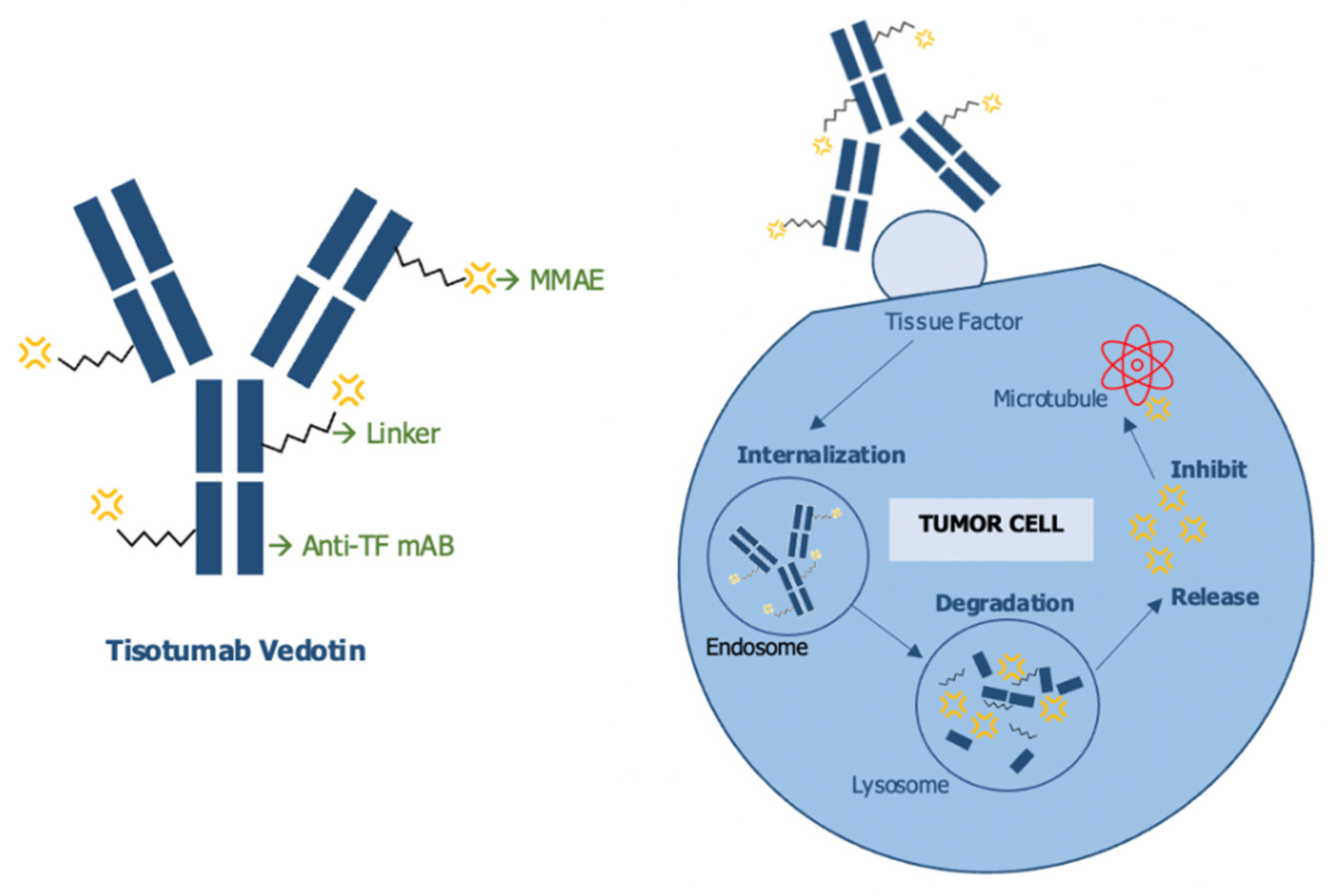
6. Antibody-Drug Conjugate (ADC): Tisotumab Vedotin
7. Vaccines
8. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [CrossRef] [PubMed]
- Pfaendler, K.S.; Tewari, K.S. Changing paradigms in the systemic treatment of advanced cervical cancer. Am. J. Obstet. Gynecol. 2016, 214, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussios, S.; Seraj, E.; Zarkavelis, G.; Petrakis, D.; Kollas, A.; Kafantari, A.; Assi, A.; Tatsi, K.; Pavlidis, N.; Pentheroudakis, G. Management of patients with recurrent/advanced cervical cancer beyond first line platinum regimens: Where do we stand? A literature review. Crit. Rev. Oncol. Hematol. 2016, 108, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Carinelli, S.; Colombo, A.; Marini, C.; Rollo, D.; Sessa, C. Cervical cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23 (Suppl. S7), vii27–vii32. [Google Scholar] [CrossRef]
- Whitney, C.W.; Sause, W.; Bundy, B.N.; Malfetano, J.H.; Hannigan, E.V.; Fowler, W.C., Jr.; Clarke-Pearson, D.L.; Liao, S.Y. Randomized comparison of fluorouracil plus cisplatin versus hydroxyurea as an adjunct to radiation therapy in stage IIB-IVA carcinoma of the cervix with negative para-aortic lymph nodes: A Gynecologic Oncology Group and Southwest Oncology Group study. J. Clin. Oncol 1999, 17, 1339–1348. [Google Scholar] [CrossRef] [Green Version]
- Ferrandina, G.; Palluzzi, E.; Gallotta, V.; Gambacorta, M.A.; Autorino, R.; Turco, L.C.; Macchia, G.; Cosentino, F.; Gui, B.; Mattoli, M.V.; et al. Neo-adjuvant platinum-based chemotherapy followed by chemoradiation and radical surgery in locally advanced cervical cancer (Lacc) patients: A phase II study. Eur. J. Surg. Oncol. 2018, 44, 1062–1068. [Google Scholar] [CrossRef]
- Tewari, K.S.; Sill, M.W.; Long, H.J., III; Penson, R.T.; Huang, H.; Ramondetta, L.M.; Landrum, L.M.; Oaknin, A.; Reid, T.J.; Leitao, M.M.; et al. Improved survival with bevacizumab in advanced cervical cancer. N. Engl. J. Med. 2014, 370, 734–743. [Google Scholar] [CrossRef] [Green Version]
- McLachlan, J.; Boussios, S.; Okines, A.; Glaessgen, D.; Bodlar, S.; Kalaitzaki, R.; Taylor, A.; Lalondrelle, S.; Gore, M.; Kaye, S.; et al. The Impact of Systemic Therapy Beyond First-line Treatment for Advanced Cervical Cancer. Clin Oncol. 2017, 29, 153–160. [Google Scholar] [CrossRef]
- SEER Cancer Stat Facts: Cervical Cancer. Available online: http://seer.cancer.gov/statfacts/html/cervix.html (accessed on 20 November 2021).
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Klempner, S.J.; Fabrizio, D.; Bane, S.; Reinhart, M.; Peoples, T.; Ali, S.M.; Sokol, E.S.; Frampton, G.; Schrock, A.B.; Anhorn, R.; et al. Tumor Mutational Burden as a Predictive Biomarker for Response to Immune Checkpoint Inhibitors: A Review of Current Evidence. Oncologist 2020, 25, e147–e159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazo, P.A. The molecular genetics of cervical carcinoma. Br. J. Cancer 1999, 80, 2008–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sun, H.; Zeng, Q.; Guo, X.J.; Wang, H.; Liu, H.H.; Dong, Z.Y. HPV-positive status associated with inflamed immune microenvironment and improved response to anti-PD-1 therapy in head and neck squamous cell carcinoma. Sci. Rep. 2019, 9, 13404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezache, L.; Paniccia, B.; Nyinawabera, A.; Nuovo, G.J. Enhanced expression of PD L1 in cervical intraepithelial neoplasia and cervical cancers. Mod. Pathol. 2015, 28, 1594–1602. [Google Scholar] [CrossRef]
- Lyford-Pike, S.; Peng, S.; Young, G.D.; Taube, J.M.; Westra, W.H.; Akpeng, B.; Bruno, T.C.; Richmon, J.D.; Wang, H.; Bishop, J.A.; et al. Evidence for a role of the PD-1:PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Res. 2013, 73, 1733–1741. [Google Scholar] [CrossRef] [Green Version]
- Enwere, E.K.; Kornaga, E.N.; Dean, M.; Koulis, T.A.; Phan, T.; Kalantarian, M.; Köbel, M.; Ghatage, P.; Magliocco, A.M.; Lees-Miller, S.P.; et al. Expression of PD-L1 and presence of CD8-positive T cells in pre-treatment specimens of locally advanced cervical cancer. Mod. Pathol. 2017, 30, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Duensing, S.; Münger, K. Mechanisms of genomic instability in human cancer: Insights from studies with human papillomavirus oncoproteins. Int. J. Cancer 2004, 109, 157–162. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef]
- Hino, R.; Kabashima, K.; Kato, Y.; Yagi, H.; Nakamura, M.; Honjo, T.; Okazaki, T.; Tokura, Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010, 116, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [Green Version]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Nasti, T.H.; Sharpe, A.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauricio, D.; Zeybek, B.; Tymon-Rosario, J.; Harold, J.; Santin, A.D. Immunotherapy in Cervical Cancer. Curr. Oncol. Rep. 2021, 23, 61. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.C.; Ros, W.; Delord, J.P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- Naumann, R.W.; Hollebecque, A.; Meyer, T.; Devlin, M.J.; Oaknin, A.; Kerger, J.; López-Picazo, J.M.; Machiels, J.P.; Delord, J.P.; Evans, T.R.J.; et al. Safety and Efficacy of Nivolumab Monotherapy in Recurrent or Metastatic Cervical, Vaginal, or Vulvar Carcinoma: Results From the Phase I/II CheckMate 358 Trial. J. Clin. Oncol. 2019, 37, 2825–2834. [Google Scholar] [CrossRef]
- Naumann, R.W.; Oaknin, A.; Meyer, T.; Lopez-Picazo, J.M.; Lao, C. LBA62- Efficacy and safety of nivolumab (Nivo) + ipilimumab (Ipi) in patients (pts) with recurrent/metastatic (R/M) cervical cancer: Results from CheckMate 358. Ann. Oncol. 2020, 30, v898–v899. [Google Scholar] [CrossRef]
- Callahan, M.K.; Odunsi, K.; Wolchok, J.D. Phase 1 study to evaluate the safety and tolerability of MEDI4736 (durvalumab, DUR) + tremelimumab (TRE) in patients with advanced solid tumors. Am. Soc. Clin. Oncol. 2017, 35, 3069. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Oaknin, A.; Monk, B.J.; Leary, A. Single-agent anti-PD-1 balstilimab or in combination with anti-CTLA-4 zalifrelimab for recurrent/metastatic (R/M) cervical cancer (CC): Preliminary results of two independent phase II trials. Ann. Oncol. 2020, 31, S1164–S1165. [Google Scholar] [CrossRef]
- Dobbs, S.P.; Hewett, P.W.; Johnson, I.R.; Carmichael, J.; Murray, J.C. Angiogenesis is associated with vascular endothelial growth factor expression in cervical intraepithelial neoplasia. Br. J. Cancer 1997, 76, 1410–1415. [Google Scholar] [CrossRef] [Green Version]
- Cooper, R.A.; Wilks, D.P.; Logue, J.P.; Davidson, S.E.; Hunter, R.D.; Roberts, S.A.; West, C.M. High tumor angiogenesis is associated with poorer survival in carcinoma of the cervix treated with radiotherapy. Clin. Cancer Res. 1998, 4, 2795–2800. [Google Scholar]
- Zhang, L.; Chen, Y.; Li, F.; Bao, L.; Liu, W. Atezolizumab and Bevacizumab Attenuate Cisplatin Resistant Ovarian Cancer Cells Progression Synergistically via Suppressing Epithelial-Mesenchymal Transition. Front. Immunol. 2019, 10, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grau, J.F.; Farinas-Madrid, L.; Oaknin, A. A randomized phase III trial of platinum chemotherapy plus paclitaxel with bevacizumab and atezolizumab versus platinum chemotherapy plus paclitaxel and bevacizumab in metastatic (stage IVB), persistent, or recurrent carcinoma of the cervix: The BEATcc study (ENGOT-Cx10/GEICO 68-C/JGOG1084/GOG-3030). Int. J. Gynecol. Cancer 2020, 30, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Lan, C.; Shen, J.; Wang, Y.; Li, J.; Liu, Z.; He, M.; Cao, X.; Ling, J.; Huang, J.; Zheng, M.; et al. Camrelizumab Plus Apatinib in Patients With Advanced Cervical Cancer (CLAP): A Multicenter, Open-Label, Single-Arm, Phase II Trial. J. Clin. Oncol. 2020, 38, 4095–4106. [Google Scholar] [CrossRef]
- Zhao, X.; Shao, C. Radiotherapy-Mediated Immunomodulation and Anti-Tumor Abscopal Effect Combining Immune Checkpoint Blockade. Cancers 2020, 12, 2762. [Google Scholar] [CrossRef] [PubMed]
- Trommer, M.; Yeo, S.Y.; Persigehl, T.; Bunck, A.; Grüll, H.; Schlaak, M.; Theurich, S.; von Bergwelt-Baildon, M.; Morgenthaler, J.; Herter, J.M.; et al. Abscopal Effects in Radio-Immunotherapy-Response Analysis of Metastatic Cancer Patients With Progressive Disease Under Anti-PD-1 Immune Checkpoint Inhibition. Front. Pharmacol. 2019, 10, 511. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.; Matulonis, U. Immunotherapy and radiation combinatorial trials in gynecologic cancer: A potential synergy? Gynecol. Oncol. 2019, 154, 236–245. [Google Scholar] [CrossRef]
- Mittica, G.; Capellero, S.; Genta, S.; Cagnazzo, C.; Aglietta, M.; Sangiolo, D.; Valabrega, G. Adoptive immunotherapy against ovarian cancer. J. Ovarian Res. 2016, 9, 30. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.F.; Lee, C.N.; Chang, M.C.; Su, Y.N.; Chen, C.A.; Hsieh, C.Y. Antigen-specific CD8+ T lymphocytes generated from a DNA vaccine control tumors through the Fas-FasL pathway. Mol. Ther. 2005, 12, 960–968. [Google Scholar] [CrossRef]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef]
- Martins, P.R.; Machado, C.M.T.; Coxir, S.A.; de Oliveira, A.J.; Moreira, T.B.; Campos, L.S.; Alcântara, R.; de Paula, S.O.C.; de Oliveira Salles, P.G.; Gollob, K.J.; et al. Cervical cancer patients that respond to chemoradiation therapy display an intense tumor infiltrating immune profile before treatment. Exp. Mol. Pathol. 2019, 111, 104314. [Google Scholar] [CrossRef] [PubMed]
- Heeren, A.M.; van Luijk, I.F.; Lakeman, J.; Pocorni, N.; Kole, J.; de Menezes, R.X.; Kenter, G.G.; Bosse, T.; de Kroon, C.D.; Jordanova, E.S. Neoadjuvant cisplatin and paclitaxel modulate tumor-infiltrating T cells in patients with cervical cancer. Cancer Immunol. Immunother. 2019, 68, 1759–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorta-Estremera, S.; Colbert, L.E.; Nookala, S.S.; Yanamandra, A.V.; Yang, G.; Delgado, A.; Mikkelson, M.; Eifel, P.; Jhingran, A.; Lilie, L.L.; et al. Kinetics of Intratumoral Immune Cell Activation During Chemoradiation for Cervical Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, Y.; Yoshimoto, Y.; Murata, K.; Noda, S.E.; Ando, K.; Ebara, T.; Okonogi, N.; Kaminuma, T.; Yamada, S.; Ikota, H.; et al. Treatment outcomes of patients with adenocarcinoma of the uterine cervix after definitive radiotherapy and the prognostic impact of tumor-infiltrating CD8+ lymphocytes in pre-treatment biopsy specimens: A multi-institutional retrospective study. J. Radiat. Res. 2020, 61, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Stevanović, S.; Draper, L.M.; Langhan, M.M.; Campbell, T.E.; Kwong, M.L.; Wunderlich, J.R.; Dudley, M.E.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J. Clin. Oncol. 2015, 33, 1543–1550. [Google Scholar] [CrossRef] [Green Version]
- Jazaeri, A.A.; Zsiros, E.; Monk, B.J. Safety and efficacy of adoptive cell transfer using autologous tumor infiltrating lymphocytes (LN-145) for treatment of recurrent, metastatic, or persistent cervical carcinoma. Am. Soc. Clin. Oncol. 2019, 37, 2538. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, A.X.J.; Chen, G.; Wu, Y.; Gu, W. Prognostic and therapeutic TILs of cervical cancer-Current advances and future perspectives. Mol. Ther. Oncolytics 2021, 22, 410–430. [Google Scholar]
- Ruf, W.; Disse, J.; Carneiro-Lobo, T.C.; Yokota, N.; Schaffner, F. Tissue factor and cell signalling in cancer progression and thrombosis. J. Thromb. Haemost. 2011, 9 (Suppl. S1), 306–315. [Google Scholar] [CrossRef] [Green Version]
- Breij, E.C.; de Goeij, B.E.; Verploegen, S.; Schuurhuis, D.H.; Amirkhosravi, A.; Francis, J.; Miller, V.B.; Houtkamp, M.; Bleeker, W.K.; Satijn, D.; et al. An antibody-drug conjugate that targets tissue factor exhibits potent therapeutic activity against a broad range of solid tumors. Cancer Res. 2014, 74, 1214–1226. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Concin, N.; Vergote, I.; de Bono, J.S.; Slomovitz, B.M.; Drew, Y.; Arkenau, H.T.; Machiels, J.P.; Spicer, J.F.; Jones, R.; et al. Tisotumab Vedotin in Previously Treated Recurrent or Metastatic Cervical Cancer. Clin. Cancer Res. 2020, 26, 1220–1228. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.L.; Lorusso, D.; Gennigens, C.; González-Martín, A.; Randall, L.; Cibula, D.; Lund, B.; Woelber, L.; Pignata, S.; Forget, F.; et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG-3023/ENGOT-cx6): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021, 22, 609–619. [Google Scholar] [CrossRef]
- Arbyn, M.; Kyrgiou, M.; Simoens, C.; Raifu, A.O.; Koliopoulos, G.; Martin-Hirsch, P.; Prendiville, W.; Paraskevaidis, E. Perinatal mortality and other severe adverse pregnancy outcomes associated with treatment of cervical intraepithelial neoplasia: Meta-analysis. BMJ 2008, 337, a1284. [Google Scholar] [CrossRef] [Green Version]
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Oldak, M.; Tolzmann, L.; Wnorowski, A.; Podgórska, M.J.; Silling, S.; Lin, R.; Hiscott, J.; Müller, C.S.; Vogt, T.; Smola, H.; et al. Differential regulation of human papillomavirus type 8 by interferon regulatory factors 3 and 7. J. Virol. 2011, 85, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Smola, S. Immunopathogenesis of HPV-Associated Cancers and Prospects for Immunotherapy. Viruses 2017, 9, 254. [Google Scholar] [CrossRef]
- Sperling, T.; Ołdak, M.; Walch-Rückheim, B.; Wickenhauser, C.; Doorbar, J.; Pfister, H.; Malejczyk, M.; Majewski, S.; Keates, A.C.; Smola, S. Human papillomavirus type 8 interferes with a novel C/EBPβ-mediated mechanism of keratinocyte CCL20 chemokine expression and Langerhans cell migration. PLoS Pathog 2012, 8, e1002833. [Google Scholar] [CrossRef]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Giraudo, E.; Inoue, M.; Hanahan, D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Investig. 2004, 114, 623–633. [Google Scholar] [CrossRef]
- Hess, S.; Smola, H.; Sandaradura De Silva, U.; Hadaschik, D.; Kube, D.; Baldus, S.E.; Flucke, U.; Pfister, H. Loss of IL-6 receptor expression in cervical carcinoma cells inhibits autocrine IL-6 stimulation: Abrogation of constitutive monocyte chemoattractant protein-1 production. J. Immunol. 2000, 165, 1939–1948. [Google Scholar] [CrossRef] [Green Version]
- Kirma, N.; Hammes, L.S.; Liu, Y.G.; Nair, H.B.; Valente, P.T.; Kumar, S.; Flowers, L.C.; Tekmal, R.R. Elevated expression of the oncogene c-fms and its ligand, the macrophage colony-stimulating factor-1, in cervical cancer and the role of transforming growth factor-beta1 in inducing c-fms expression. Cancer Res. 2007, 67, 1918–1926. [Google Scholar] [CrossRef] [Green Version]
- Pander, J.; Heusinkveld, M.; van der Straaten, T.; Jordanova, E.S.; Baak-Pablo, R.; Gelderblom, H.; Morreau, H.; van der Burg, S.H.; Guchelaar, H.J.; van Hall, T. Activation of tumor-promoting type 2 macrophages by EGFR-targeting antibody cetuximab. Clin Cancer Res. 2011, 17, 5668–5673. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, A.; Nonn, M.; Sehr, P.; Schreckenberger, C.; Pawlita, M.; Dürst, M.; Schneider, A.; Kaufmann, A.M. Dendritic cell-based tumor vaccine for cervical cancer II: Results of a clinical pilot study in 15 individual patients. J. Cancer Res. Clin. Oncol. 2003, 129, 521–530. [Google Scholar] [CrossRef]
- Santin, A.D.; Bellone, S.; Palmieri, M.; Zanolini, A.; Ravaggi, A.; Siegel, E.R.; Roman, J.J.; Pecorelli, S.; Cannon, M.J. Human papillomavirus type 16 and 18 E7-pulsed dendritic cell vaccination of stage IB or IIA cervical cancer patients: A phase I escalating-dose trial. J. Virol. 2008, 82, 1968–1979. [Google Scholar] [CrossRef] [Green Version]
- Rahma, O.E.; Herrin, V.E.; Ibrahim, R.A.; Toubaji, A.; Bernstein, S.; Dakheel, O.; Steinberg, S.M.; Abu Eid, R.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Pre-immature dendritic cells (PIDC) pulsed with HPV16 E6 or E7 peptide are capable of eliciting specific immune response in patients with advanced cervical cancer. J. Transl. Med. 2014, 12, 353. [Google Scholar] [CrossRef] [Green Version]
- Kenter, G.G.; Welters, M.J.; Valentijn, A.R.; Lowik, M.J.; Berends-van der Meer, D.M.; Vloon, A.P.; Essahsah, F.; Fathers, L.M.; Offringa, R.; Drijfhout, J.W.; et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N. Engl. J. Med. 2009, 361, 1838–1847. [Google Scholar] [CrossRef] [Green Version]
- Petit, R.G.; Mehta, A.; Jain, M.; Gupta, S.; Nagarkar, R.; Kumar, V.; Premkumar, S.; Neve, R.; John, S.; Basu, P. ADXS11-001 immunotherapy targeting HPV-E7: Final results from a Phase II study in Indian women with recurrent cervical cancer. J. Immunother. Cancer 2014, 2, P92. [Google Scholar] [CrossRef] [Green Version]
- Kawana, K.; Adachi, K.; Kojima, S.; Taguchi, A.; Tomio, K.; Yamashita, A.; Nishida, H.; Nagasaka, K.; Arimoto, T.; Yokoyama, T.; et al. Oral vaccination against HPV E7 for treatment of cervical intraepithelial neoplasia grade 3 (CIN3) elicits E7-specific mucosal immunity in the cervix of CIN3 patients. Vaccine 2014, 32, 6233–6239. [Google Scholar] [CrossRef]
- Valdez Graham, V.; Sutter, G.; José, M.V.; García-Carranca, A.; Erfle, V.; Moreno Mendoza, N.; Merchant, H.; Rosales, R. Human tumor growth is inhibited by a vaccinia virus carrying the E2 gene of bovine papillomavirus. Cancer 2000, 88, 1650–1662. [Google Scholar] [CrossRef]
- Rosales, R.; López-Contreras, M.; Rosales, C.; Magallanes-Molina, J.R.; Gonzalez-Vergara, R.; Arroyo-Cazarez, J.M.; Ricardez-Arenas, A.; Del Follo-Valencia, A.; Padilla-Arriaga, S.; Guerrero, M.V.; et al. Regression of human papillomavirus intraepithelial lesions is induced by MVA E2 therapeutic vaccine. Hum. Gene Ther. 2014, 25, 1035–1049. [Google Scholar] [CrossRef]
- Kaufmann, A.M.; Stern, P.L.; Rankin, E.M.; Sommer, H.; Nuessler, V.; Schneider, A.; Adams, M.; Onon, T.S.; Bauknecht, T.; Wagner, U.; et al. Safety and immunogenicity of TA-HPV, a recombinant vaccinia virus expressing modified human papillomavirus (HPV)-16 and HPV-18 E6 and E7 genes, in women with progressive cervical cancer. Clin. Cancer Res. 2002, 8, 3676–3685. [Google Scholar]
- Brun, J.L.; Dalstein, V.; Leveque, J.; Mathevet, P.; Raulic, P.; Baldauf, J.J.; Scholl, S.; Huynh, B.; Douvier, S.; Riethmuller, D.; et al. Regression of high-grade cervical intraepithelial neoplasia with TG4001 targeted immunotherapy. Am. J. Obstet. Gynecol. 2011, 204, 169.e1–169.e8. [Google Scholar] [CrossRef] [PubMed]
- Barra, F.; Della Corte, L.; Noberasco, G.; Foreste, V.; Riemma, G.; Di Filippo, C.; Bifulco, G.; Orsi, A.; Icardi, G.; Ferrero, S. Advances in therapeutic vaccines for treating human papillomavirus-related cervical intraepithelial neoplasia. J. Obstet. Gynaecol. Res. 2020, 46, 989–1006. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, B.; Morrow, M.P.; Hutnick, N.A.; Shin, T.H.; Lucke, C.E.; Weiner, D.B. Clinical applications of DNA vaccines: Current progress. Clin. Infect. Dis. 2011, 53, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- INOVIO Announces Positive Results from REVEAL 1, a Phase 3 Pivotal Trial Evaluating VGX-3100, Its DNA-Based HPV Immunotherapy for the Treatment of High-Grade Precancerous Cervical Dysplasia Caused by HPV-16 and/or HPV-18; CISION ps Newswire: New York, NY, USA, 2021.
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Tan, X.; Hu, X.; Zhou, M.; Yan, J.; Ding, C. Tumor Microenvironment following Gemcitabine Treatment Favors Differentiation of Immunosuppressive Ly6C(high) Myeloid Cells. J. Immunol. 2020, 204, 212–223. [Google Scholar] [CrossRef]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.M.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Trial | Phase | Number of Patients | Setting | Drugs and Schedule | Primary Endpoint |
|---|---|---|---|---|---|
| NCT03556839 (BEATcc) | III | 404 | Persistent Recurrent Metastatic | Arm A: Cisplatin 50 mg/m2 or carboplatin AUC 5 + paclitaxel 175 mg/m2 + bevacizumab 15 mg/kg q3W. Patients who achieve a CR after ≥6 cycles may be allowed to continue bevacizumab; Arm B: Cisplatin 50 mg/m2 or carboplatin AUC 5 + paclitaxel 175 mg/m2 + bevacizumab 15 mg/kg + atezolizumab 1200 mg q3W. Patients who achieve a CR after ≥6 cycles may be allowed to continue bevacizumab plus atezolizumab | OS |
| NCT04221945 (ENGOT-cx11/KEYNOTE-A18) | III | 980 | Locally advanced | Pembrolizumab 200 mg or placebo q3w for 5 cycles + CRT (weekly cisplatin 40 mg/m2 + external beam radiotherapy followed by brachytherapy) followed by 15 cycles of pembrolizumab 400 mg or placebo q6w | PFS and OS |
| NCT03830866 (CALLA) | III | 770 | Locally advanced | External beam radiotherapy with cisplatin (40 mg/m2) or carboplatin (AUC 2) once a week for 5 weeks, followed by brachytherapy, with durvalumab 1500 mg or placebo q4w for 24 cycles | PFS |
| NCT03104699 | II | 211 | Locally advanced Recurrent Metastatic | Balstilimab 3 mg/kg q2w up to 2 years | ORR |
| NCT03495882 | II | 154 | Locally advanced Recurrent Metastatic | Balstilimab 3 mg/kg q2w in combination with Zalifrelimab 1 mg/kg q6w up to 2 years | ORR |
| NCT03257267 (EMPOWER-GOG 3016/ENGOT-cx9) | III | 608 | Recurrent Metastatic | Experimental arm: Cemiplimab 350 mg intravenous administration every 3 weeks Investigator Choice Chemotherapy: - pemetrexed 500 mg/m2 q3w - topotecan 1 mg/m2 daily ×5 days, q3w - irinotecan 100 mg/m2 days 1, 8, 15, and 22, followed by 2 weeks rest, for 42 days (6-week cycle) - gemcitabine 1000 mg/m2 days 1 and 8, q3w - vinorelbine 30 mg/m2 days 1 and 8, q3w. Treatments will be given IV for up to 96 weeks | OS |
| NCT04238988 (CERV-3) | II | 45 | Locally advanced | Three cycles of NACT with carboplatin AUC 5, paclitaxel 175 mg/m2 and pembrolizumab 200 mg q3w, then surgery, then adjuvant carboplatin and paclitaxel in combination with pembrolizumab, followed by pembrolizumab 200 mg q3w for up to 35 cycles (only high risk) | 2-years PFS |
| NCT03635567 (Keynote-826) | III | 600 | Persistent Recurrent Metastatic | Investigator Choice Chemotherapy: - paclitaxel 175 mg/m2 + cisplatin 50 mg/m2 - carboplatin AUC 5, with or Without bevacizumab 15 mg/kg) + pembrolizumab 200 mg or placebo q3w until disease progression, unacceptable toxicity or patient withdrawal for up to 35 cycles | PFS and OS |
| NCT04300647 (SKYSCRAPER-04) | II | 172 | Recurrent Metastatic | Atezolizumab 1200 mg q3w alone or in combination with tiragolumab 600 mg q3w | ORR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turinetto, M.; Valsecchi, A.A.; Tuninetti, V.; Scotto, G.; Borella, F.; Valabrega, G. Immunotherapy for Cervical Cancer: Are We Ready for Prime Time? Int. J. Mol. Sci. 2022, 23, 3559. https://doi.org/10.3390/ijms23073559
Turinetto M, Valsecchi AA, Tuninetti V, Scotto G, Borella F, Valabrega G. Immunotherapy for Cervical Cancer: Are We Ready for Prime Time? International Journal of Molecular Sciences. 2022; 23(7):3559. https://doi.org/10.3390/ijms23073559
Chicago/Turabian StyleTurinetto, Margherita, Anna A. Valsecchi, Valentina Tuninetti, Giulia Scotto, Fulvio Borella, and Giorgio Valabrega. 2022. "Immunotherapy for Cervical Cancer: Are We Ready for Prime Time?" International Journal of Molecular Sciences 23, no. 7: 3559. https://doi.org/10.3390/ijms23073559
APA StyleTurinetto, M., Valsecchi, A. A., Tuninetti, V., Scotto, G., Borella, F., & Valabrega, G. (2022). Immunotherapy for Cervical Cancer: Are We Ready for Prime Time? International Journal of Molecular Sciences, 23(7), 3559. https://doi.org/10.3390/ijms23073559