
Interdependence of Angiogenesis and Arteriogenesis in Development and Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
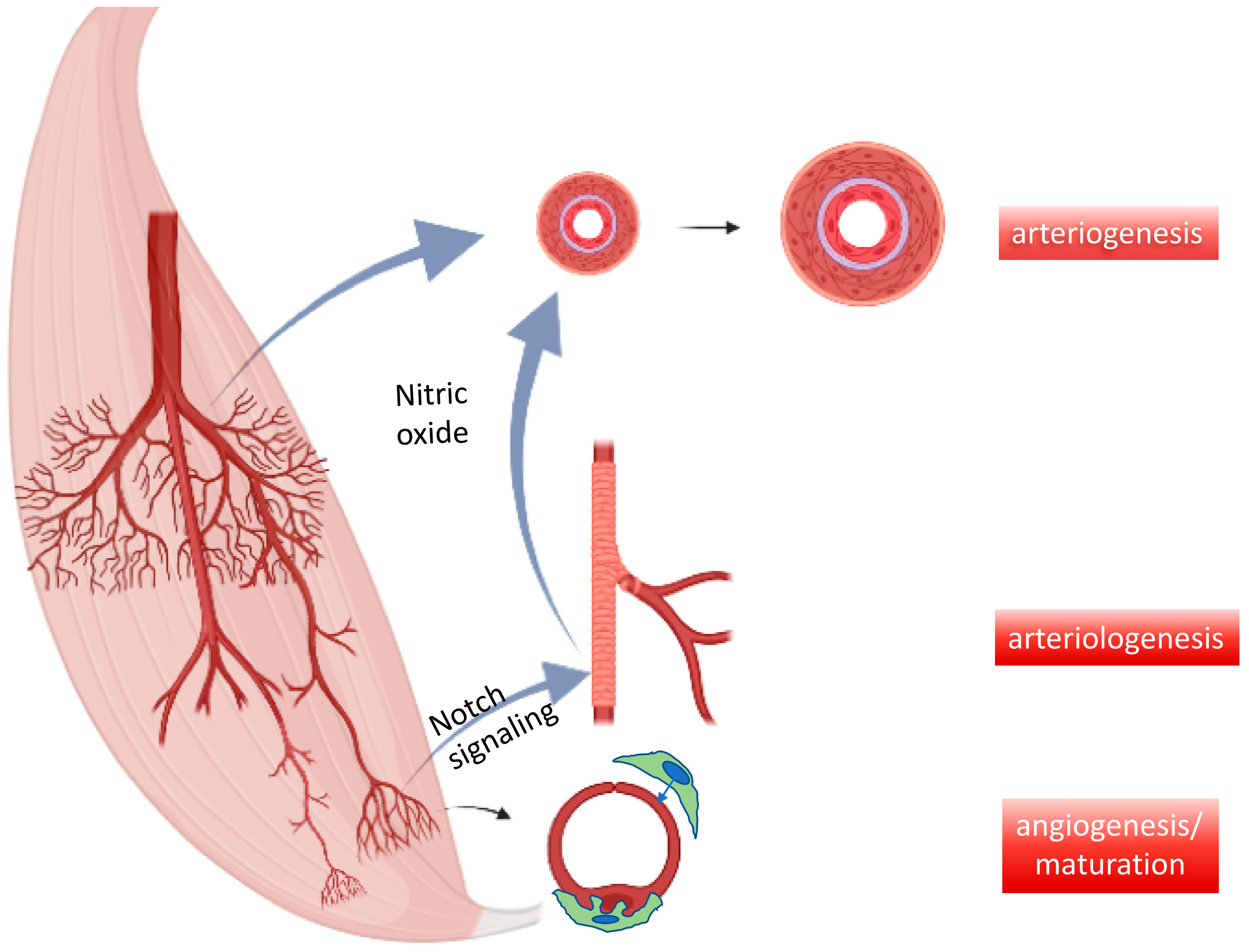
:1. Introduction
2. An Underestimated Problem—Capillary Rarefaction in Vascular Disease
3. Angiogenic Vessel Building—Building back Better-Perfused Capillaries and Arterioles
4. Capillary Maturation and De Novo Arteriologenesis—Linking Angiogenesis and Arteriogenesis
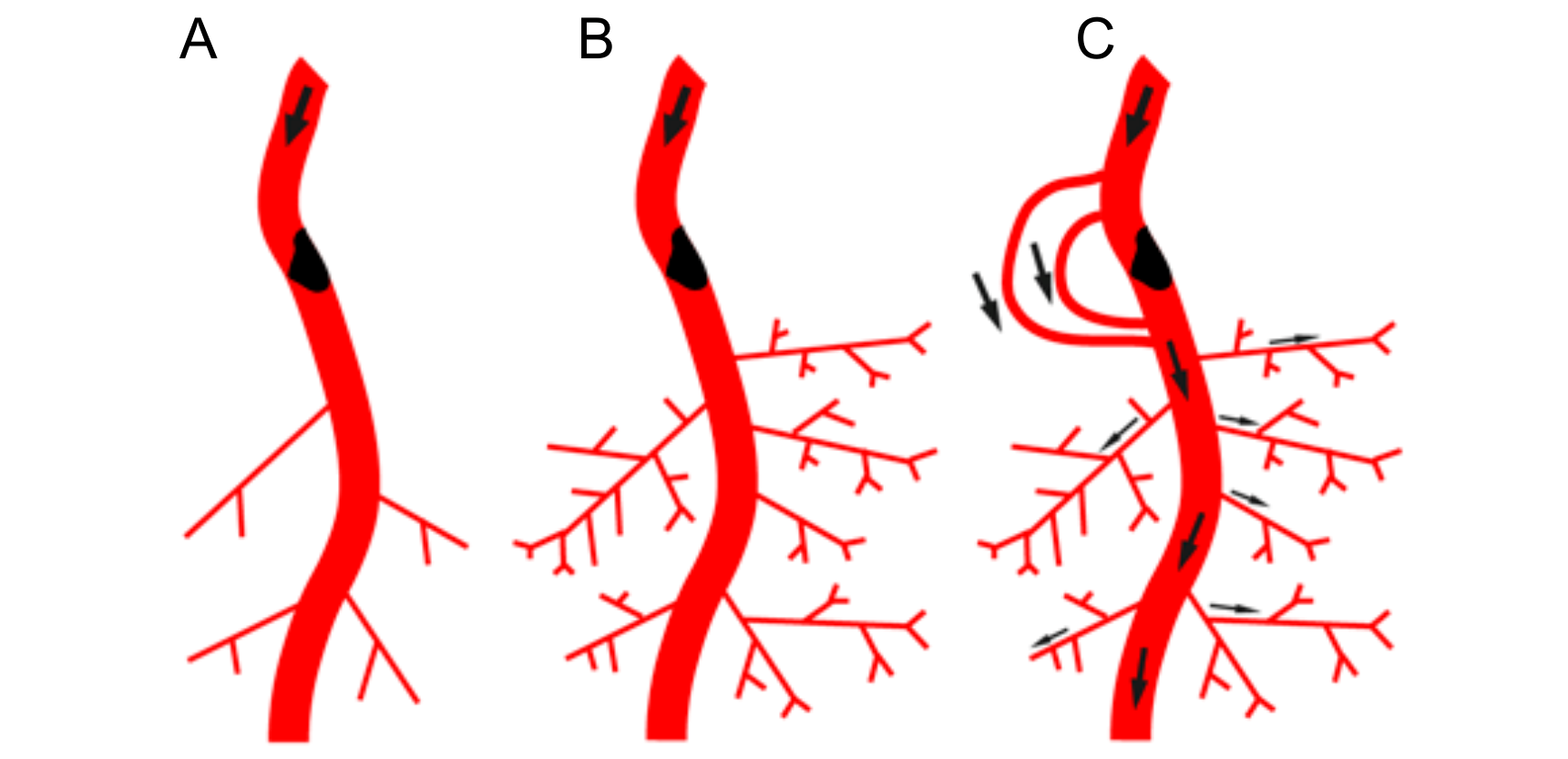
5. Arteriogenesis in the Developing Vasculature
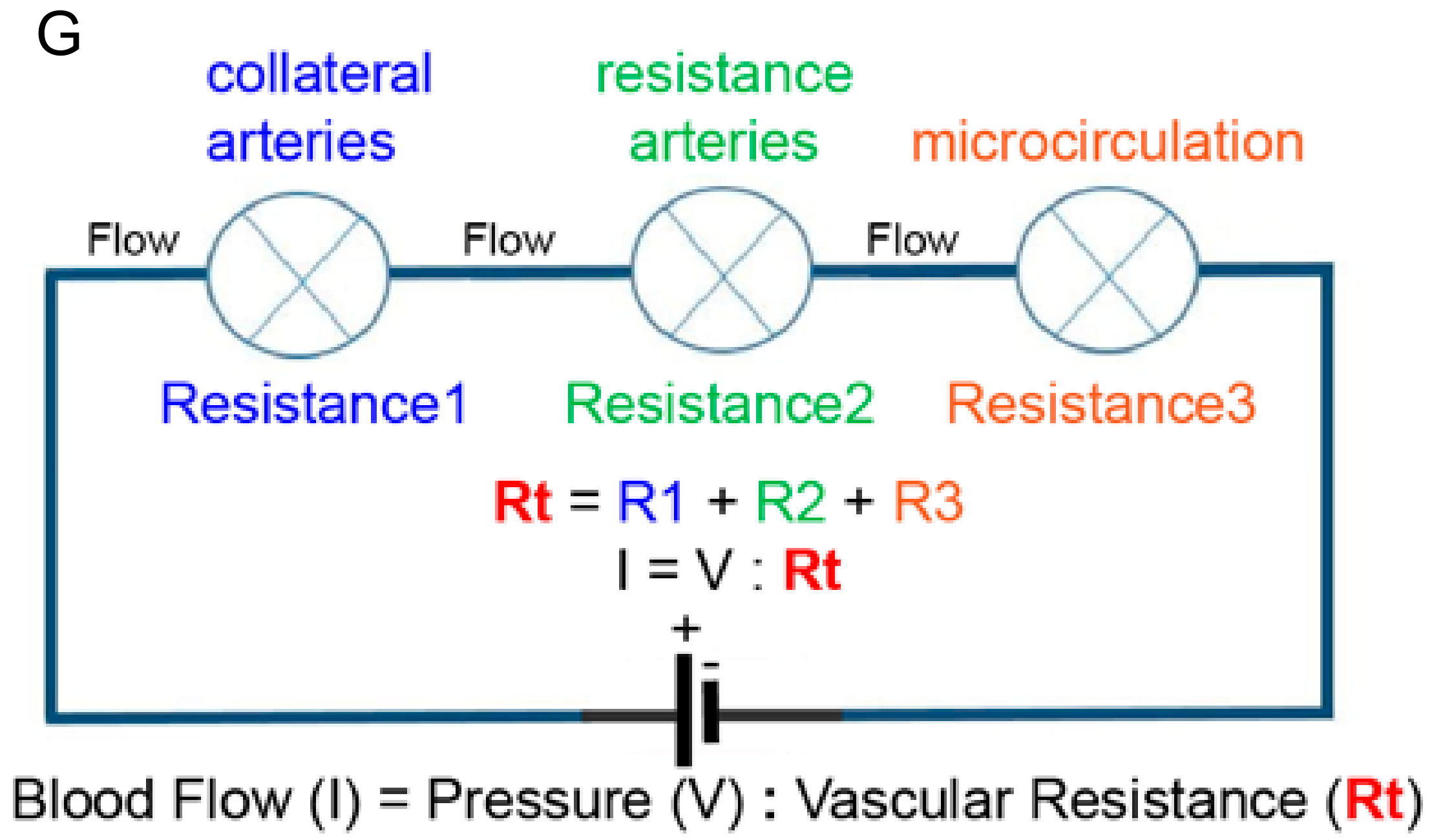
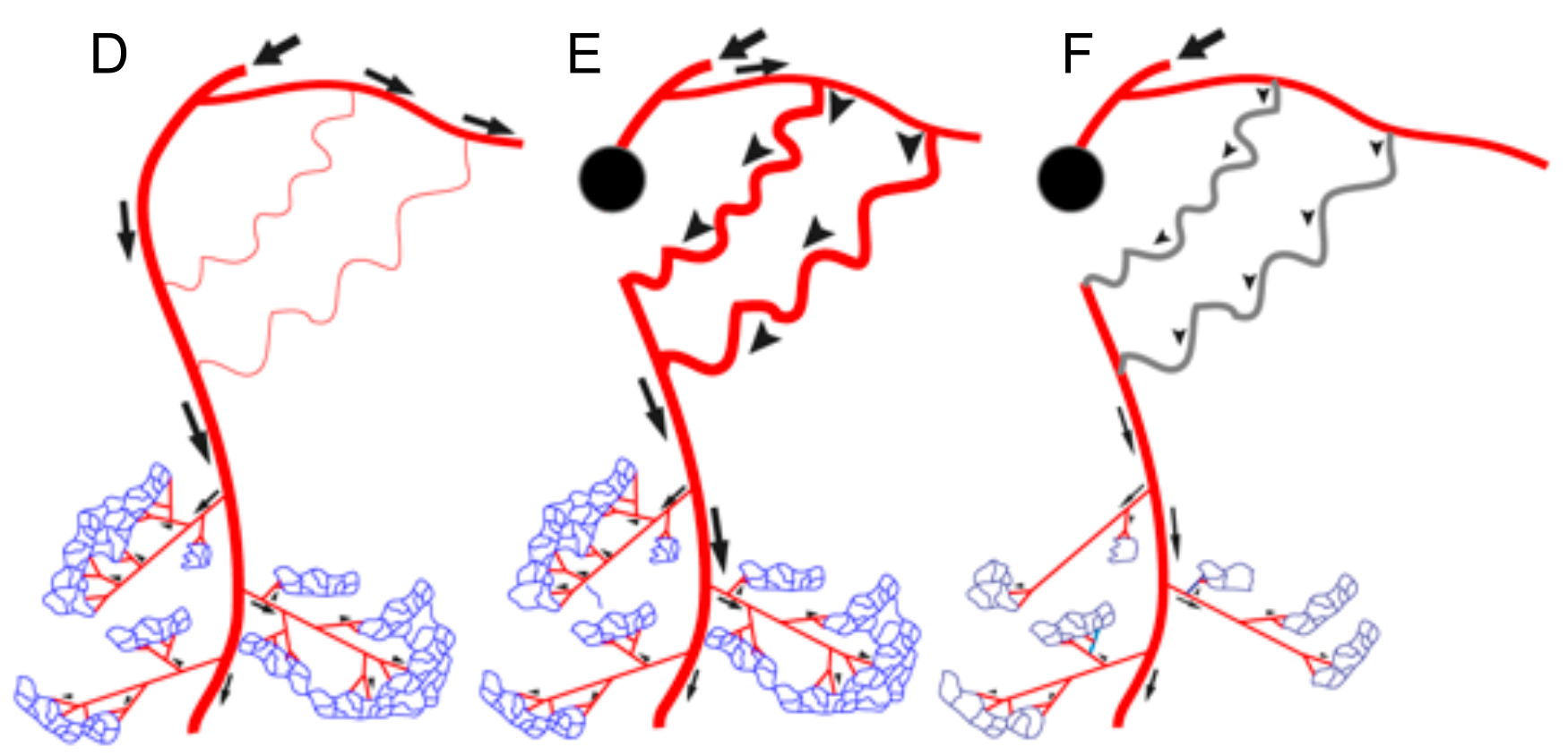
6. How Will Distal Microvessel Growth Induce Proximal Collateral Growth?
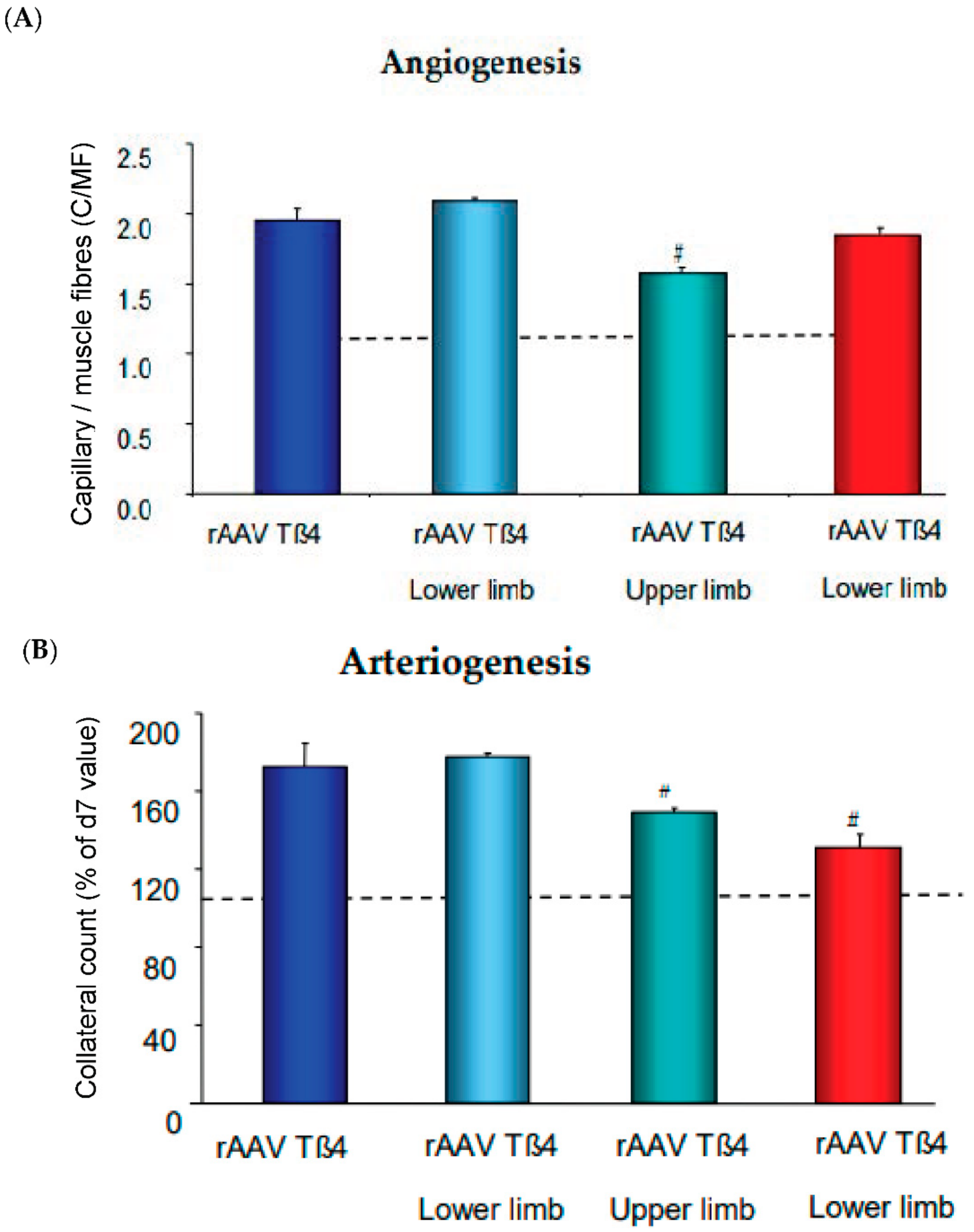
7. Angiogenesis, Maturation, and Arteriogenesis in Therapeutic Approaches
8. The Concept of Retrograde Communication and Arterial Growth
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sherman, T.F. On connecting large vessels to small. The meaning of Murray’s law. J. Gen. Physiol. 1981, 78, 431–453. [Google Scholar] [CrossRef] [Green Version]
- Faber, J.E.; Chilian, W.M.; Deindl, E.; van Royen, N.; Simons, M. A brief etymology of the collateral circulation. Arter. Thromb. Vasc. Biol. 2014, 34, 1854–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinkel, R.; Trenkwalder, T.; Petersen, B.; Husada, W.; Gesenhues, F.; Lee, S.; Hannappel, E.; Bock-Marquette, I.; Theisen, D.; Leitner, L.; et al. MRTF-A controls vessel growth and maturation by increasing the expression of CCN1 and CCN2. Nat. Commun. 2014, 5, 3970. [Google Scholar] [CrossRef] [Green Version]
- Klems, A.; van Rijssel, J.; Ramms, A.S.; Wild, R.; Hammer, J.; Merkel, M.; Derenbach, L.; Préau, L.; Hinkel, R.; Suarez-Martinez, I.; et al. The GEF Trio controls endothelial cell size and arterial remodeling downstream of Vegf signaling in both zebrafish and cell models. Nat. Commun. 2020, 11, 5319. [Google Scholar] [CrossRef] [PubMed]
- Red-Horse, K.; Siekmann, A.F. Veins and Arteries Build Hierarchical Branching Patterns Differently: Bottom-Up versus Top-Down. Bioessays 2019, 41, e1800198. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Arpino, J.-M.; Lee, J.J.; Pickering, J.G. Regenerated Microvascular Networks in Ischemic Skeletal Muscle. Front. Physiol. 2021, 12, 662073. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Fioretto, P.; Dodson, P.M. Does microvascular disease predict macrovascular events in type 2 diabetes? Atherosclerosis 2011, 218, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef]
- Hinkel, R.; Hoewe, A.; Renner, S.; Ng, J.; Lee, S.; Klett, K.; Kaczmarek, V.; Moretti, A.; Laugwitz, K.L.; Skroblin, P.; et al. Diabetes Mellitus-Induced Microvascular Destabilization in the Myocardium. J. Am. Coll. Cardiol. 2017, 69, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Schoos, M.M.; Dangas, G.D.; Mehran, R.; Kirtane, A.J.; Yu, J.; Litherland, C.; Clemmensen, P.; Stuckey, T.D.; Witzenbichler, B.; Weisz, G.; et al. Impact of Hemoglobin A1c Levels on Residual Platelet Reactivity and Outcomes After Insertion of Coronary Drug-Eluting Stents (from the ADAPT-DES Study). Am. J. Cardiol. 2016, 117, 192–200. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, T.; Bähr, A.; Howe, A.; Klett, K.; Husada, W.; Weber, C.; Laugwitz, K.-L.; Kupatt, C.; Hinkel, R. Tβ4 Increases Neovascularization and Cardiac Function in Chronic Myocardial Ischemia of Normo- and Hypercholesterolemic Pigs. Mol. Ther. 2018, 26, 1706–1714. [Google Scholar] [CrossRef] [PubMed]
- Kureishi, Y.; Luo, Z.; Shiojima, I.; Bialik, A.; Fulton, D.; Lefer, D.J.; Sessa, W.C.; Walsh, K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat. Med. 2000, 6, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Camici, P.G.; Tschöpe, C.; Di Carli, M.F.; Rimoldi, O.; Van Linthout, S. Coronary microvascular dysfunction in hypertrophy and heart failure. Cardiovasc. Res. 2020, 116, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Ramanujam, D.; Schon, A.P.; Beck, C.; Vaccarello, P.; Felician, G.; Dueck, A.; Esfandyari, D.; Meister, G.; Meitinger, T.; Schulz, C.; et al. MicroRNA-21-Dependent Macrophage-to-Fibroblast Signaling Determines the Cardiac Response to Pressure Overload. Circulation 2021, 143, 1513–1525. [Google Scholar] [CrossRef]
- Hinkel, R.; Ramanujam, D.; Kaczmarek, V.; Howe, A.; Klett, K.; Beck, C.; Dueck, A.; Thum, T.; Laugwitz, K.L.; Maegdefessel, L.; et al. AntimiR-21 Prevents Myocardial Dysfunction in a Pig Model of Ischemia/Reperfusion Injury. J. Am. Coll. Cardiol. 2020, 75, 1788–1800. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Zhao, L. Risk of hypertension with anti-VEGF monoclonal antibodies in cancer patients: A systematic review and meta-analysis of 105 phase II/III randomized controlled trials. J. Chemother. 2021. epub ahead of print. [Google Scholar] [CrossRef]
- Zhao, E.; Barber, J.; Sen, C.K.; Arciero, J. Modeling acute and chronic vascular responses to a major arterial occlusion. Microcirculation 2022, 29, e12738. [Google Scholar] [CrossRef] [PubMed]
- Kamei, M.; Saunders, W.B.; Bayless, K.J.; Dye, L.; Davis, G.E.; Weinstein, B.M. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature 2006, 442, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Gebala, V.; Collins, R.; Geudens, I.; Phng, L.K.; Gerhardt, H. Blood flow drives lumen formation by inverse membrane blebbing during angiogenesis in vivo. Nat. Cell Biol. 2016, 18, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Muhl, L.; Burmakin, M.; Wang, Y.; Duchez, A.C.; Betsholtz, C.; Arthur, H.M.; Jakobsson, L. Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat. Cell Biol. 2017, 19, 639–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, C.A.; Jones, M.L.; Bernabeu, M.O.; Vion, A.C.; Barbacena, P.; Fan, J.; Mathivet, T.; Fonseca, C.G.; Ragab, A.; Yamaguchi, T.P.; et al. Non-canonical Wnt signalling modulates the endothelial shear stress flow sensor in vascular remodelling. Elife 2016, 5, e07727. [Google Scholar] [CrossRef] [PubMed]
- Ola, R.; Künzel, S.H.; Zhang, F.; Genet, G.; Chakraborty, R.; Pibouin-Fragner, L.; Martin, K.; Sessa, W.; Dubrac, A.; Eichmann, A. SMAD4 Prevents Flow Induced Arteriovenous Malformations by Inhibiting Casein Kinase 2. Circulation 2018, 138, 2379–2394. [Google Scholar] [CrossRef] [PubMed]
- Ola, R.; Dubrac, A.; Han, J.; Zhang, F.; Fang, J.S.; Larrivée, B.; Lee, M.; Urarte, A.A.; Kraehling, J.R.; Genet, G.; et al. PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat. Commun. 2016, 7, 13650. [Google Scholar] [CrossRef] [PubMed]
- Rochon, E.R.; Menon, P.G.; Roman, B.L. Alk1 controls arterial endothelial cell migration in lumenized vessels. Development 2016, 143, 2593–2602. [Google Scholar] [PubMed] [Green Version]
- Laux, D.W.; Young, S.; Donovan, J.P.; Mansfield, C.J.; Upton, P.D.; Roman, B.L. Circulating Bmp10 acts through endothelial Alk1 to mediate flow-dependent arterial quiescence. Development 2013, 140, 3403–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsina-Sanchís, E.; García-Ibáñez, Y.; Figueiredo, A.M.; Riera-Domingo, C.; Figueras, A.; Matias-Guiu, X.; Casanovas, O.; Botella, L.M.; Pujana, M.A.; Riera-Mestre, A.; et al. ALK1 Loss Results in Vascular Hyperplasia in Mice and Humans Through PI3K Activation. Arter. Thromb. Vasc. Biol. 2018, 38, 1216–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bry, M.; Kivelä, R.; Holopainen, T.; Anisimov, A.; Tammela, T.; Soronen, J.; Silvola, J.; Saraste, A.; Jeltsch, M.; Korpisalo, P.; et al. Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation 2010, 122, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Kivelä, R.; Bry, M.; Robciuc, M.R.; Räsänen, M.; Taavitsainen, M.; Silvola, J.M.; Saraste, A.; Hulmi, J.J.; Anisimov, A.; Mäyränpää, M.I.; et al. VEGF-B-induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO Mol. Med. 2014, 6, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Lähteenvuo, J.E.; Lähteenvuo, M.T.; Kivelä, A.; Rosenlew, C.; Falkevall, A.; Klar, J.; Heikura, T.; Rissanen, T.T.; Vähäkangas, E.; Korpisalo, P.; et al. Vascular endothelial growth factor-B induces myocardium-specific angiogenesis and arteriogenesis via vascular endothelial growth factor receptor-1- and neuropilin receptor-1-dependent mechanisms. Circulation 2009, 119, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Dewerchin, M.; Carmeliet, P. PlGF: A multitasking cytokine with disease-restricted activity. Cold Spring Harb. Perspect. Med. 2012, 2, a011056. [Google Scholar] [CrossRef] [PubMed]
- Ylä-Herttuala, S.; Baker, A.H. Cardiovascular Gene Therapy: Past, Present, and Future. Mol. Ther. 2017, 25, 1095–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, C.R.; Banfi, A.; Glazer, N.L.; Thurston, G.; Springer, M.L.; Kraft, P.E.; McDonald, D.M.; Blau, H.M. Microenvironmental VEGF concentration, not total dose, determines a threshold between normal and aberrant angiogenesis. J. Clin. Investig. 2004, 113, 516–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, M.; Kumar, S.; Sharife, H.; Volinsky, E.; Gileles-Hillel, A.; Licht, T.; Permyakova, A.; Hinden, L.; Azar, S.; Friedmann, Y.; et al. Counteracting age-related VEGF signaling insufficiency promotes healthy aging and extends life span. Science 2021, 373, eabc8479. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, T.; Horstkotte, J.; Schwab, C.; Pfetsch, V.; Weinmann, K.; Dietzel, S.; Rohwedder, I.; Hinkel, R.; Gross, L.; Lee, S.; et al. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J. Clin. Investig. 2013, 123, 3436–3445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arboleda-Velasquez, J.F.; Manent, J.; Lee, J.H.; Tikka, S.; Ospina, C.; Vanderburg, C.R.; Frosch, M.P.; Rodríguez-Falcón, M.; Villen, J.; Gygi, S.; et al. Hypomorphic Notch 3 alleles link Notch signaling to ischemic cerebral small-vessel disease. Proc. Natl. Acad. Sci. USA 2011, 108, E128–E135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Lagos-Quintana, M.; Liu, D.; Shi, Y.; Helker, C.; Herzog, W.; le Noble, F. miR-30a regulates endothelial tip cell formation and arteriolar branching. Hypertension 2013, 62, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Gaengel, K.; Niaudet, C.; Hagikura, K.; Siemsen, B.; Muhl, L.; Hofmann, J.-A.; Ebarasi, L.; Nystr+Âm, S.; Rymo, S.; Chen, L.-A.; et al. The Sphingosine-1-Phosphate Receptor S1PR1 Restricts Sprouting Angiogenesis by Regulating the Interplay between VE-Cadherin and VEGFR2. Dev. Cell 2012, 23, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupatt, C.; Hinkel, R.; Pfosser, A.; El-Aouni, C.; Wuchrer, A.; Fritz, A.; Globisch, F.; Thormann, M.; Horstkotte, J.; Lebherz, C.; et al. Cotransfection of Vascular Endothelial Growth Factor-A and Platelet-Derived Growth Factor-B Via Recombinant Adeno-Associated Virus Resolves Chronic Ischemic Malperfusion: Role of Vessel Maturation. J. Am. Coll. Cardiol. 2010, 56, 414–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsic, N.; Zentilin, L.; Zacchigna, S.; Santoro, D.; Stanta, G.; Salvi, A.; Sinagra, G.; Giacca, M. Induction of functional neovascularization by combined VEGF and angiopoietin-1 gene transfer using AAV vectors. Mol. Ther. 2003, 7, 450–459. [Google Scholar] [CrossRef]
- Red-Horse, K.; Das, S. New Research Is Shining Light on How Collateral Arteries Form in the Heart: A Future Therapeutic Direction? Curr. Cardiol. Rep. 2021, 23, 30. [Google Scholar] [CrossRef]
- Lucitti, J.L.; Mackey, J.K.; Morrison, J.C.; Haigh, J.J.; Adams, R.H.; Faber, J.E. Formation of the collateral circulation is regulated by vascular endothelial growth factor-A and a disintegrin and metalloprotease family members 10 and 17. Circ. Res. 2012, 111, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Rzechorzek, W.; Zhang, H.; Buckley, B.K.; Hua, K.; Pomp, D.; Faber, J.E. Aerobic exercise prevents rarefaction of pial collaterals and increased stroke severity that occur with aging. J. Cereb. Blood Flow Metab. 2017, 37, 3544–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Prabhakar, P.; Sealock, R.; Faber, J.E. Wide genetic variation in the native pial collateral circulation is a major determinant of variation in severity of stroke. J. Cereb. Blood Flow Metab. 2010, 30, 923–934. [Google Scholar] [CrossRef]
- Lucitti, J.L.; Sealock, R.; Buckley, B.K.; Zhang, H.; Xiao, L.; Dudley, A.C.; Faber, J.E. Variants of Rab GTPase-Effector Binding Protein-2 Cause Variation in the Collateral Circulation and Severity of Stroke. Stroke 2016, 47, 3022–3031. [Google Scholar] [CrossRef] [PubMed]
- Clayton, J.A.; Chalothorn, D.; Faber, J.E. Vascular endothelial growth factor-A specifies formation of native collaterals and regulates collateral growth in ischemia. Circ. Res. 2008, 103, 1027–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buschmann, I.; Pries, A.; Styp-Rekowska, B.; Hillmeister, P.; Loufrani, L.; Henrion, D.; Shi, Y.; Duelsner, A.; Hoefer, I.; Gatzke, N.; et al. Pulsatile shear and Gja5 modulate arterial identity and remodeling events during flow-driven arteriogenesis. Development 2010, 137, 2187–2196. [Google Scholar] [CrossRef] [Green Version]
- Cristofaro, B.; Shi, Y.; Faria, M.; Suchting, S.; Leroyer, A.S.; Trindade, A.; Duarte, A.; Zovein, A.C.; Iruela-Arispe, M.L.; Nih, L.R.; et al. Dll4-Notch signaling determines the formation of native arterial collateral networks and arterial function in mouse ischemia models. Development 2013, 140, 1720–1729. [Google Scholar] [CrossRef] [Green Version]
- Faber, J.E.; Storz, J.F.; Cheviron, Z.A.; Zhang, H. High-altitude rodents have abundant collaterals that protect against tissue injury after cerebral, coronary and peripheral artery occlusion. J. Cereb. Blood Flow Metab. 2021, 41, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Schierling, W.; Troidl, K.; Troidl, C.; Schmitz-Rixen, T.; Schaper, W.; Eitenmüller, I.K. The Role of Angiogenic Growth Factors in Arteriogenesis. J. Vasc. Res. 2009, 46, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Heil, M.; Ziegelhoeffer, T.; Wagner, S.; Fernandez, B.; Helisch, A.; Martin, S.; Tribulova, S.; Kuziel, W.A.; Bachmann, G.; Schaper, W. Collateral artery growth (arteriogenesis) after experimental arterial occlusion is impaired in mice lacking CC-chemokine receptor-2. Circ. Res. 2004, 94, 671–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Royen, N.; Hoefer, I.; Bottinger, M.; Hua, J.; Grundmann, S.; Voskuil, M.; Bode, C.; Schaper, W.; Buschmann, I.; Piek, J.J. Local Monocyte Chemoattractant Protein-1 Therapy Increases Collateral Artery Formation in Apolipoprotein E-Deficient Mice but Induces Systemic Monocytic CD11b Expression, Neointimal Formation, and Plaque Progression. Circ. Res. 2003, 92, 218–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troidl, C.; Jung, G.; Troidl, K.; Hoffmann, J.; Mollmann, H.; Nef, H.; Schaper, W.; Hamm, C.W.; Schmitz-Rixen, T. The temporal and spatial distribution of macrophage subpopulations during arteriogenesis. Curr. Vasc. Pharmacol. 2013, 11, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Götze, A.M.; Schubert, C.; Jung, G.; Dörr, O.; Liebetrau, C.; Hamm, C.W.; Schmitz-Rixen, T.; Troidl, C.; Troidl, K. IL10 Alters Peri-Collateral Macrophage Polarization and Hind-Limb Reperfusion in Mice after Femoral Artery Ligation. Int. J. Mol. Sci. 2020, 21, 2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauriol, J.; Keith, K.; Jaffr, F.; Couvillon, A.; Saci, A.; Goonasekera, S.A.; McCarthy, J.R.; Kessinger, C.W.; Wang, J.; Ke, Q.; et al. RhoA signaling in cardiomyocytes protects against stress-induced heart failure but facilitates cardiac fibrosis. Sci. Signal. 2014, 7, ra100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorn, T.; Kornherr, J.; Parrotta, E.I.; Zawada, D.; Ayetey, H.; Santamaria, G.; Iop, L.; Mastantuono, E.; Sinnecker, D.; Goedel, A.; et al. Interplay of cell-cell contacts and RhoA/MRTF-A signaling regulates cardiomyocyte identity. EMBO J. 2018, 37, e98133. [Google Scholar] [CrossRef] [PubMed]
- Eitenmuller, I.; Volger, O.; Kluge, A.; Troidl, K.; Barancik, M.; Cai, W.J.; Heil, M.; Pipp, F.; Fischer, S.; Horrevoets, A.J.G.; et al. The Range of Adaptation by Collateral Vessels After Femoral Artery Occlusion. Circ. Res. 2006, 99, 656–662. [Google Scholar] [CrossRef]
- Limbourg, A.; Ploom, M.; Elligsen, D.; Sorensen, I.; Ziegelhoeffer, T.; Gossler, A.; Drexler, H.; Limbourg, F.P. Notch Ligand Delta-Like 1 Is Essential for Postnatal Arteriogenesis. Circ. Res. 2007, 100, 363–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Cai, B.; Wu, X.; Peng, S.; Gan, L.; Huang, D.; Liu, G.; Dong, L.; Xiao, L.; Liu, J.; et al. microRNA-352 regulates collateral vessel growth induced by elevated fluid shear stress in the rat hind limb. Sci. Rep. 2017, 7, 6643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Z.; van Mil, A.; Brandt, M.M.; Grundmann, S.; Hoefer, I.; Smits, M.; El Azzouzi, H.; Fukao, T.; Cheng, C.; Doevendans, P.A.; et al. MicroRNA-132/212 family enhances arteriogenesis after hindlimb ischaemia through modulation of the Ras-MAPK pathway. J. Cell Mol. Med. 2015, 19, 1994–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landskroner-Eiger, S.; Qiu, C.; Perrotta, P.; Siragusa, M.; Lee, M.Y.; Ulrich, V.; Luciano, A.K.; Zhuang, Z.W.; Corti, F.; Simons, M.; et al. Endothelial miR-17∼92 cluster negatively regulates arteriogenesis via miRNA-19 repression of WNT signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 12812–12817. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
le Noble, F.; Kupatt, C. Interdependence of Angiogenesis and Arteriogenesis in Development and Disease. Int. J. Mol. Sci. 2022, 23, 3879. https://doi.org/10.3390/ijms23073879
le Noble F, Kupatt C. Interdependence of Angiogenesis and Arteriogenesis in Development and Disease. International Journal of Molecular Sciences. 2022; 23(7):3879. https://doi.org/10.3390/ijms23073879
Chicago/Turabian Stylele Noble, Ferdinand, and Christian Kupatt. 2022. "Interdependence of Angiogenesis and Arteriogenesis in Development and Disease" International Journal of Molecular Sciences 23, no. 7: 3879. https://doi.org/10.3390/ijms23073879
APA Stylele Noble, F., & Kupatt, C. (2022). Interdependence of Angiogenesis and Arteriogenesis in Development and Disease. International Journal of Molecular Sciences, 23(7), 3879. https://doi.org/10.3390/ijms23073879