
Single Cell Self-Paced Clustering with Transcriptome Sequencing Data


Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Data Preprocessing
- Step 1:
- Genes with no count in any cell were filtered out.
- Step 2:
- We filtered genes that were not expressed in almost all cells.
- Step 3:
- The top N high variable genes (HVGs) were selected. One thousand highly variable genes were selected by default. In Section 3.2. We discuss the influence of different N values for the experimental accuracy.
- Step 4:
- The last step was to take the log transform and scale of the read counts, so that count values follow unit variance and zero mean.
2.3. scSPaC Model
2.4. Optimization
- Step 1:
- Fix , update and .
- Step 2:
- Fix and , update .
2.5. Evaluation Metrics
2.5.1. ARI
2.5.2. Purity
2.5.3. NMI
3. Results and Discussion
3.1. Experimental Performance on All Datasets
- K-means [48], the classical K-means algorithm.
- NMF [35], the standard NMF clustering with Frobenius norm (F-norm).
- ONMF [49], the orthogonal NMF for clustering.
- -NMF [36], the sparse NMF clustering with -norm.
- Scanpy [34] is a Python-based toolkit for analyzing single cell gene expression data. Scanpy was downloaded from https://github.com/theislab/scanpy (accessed on 3 March 2022). It includes clustering and is used as the comparison algorithm in the experiment. We ran Scanpy with default parameters, for example, and .
- Seurat3 [50] is a graph-based clustering tool. For all datasets, Seurat was performed with default parameters and downloaded from https://github.com/satijalab/seurat (accessed on 3 March 2022). We set the number of neighbors to 20 and the cluster resolution to 0.8, and used the function and 0.05 (the bound of P-value) to determine the number of principal components.
- SC3 [51] is a single cell cluster tool combining multiple clustering solutions through a consensus approach. SC3 was downloaded from https://github.com/hemberg-lab/SC3 (accessed on 3 March 2022) and ran with default parameters. For example, , , , and .
3.2. Different Numbers of Variable Genes Were Selected for Comparison
3.3. Accuracy in Estimating the Number of Clusters
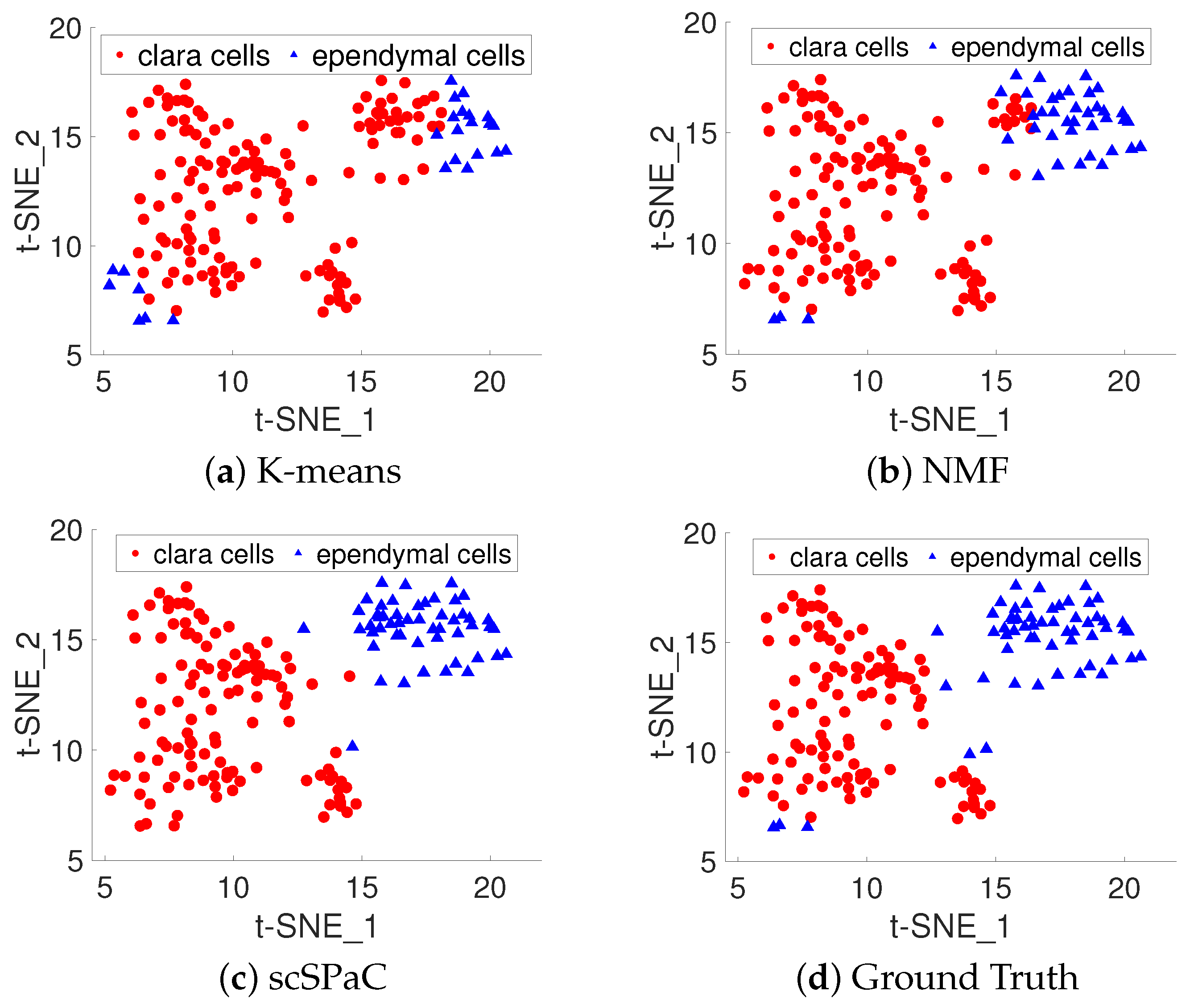
3.4. Clustering Pulmonary Alveolar Type II, Clara and Ependymal Cells of Human ScRNA-seq Data
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NMF | Nonnegative Matrix Factorization |
| SPL | Self-Paced Learning |
| t-SNE | t-Distributed Stochastic Neighbor Embedding |
| HVGs | High Variable Genes |
References
- Tsoucas, D.; Yuan, G.C. Recent progress in single-cell cancer genomics. Curr. Opin. Genet. Dev. 2017, 42, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. Non-genetic heterogeneity of cells in development: More than just noise. Development 2009, 136, 3853–3862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Liu, J.; Lu, Q.; Riggs, A.D.; Wu, X. SAIC: An iterative clustering approach for analysis of single cell RNA-seq data. BMC Genom. 2017, 18, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marco, E.; Karp, R.L.; Guo, G.; Robson, P.; Hart, A.H.; Trippa, L.; Yuan, G.C. Bifurcation analysis of single-cell gene expression data reveals epigenetic landscape. Proc. Natl. Acad. Sci. USA 2014, 111, E5643–E5650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mieth, B.; Hockley, J.R.; Görnitz, N.; Vidovic, M.M.C.; Müller, K.R.; Gutteridge, A.; Ziemek, D. Using transfer learning from prior reference knowledge to improve the clustering of single-cell RNA-Seq data. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.; Troup, M.; Ho, J.W. CIDR: Ultrafast and accurate clustering through imputation for single-cell RNA-seq data. Genome Biol. 2017, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Lei, J.; Klei, L.; Devlin, B.; Roeder, K. Semisoft clustering of single-cell data. Proc. Natl. Acad. Sci. USA 2019, 116, 466–471. [Google Scholar] [CrossRef] [Green Version]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Von Luxburg, U. A tutorial on spectral clustering. Stat. Comput. 2007, 17, 395–416. [Google Scholar] [CrossRef]
- Hu, Y.; Li, B.; Chen, F.; Qu, K. Single-cell data clustering based on sparse optimization and low-rank matrix factorization. G3 2021, 11, 1–7. [Google Scholar] [CrossRef]
- Wang, B.; Zhu, J.; Pierson, E.; Ramazzotti, D.; Batzoglou, S. Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat. Methods 2017, 14, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Zhao, H. Spectral clustering based on learning similarity matrix. Bioinformatics 2018, 34, 2069–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.P.; Packer, B.; Koller, D. Self-paced learning for latent variable models. In Proceedings of the Conference on Advances in Neural Information Processing Systems, Vancouver, BC, Canada, 6–11 December 2010; pp. 1189–1197. [Google Scholar]
- Bengio, Y.; Louradour, J.; Collobert, R.; Weston, J. Curriculum learning. In Proceedings of the 26th International Conference on Machine Learning, Montreal, QC, Canada, 14–18 June 2009; pp. 41–48. [Google Scholar]
- Kumar, M.P.; Turki, H.; Preston, D.; Koller, D. Learning specific-class segmentation from diverse data. In Proceedings of the IEEE International Conference on Computer Vision, Barcelona, Spain, 6–13 November 2011; pp. 1800–1807. [Google Scholar]
- Jiang, L.; Meng, D.; Zhao, Q.; Shan, S.; Hauptmann, A.G. Self-Paced Curriculum Learning. In Proceedings of the 29th AAAI Conference on Artificial Intelligence, Austin, TX, USA, 25–30 January 2015; pp. 2694–2900. [Google Scholar]
- Tang, K.; Ramanathan, V.; Li, F.F.; Koller, D. Shifting Weights: Adapting Object Detectors from Image to Video. In Proceedings of the Conference on Advances in Neural Information Processing Systems, Stateline, NV, USA, 3–8 December 2012; pp. 647–655. [Google Scholar]
- Huang, Z.; Ren, Y.; Pu, X.; He, L. Non-Linear Fusion for Self-Paced Multi-View Clustering. In Proceedings of the 29th ACM International Conference on Multimedia, Online, 20–24 October 2021; pp. 3211–3219. [Google Scholar]
- Ren, Y.; Zhao, P.; Xu, Z.; Yao, D. Balanced Self-Paced Learning with Feature Corruption. In Proceedings of the International Joint Conference on Neural Networks, Anchorage, AK, USA, 14–19 May 2017; pp. 2064–2071. [Google Scholar]
- Ghasedi, K.; Wang, X.; Deng, C.; Huang, H. Balanced self-paced learning for generative adversarial clustering network. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Long Beach, CA, USA, 16–20 June 2019; pp. 4391–4400. [Google Scholar]
- Zheng, W.; Zhu, X.; Wen, G.; Zhu, Y.; Yu, H.; Gan, J. Unsupervised feature selection by self-paced learning regularization. Pattern Recognit. Lett. 2020, 132, 4–11. [Google Scholar] [CrossRef]
- Ren, Y.; Que, X.; Yao, D.; Xu, Z. Self-paced multi-task clustering. Neurocomputing 2019, 350, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Wen, G.; Gan, J.; Zheng, W.; Lei, C. Self-paced learning for k-means clustering algorithm. Pattern Recognit. Lett. 2020, 132, 69–75. [Google Scholar] [CrossRef]
- Huang, Z.; Ren, Y.; Pu, X.; Pan, L.; Yao, D.; Yu, G. Dual self-paced multi-view clustering. Neural Netw. 2021, 140, 184–192. [Google Scholar] [CrossRef]
- Zappia, L.; Phipson, B.; Oshlack, A. Splatter: Simulation of single-cell RNA sequencing data. Genome Biol. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Baron, M.; Veres, A.; Wolock, S.L.; Faust, A.L.; Gaujoux, R.; Vetere, A.; Ryu, J.H.; Wagner, B.K.; Shen-Orr, S.S.; Klein, A.M.; et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter-and intra-cell population structure. Cell Syst. 2016, 3, 346–360. [Google Scholar] [CrossRef] [Green Version]
- Kolodziejczyk, A.A.; Kim, J.K.; Tsang, J.C.; Ilicic, T.; Henriksson, J.; Natarajan, K.N.; Tuck, A.C.; Gao, X.; Bühler, M.; Liu, P.; et al. Single cell RNA-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell 2015, 17, 471–485. [Google Scholar] [CrossRef] [Green Version]
- Pollen, A.A.; Nowakowski, T.J.; Shuga, J.; Wang, X.; Leyrat, A.A.; Lui, J.H.; Li, N.; Szpankowski, L.; Fowler, B.; Chen, P.; et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotechnol. 2014, 32, 1053–1058. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Goolam, M.; Scialdone, A.; Graham, S.J.; Macaulay, I.C.; Jedrusik, A.; Hupalowska, A.; Voet, T.; Marioni, J.C.; Zernicka-Goetz, M. Heterogeneity in Oct4 and Sox2 targets biases cell fate in 4-cell mouse embryos. Cell 2016, 165, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; La Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lake, B.B.; Zhang, K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 2019, 37, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Svensson, V.; Natarajan, K.N.; Ly, L.H.; Miragaia, R.J.; Labalette, C.; Macaulay, I.C.; Cvejic, A.; Teichmann, S.A. Power analysis of single-cell RNA-sequencing experiments. Nat. Methods 2017, 14, 381–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 1–5. [Google Scholar]
- Lee, D.D.; Seung, H.S. Algorithms for non-negative matrix factorization. In Proceedings of the Conference on Advances in Neural Information Processing Systems, Vancouver, BC, Canada, 3–8 December 2001; pp. 556–562. [Google Scholar]
- Kong, D.; Ding, C.; Huang, H. Robust nonnegative matrix factorization using l21-norm. In Proceedings of the International on Conference on Information and Knowledge Management, Glasgow, Scotland, UK, 24–28 October 2011; pp. 673–682. [Google Scholar]
- Gao, H.; Nie, F.; Cai, W.; Huang, H. Robust Capped Norm Nonnegative Matrix Factorization. In Proceedings of the International on Conference on Information and Knowledge Management, Melbourne, Australia, 18–23 October 2015; pp. 871–880. [Google Scholar]
- Zhu, X.; Zhang, Z. Improved self-paced learning framework for nonnegative matrix factorization. Pattern Recognit. Lett. 2017, 97, 1–7. [Google Scholar] [CrossRef]
- Huang, S.; Zhao, P.; Ren, Y.; Li, T.; Xu, Z. Self-paced and soft-weighted nonnegative matrix factorization for data representation. Knowl.-Based Syst. 2019, 164, 29–37. [Google Scholar] [CrossRef]
- Jiang, L.; Meng, D.; Mitamura, T.; Hauptmann, A.G. Easy samples first: Self-paced reranking for zero-example multimedia search. In Proceedings of the 22nd ACM International Conference on Multimedia, Seoul, Korea, 13–21 August 2014; pp. 547–556. [Google Scholar]
- Rand, W.M. Objective criteria for the evaluation of clustering methods. J. Am. Stat. Assoc. 1971, 66, 846–850. [Google Scholar] [CrossRef]
- Hubert, L.; Arabie, P. Comparing partitions. J. Classif. 1985, 2, 193–218. [Google Scholar] [CrossRef]
- Schütze, H.; Manning, C.D.; Raghavan, P. Introduction to Information Retrieval; Cambridge University Press: Cambridge, UK, 2008; Volume 39. [Google Scholar]
- Strehl, A.; Ghosh, J. Cluster ensembles—A knowledge reuse framework for combining multiple partitions. J. Mach. Learn. Res. 2002, 3, 583–617. [Google Scholar]
- Qi, R.; Ma, A.; Ma, Q.; Zou, Q. Clustering and classification methods for single-cell RNA-sequencing data. Briefings Bioinform. 2020, 21, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Dong, X.; Freytag, S.; Lê Cao, K.A.; Su, S.; JalalAbadi, A.; Amann-Zalcenstein, D.; Weber, T.S.; Seidi, A.; Jabbari, J.S.; et al. Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat. Methods 2019, 16, 479–487. [Google Scholar] [CrossRef]
- Li, B.; Gould, J.; Yang, Y.; Sarkizova, S.; Tabaka, M.; Ashenberg, O.; Rosen, Y.; Slyper, M.; Kowalczyk, M.S.; Villani, A.C.; et al. Cumulus provides cloud-based data analysis for large-scale single-cell and single-nucleus RNA-seq. Nat. Methods 2020, 17, 793–798. [Google Scholar] [CrossRef]
- MacQueen, J. Some Methods for Classification and Analysis of Multivariate Observations. In Proceedings of the 5th Berkeley Symposium on Mathematical Statistics and Probability, Berkeley, CA, USA, 21 June–18 July 1965; pp. 281–297. [Google Scholar]
- Ding, C.; Li, T.; Peng, W.; Park, H. Orthogonal nonnegative matrix t-factorizations for clustering. In Proceedings of the 12th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 20–23 August 2006; pp. 126–135. [Google Scholar]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiselev, V.Y.; Kirschner, K.; Schaub, M.T.; Andrews, T.; Yiu, A.; Chandra, T.; Natarajan, K.N.; Reik, W.; Barahona, M.; Green, A.R.; et al. SC3: Consensus clustering of single-cell RNA-seq data. Nat. Methods 2017, 14, 483–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, S.H.; Sham, P.C.; Wang, J. Evaluation of tools for highly variable gene discovery from single-cell RNA-seq data. Briefings Bioinform. 2019, 20, 1583–1589. [Google Scholar] [CrossRef]
- Franzén, O.; Gan, L.M.; Björkegren, J.L. PanglaoDB: A web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, 2019, baz046. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Hume, A.J.; Abo, K.M.; Werder, R.B.; Villacorta-Martin, C.; Alysandratos, K.D.; Beermann, M.L.; Simone-Roach, C.; Lindstrom-Vautrin, J.; Olejnik, J.; et al. SARS-CoV-2 infection of pluripotent stem cell-derived human lung alveolar type 2 cells elicits a rapid epithelial-intrinsic inflammatory response. Cell Stem Cell 2020, 27, 962–973. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, F.; Lane, N.D.; Shu, Y.; Zeng, X.; Fang, B.; Yan, S.; Xu, H. Deep learning in the era of edge computing: Challenges and opportunities. Fog Comput. Theory Pract. 2020, 67–78. [Google Scholar] [CrossRef]
- Janiesch, C.; Zschech, P.; Heinrich, K. Machine learning and deep learning. Electron. Mark. 2021, 31, 685–695. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | # Clusters | # Cells | # Genes | Cluster Size | Reference |
|---|---|---|---|---|---|
| simulated data | 2 | 200 | 22002 | Splatter [25] | |
| baron | 14 | 1937 | 20125 | GSE84133 [26] | |
| kolodziejczyk | 3 | 704 | 32316 | E−MTAB−2600 [27] | |
| pollen | 11 | 301 | 20367 | SRP041736 [28] | |
| rca | 7 | 561 | 20949 | GSE81861 [29] | |
| goolam | 5 | 124 | 26670 | E−MTAB−3321 [30] | |
| zeisel | 9 | 3005 | 13845 | GSE60361 [31] | |
| cell lines | 4 | 1047 | 18666 | GSE126074 [32] |
| Datasets | ARI | Purity | NMI |
|---|---|---|---|
| K-means | 0.45 ± 0.93 | 52.45 ± 2.96 | 0.89 ± 1.07 |
| NMF | 9.92 ± 9.72 | 64.03 ± 7.78 | 8.20 ± 7.30 |
| ONMF | 0.47 ± 1.01 | 52.50 ± 3.00 | 1.00 ± 1.27 |
| -NMF | 0.64 ± 0.93 | 53.78 ± 2.74 | 1.29 ± 1.18 |
| Seurat | 0.00 ± 0.00 | 54.83 ± 0.06 | 0.10 ± 0.01 |
| Scanpy | 0.20 ± 0.00 | 57.52 ± 0.08 | 3.67 ± 0.13 |
| SC3 | 10.79 ± 0.95 | 63.68 ± 5.72 | 9.26 ± 1.09 |
| scSPaC | 26.69 ± 15.44 | 74.35 ± 9.11 | 22.02 ± 12.16 |
| sscSPaC | 10.89 ± 10.40 | 64.70 ± 8.01 | 10.47 ± 8.67 |
| Datasets | Baron | Goolam | Kolodziejczyk | Pollen | Rca | Zeisel | Cell Line |
|---|---|---|---|---|---|---|---|
| K-means | 35.96 ± 4.44 | 15.73 ± 3.83 | 28.56 ± 15.33 | 62.55 ± 10.25 | 3.00 ± 0.22 | 10.12 ± 3.02 | 81.46 ± 4.36 |
| NMF | 49.73 ± 9.03 | 13.26 ± 6.60 | 37.38 ± 6.56 | 79.39 ± 4.88 | 11.33 ± 0.61 | 24.21 ± 2.96 | 79.85 ± 1.98 |
| ONMF | 50.03 ± 11.03 | 22.16 ± 4.48 | 40.73 ± 3.26 | 77.50 ± 4.51 | 6.83 ± 0.23 | 24.54 ± 4.89 | 80.29 ± 3.75 |
| -NMF | 43.21 ± 4.16 | 33.61 ± 5.34 | 39.48 ± 2.23 | 76.66 ± 4.92 | 7.70 ± 0.98 | 35.83 ± 4.17 | 82.53 ± 4.26 |
| Seurat | 61.82 ± 0.18 | 47.63 ± 0.08 | 50.97 ± 0.82 | 81.82 ± 0.12 | 52.41 ± 0.08 | 52.73 ± 0.82 | 69.73 ± 0.12 |
| Scanpy | 74.91 ± 0.24 | 54.25 ± 0.16 | 45.37 ± 1.22 | 84.91 ± 0.10 | 54.5 ± 0.16 | 48.46 ± 0.92 | 82.61 ± 0.10 |
| SC3 | 79.62 ± 3.44 | 57.52 ± 2.38 | 47.57 ± 3.64 | 91.62 ± 3.93 | 59.8 ± 3.30 | 49.78 ± 2.88 | 88.36 ± 5.14 |
| scSPaC | 83.57 ± 8.00 | 58.43 ± 4.78 | 48.90 ± 2.55 | 88.16 ± 3.73 | 57.02 ± 1.75 | 62.57 ± 3.39 | 91.71 ± 3.68 |
| sscSPaC | 78.84 ± 2.70 | 60.6 ± 4.97 | 51.48 ± 2.52 | 89.27 ± 5.40 | 58.49 ± 3.59 | 64.75 ± 2.09 | 90.37 ± 5.09 |
| Datasets | Baron | Goolam | Kolodziejczyk | Pollen | Rca | Zeisel | Cell Line |
|---|---|---|---|---|---|---|---|
| K-means | 71.95 ± 2.18 | 57.66 ± 2.82 | 62.66 ± 9.25 | 77.54 ± 7.79 | 30.42 ± 0.19 | 49.57 ± 2.75 | 86.43 ± 0.12 |
| NMF | 82.56 ± 2.85 | 59.23 ± 2.79 | 68.27 ± 3.31 | 90.02 ± 3.18 | 31.37 ± 0.54 | 60.69 ± 2.46 | 81.74 ± 0.1 |
| ONMF | 80.92 ± 4.01 | 59.23 ± 1.95 | 69.49 ± 1.4 | 88.34 ± 3.8 | 31.01 ± 0.4 | 58.34 ± 2.26 | 82.18 ± 0.01 |
| -NMF | 92.35 ± 1.47 | 70.85 ± 4.16 | 69.22 ± 0.94 | 91.01 ± 1.74 | 32.07 ± 1.13 | 66.3 ± 2.64 | 87.81 ± 0.1 |
| Seurat | 86.15 ± 0.26 | 72.18 ± 0.04 | 81.36 ± 0.02 | 86.15 ± 0.17 | 72.91 ± 0.01 | 51.99 ± 0.02 | 79.52 ± 0.04 |
| Scanpy | 87.89 ± 0.06 | 75.63 ± 0.64 | 76.44 ± 0.1 | 93.69 ± 0.06 | 78.59 ± 0.64 | 50.68 ± 0.1 | 88.41 ± 0.03 |
| SC3 | 90.72 ± 2.28 | 76.59 ± 2.76 | 78.13 ± 3.51 | 94.95 ± 2.76 | 86.83 ± 1.08 | 78.14 ± 3.01 | 92.75 ± 0.09 |
| scSPaC | 93.26 ± 2.42 | 78.39 ± 1.88 | 79.03 ± 3.48 | 96.21 ± 2.05 | 83.22 ± 2.07 | 89.05 ± 1.91 | 93.94 ± 1.58 |
| sscSPaC | 92.94 ± 1.39 | 83.14 ± 3.7 | 81.85 ± 4.16 | 95.83 ± 4.08 | 84.85 ± 1.92 | 87.81 ± 2.28 | 93.18 ± 1.45 |
| Datasets | Baron | Goolam | Kolodziejczyk | Pollen | Rca | Zeisel | Cell Line |
|---|---|---|---|---|---|---|---|
| K-means | 42.77 ± 3.74 | 20.2 ± 5.43 | 32.85 ± 16.3 | 80.57 ± 6.05 | 1.39 ± 0.19 | 19.15 ± 3.56 | 79.47 ± 2.39 |
| NMF | 62.11 ± 4.29 | 17.34 ± 6.42 | 42.43 ± 5.59 | 91.09 ± 2.4 | 2.62 ± 0.72 | 35.53 ± 2.22 | 80.81 ± 3.73 |
| ONMF | 60.77 ± 4.89 | 16.07 ± 3.87 | 44.33 ± 2.65 | 89.94 ± 2.84 | 2.15 ± 0.5 | 33.48 ± 2.86 | 80.45 ± 2.11 |
| -NMF | 64.75 ± 1.93 | 51.95 ± 4.02 | 44.15 ± 1.78 | 91.61 ± 1.80 | 5.98 ± 1.38 | 38.76 ± 2.34 | 84.86 ± 2.91 |
| Seurat | 61.57 ± 0.23 | 43.23 ± 0.07 | 51.54 ± 0.02 | 86.11 ± 0.07 | 38.92 ± 0.04 | 52.03 ± 0.02 | 63.62 ± 0.07 |
| Scanpy | 73.98 ± 0.22 | 54.9 ± 0.07 | 49.56 ± 0.03 | 89.33 ± 0.12 | 36.02 ± 0.03 | 44.25 ± 0.03 | 80.46 ± 0.12 |
| SC3 | 80.23 ± 2.72 | 56.59 ± 3.13 | 52.67 ± 6.64 | 91.25 ± 3.4 | 52.63 ± 3.57 | 50.01 ± 4.28 | 82.75 ± 3.14 |
| scSPaC | 79.82 ± 3.48 | 59.02 ± 5.48 | 53.69 ± 3.35 | 89.09 ± 1.86 | 51.70 ± 0.41 | 63.97 ± 2.12 | 89.96 ± 4.51 |
| sscSPaC | 81.94 ± 2.63 | 58.48 ± 3.78 | 56.81 ± 4.24 | 91.42 ± 5.13 | 54.96 ± 4.14 | 63.41 ± 2.68 | 87.23 ± 3.37 |
| ARI around Evaluate K by Scanpy (K ) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Datasets | Ref. K | Evaluate K by Scanpy | Best K by scSPaC | K − 3 | K − 2 | K − 1 | K | K + 1 | K + 2 | K + 3 |
| simulated data | 2 | 2 | 2 | – | – | – | 0.2662 | 0.2448 | 0.2567 | 0.2489 |
| baron | 14 | 13 | 11 | 0.7808 | 0.8357 | 0.8319 | 0.8094 | 0.7862 | 0.8249 | 0.7727 |
| goolam | 5 | 5 | 5 | 0.4227 | 0.4518 | 0.4615 | 0.5843 | 0.5758 | 0.5661 | 0.5732 |
| Kolodziejczyk | 3 | 8 | 5 | 0.4890 | 0.4863 | 0.4875 | 0.4671 | 0.4679 | 0.4628 | 0.4605 |
| pollen | 11 | 8 | 10 | 0.7098 | 0.7172 | 0.7893 | 0.8764 | 0.8753 | 0.8816 | 0.8612 |
| Rca | 7 | 9 | 8 | 0.5475 | 0.5419 | 0.5702 | 0.5671 | 0.5623 | 0.5453 | 0.5286 |
| zeisel | 9 | 13 | 10 | 0.6257 | 0.6246 | 0.6241 | 0.6078 | 0.5793 | 0.5641 | 0.5632 |
| cell line | 4 | 4 | 4 | – | 0.5468 | 0.7025 | 0.9171 | 0.9043 | 0.9102 | 0.8954 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, P.; Xu, Z.; Chen, J.; Ren, Y.; King, I. Single Cell Self-Paced Clustering with Transcriptome Sequencing Data. Int. J. Mol. Sci. 2022, 23, 3900. https://doi.org/10.3390/ijms23073900
Zhao P, Xu Z, Chen J, Ren Y, King I. Single Cell Self-Paced Clustering with Transcriptome Sequencing Data. International Journal of Molecular Sciences. 2022; 23(7):3900. https://doi.org/10.3390/ijms23073900
Chicago/Turabian StyleZhao, Peng, Zenglin Xu, Junjie Chen, Yazhou Ren, and Irwin King. 2022. "Single Cell Self-Paced Clustering with Transcriptome Sequencing Data" International Journal of Molecular Sciences 23, no. 7: 3900. https://doi.org/10.3390/ijms23073900
APA StyleZhao, P., Xu, Z., Chen, J., Ren, Y., & King, I. (2022). Single Cell Self-Paced Clustering with Transcriptome Sequencing Data. International Journal of Molecular Sciences, 23(7), 3900. https://doi.org/10.3390/ijms23073900