
Disease Modeling of Rare Neurological Disorders in Zebrafish



Abstract
:1. Introduction
2. Model Organisms for Rare Disease Research
2.1. Caenorhabditis elegans (C. elegans)
2.2. Fruit Fly (Drosophila melanogaster)
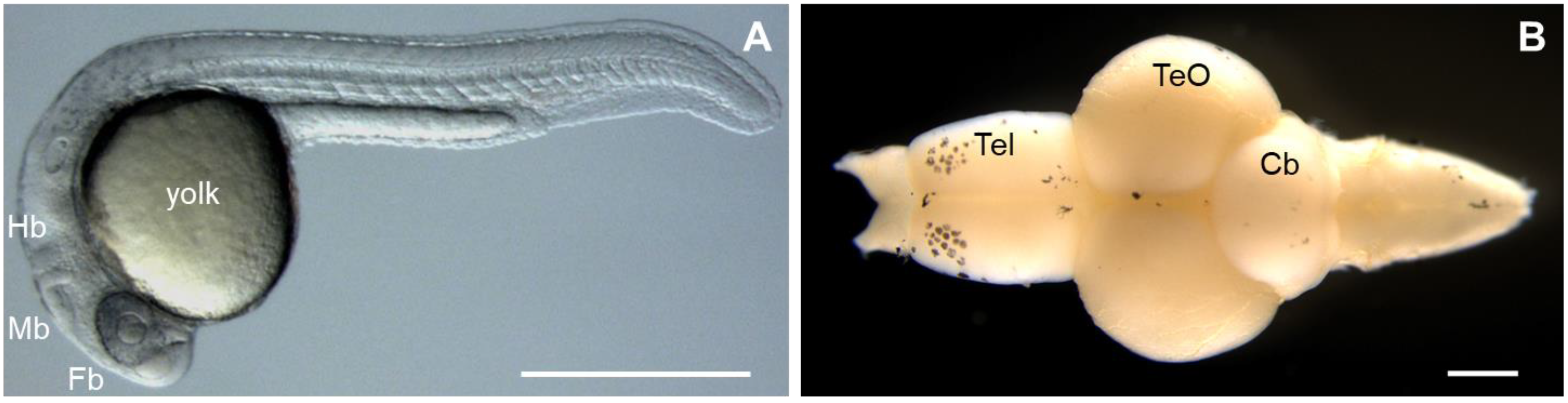
2.3. Zebrafish (Danio rerio)
3. Zebrafish Models for Rare Neurological Disorders
3.1. Kallmann Syndrome (WDR11)
3.2. Potocki–Shaffer Syndrome (PHF21A)
3.3. Miles–Carpenter Syndrome (ZC4H2)
3.4. The 12q24.31 Microdeletion Syndrome (KDM2B)
3.5. Down Syndrome and Autism (DYRK1A)
3.6. The 12q14.1 Deletion Syndrome (SAM2)
3.7. Armfield XLID Syndrome (FAM50A)
3.8. Leukodystrophy (Hypomyelination)
3.8.1. Leukodystrophy with Vanishing White Matter (VWM)/Childhood Ataxia with CNS Hypomyelination (CACH)
3.8.2. Charcot–Marie–Tooth Diseases (CMT)
3.9. Amyotrophic Lateral Sclerosis (Lou Gehrig’s Disease)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wangler, M.F.; Yamamoto, S.; Chao, H.T.; Posey, J.E.; Westerfield, M.; Postlethwait, J.; Members of the Undiagnosed Diseases Network (UDN); Hieter, P.; Boycott, K.M.; Campeau, P.M.; et al. Model Organisms Facilitate Rare Disease Diagnosis and Therapeutic Research. Genetics 2017, 207, 9–27. [Google Scholar] [CrossRef] [PubMed]
- The Lancet Neurology. Rare neurological diseases: A united approach is needed. Lancet Neurol. 2011, 10, 109. [Google Scholar] [CrossRef]
- Field, M.J.; Boat, T.F. Rare Diseases and Orphan Products: Accelerating Research and Development; National Academies Press: Washington, DC, USA, 2010; pp. 51–69. [Google Scholar]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell. Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- National Research Council (US) Committee on New and Emerging Models in Biomedical and Behavioral Research. Biomedical Models and Resources: Current Needs and Future Opportunities; National Academies Press: Washington, DC, USA, 1998; pp. 24–39.
- Bellen, H.J.; Tong, C.; Tsuda, H. 100 years of Drosophila research and its impact on vertebrate neuroscience: A history lesson for the future. Nat. Rev. Neurosci. 2010, 11, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, R.L.; Ricci, C.; Melone, G. A cladistic analysis of pseudocoelomate (aschelminth) morphology. Invertebr. Biol. 1996, 115, 104–112. [Google Scholar] [CrossRef]
- Schafer, W.R. Deciphering the neural and molecular mechanisms of C. elegans behavior. Curr. Biol. 2005, 15, R723–R729. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science Taylor and Francis Group: New York, NY, USA, 2015. [Google Scholar]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Boycott, K.M.; Rath, A.; Chong, J.X.; Hartley, T.; Alkuraya, F.S.; Baynam, G.; Brookes, A.J.; Brudno, M.; Carracedo, A.; den Dunnen, J.T.; et al. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am. J. Hum. Genet. 2017, 100, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Girard, L.R.; Fiedler, T.J.; Harris, T.W.; Carvalho, F.; Antoshechkin, I.; Han, M.; Sternberg, P.W.; Stein, L.D.; Chalfie, M. WormBook: The online review of Caenorhabditis elegans biology. Nucleic Acids Res. 2007, 35, D472–D475. [Google Scholar] [CrossRef]
- Lai, C.H.; Chou, C.Y.; Ch’ang, L.Y.; Liu, C.S.; Lin, W. Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res. 2000, 10, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenyon, C. A conserved regulatory system for aging. Cell 2001, 105, 165–168. [Google Scholar] [CrossRef] [Green Version]
- O’Kane, C.J. Modelling human diseases in Drosophila and Caenorhabditis. Semin. Cell Dev. Biol. 2003, 14, 3–10. [Google Scholar] [CrossRef]
- Schulenburg, H.; Kurz, C.L.; Ewbank, J.J. Evolution of the innate immune system: The worm perspective. Immunol. Rev. 2004, 198, 36–58. [Google Scholar] [CrossRef]
- Riessland, M.; Kaczmarek, A.; Schneider, S.; Swoboda, K.J.; Löhr, H.; Bradler, C.; Grysko, V.; Dimitriadi, M.; Hosseinibarkooie, S.; Torres-Benito, L.; et al. Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis. Am. J. Hum. Genet. 2017, 100, 297–315. [Google Scholar] [CrossRef] [Green Version]
- Bellen, H.J.; Yamamoto, S. Morgan’s legacy: Fruit flies and the functional annotation of conserved genes. Cell 2015, 163, 12–14. [Google Scholar] [CrossRef] [Green Version]
- Wangler, M.F.; Yamamoto, S.; Bellen, H.J. Fruit flies in biomedical research. Genetics 2015, 199, 639–653. [Google Scholar] [CrossRef]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Al-Ouran, R.; Hu, Y.; Kim, S.Y.; Wan, Y.W.; Wangler, M.F.; Yamamoto, S.; Chao, H.T.; Comjean, A.; Mohr, S.E.; et al. MARRVEL: Integration of Human and Model Organism Genetic Resources to Facilitate Functional Annotation of the Human Genome. Am. J. Hum. Genet. 2017, 100, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Flockhart, I.; Vinayagam, A.; Bergwitz, C.; Berger, B.; Perrimon, N.; Mohr, S.E. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinform. 2011, 12, 357. [Google Scholar] [CrossRef] [Green Version]
- Bellen, H.J.; Wangler, M.F.; Yamamoto, S. The fruit fly at the interface of diagnosis and pathogenic mechanisms of rare and common human diseases. Hum. Mol. Genet. 2019, 28, R207–R214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, N.; Chung, H.; Jolly, A.; Withers, M.; Tepe, B.; Arenkiel, B.R.; Shah, P.S.; Krogan, N.J.; Aydin, H.; Geckinli, B.B.; et al. Mutations in ANKLE2, a ZIKA Virus Target, Disrupt an Asymmetric Cell Division Pathway in Drosophila Neuroblasts to Cause Microcephaly. Dev. Cell. 2019, 51, 713–729. [Google Scholar] [CrossRef] [PubMed]
- Detrich, H.W., III; Westerfield, M.; Zon, L.I. Overview of the zebrafish system. Methods Cell Biol. 1998, 59, 3–10. [Google Scholar]
- Eisen, J.S. History of zebrafish research. In The Zebrafish in Biomedical Research; Academic Press: Cambridge, MA, USA, 2020; pp. 3–14. [Google Scholar]
- Kimmel, C.B. Genetics and early development of zebrafish. Trends Genet. 1989, 5, 283–288. [Google Scholar] [CrossRef]
- Solnica-Krezel, L.; Stemple, D.L.; Driever, W. Transparent things: Cell fates and cell movements during early embryogenesis of zebrafish. BioEssays 1995, 17, 931–939. [Google Scholar] [CrossRef]
- Cooper, M.S.; D’Amico, L.A.; Henry, C.A. Analyzing morphogenetic cell behaviors in vitally stained zebrafish embryos. Methods Mol. Biol. 1999, 122, 185–204. [Google Scholar]
- Spitsbergen, J.M.; Kent, M.L. The state of the art of the zebrafish model for toxicology and toxicologic pathology research—Advantages and current limitations. Toxicol. Pathol. 2003, 31, 62–87. [Google Scholar]
- Carney, T.J.; Mosimann, C. Switch and Trace: Recombinase Genetics in Zebrafish. Trends Genet. 2018, 34, 362–378. [Google Scholar] [CrossRef]
- Adamson, K.I.; Sheridan, E.; Grierson, A.J. Use of zebrafish models to investigate rare human disease. J. Med. Genet. 2018, 55, 641–649. [Google Scholar] [CrossRef]
- Ahmad, G.; Amiji, M. Use of CRISPR/Cas9 gene-editing tools for developing models in drug discovery. Drug Discov. Today 2018, 23, 519–533. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, T.; Lyon, A.N.; Pineda, R.H.; Wang, C.; Janssen, P.M.; Canan, B.D.; Burghes, A.H.; Beattie, C.E. A genetic model of amyotrophic lateral sclerosis in zebrafish displays phenotypic hallmarks of motoneuron disease. Dis. Models Mech. 2010, 3, 652–662. [Google Scholar] [CrossRef] [Green Version]
- Bourefis, A.R.; Campanari, M.L.; Buee-Scherrer, V.; Kabashi, E. Functional characterization of a FUS mutant zebrafish line as a novel genetic model for ALS. Neurobiol. Dis. 2020, 142, 104935. [Google Scholar] [CrossRef] [PubMed]
- Hewamadduma, C.A.; Grierson, A.J.; Ma, T.P.; Pan, L.; Moens, C.B.; Ingham, P.W.; Ramesh, T.; Shaw, P.J. Tardbpl splicing rescues motor neuron and axonal development in a mutant tardbp zebrafish. Hum. Mol. Genet. 2013, 22, 2376–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, M.P.; Higginbottom, A.; McGown, A.; Castelli, L.M.; James, E.; Hautbergue, G.M.; Shaw, P.J.; Ramesh, T.M. Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol. Commun. 2018, 6, 125. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Khan, K.; Armfield-Uhas, K.; Srikanth, S.; Thompson, N.A.; Pardo, M.; Yu, L.; Norris, J.W.; Peng, Y.; Gripp, K.W.; et al. Mutations in FAM50A suggest that Armfield XLID syndrome is a spliceosomopathy. Nat. Commun. 2020, 11, 3698. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.H.; Cho, H.J.; Han, E.; Hong, T.I.; Ariyasiri, K.; Choi, J.H.; Hwang, K.S.; Jeong, Y.M.; Yang, S.Y.; Yu, K.; et al. Zebrafish knockout of Down syndrome gene, DYRK1A, shows social impairments relevant to autism. Mol. Autism 2017, 8, 50. [Google Scholar] [CrossRef]
- Kim, H.G.; Ahn, J.W.; Kurth, I.; Ullmann, R.; Kim, H.T.; Kulharya, A.; Ha, K.S.; Itokawa, Y.; Meliciani, I.; Wenzel, W.; et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 2010, 87, 465–479. [Google Scholar] [CrossRef] [Green Version]
- Deml, B.; Reis, L.M.; Muheisen, S.; Bick, D.; Semina, E.V. EFTUD2 deficiency in vertebrates: Identification of a novel human mutation and generation of a zebrafish model. Birth Defects Res. A Clin. Mol. Teratol. 2015, 103, 630–640. [Google Scholar] [CrossRef] [Green Version]
- May, M.; Hwang, K.S.; Miles, J.; Williams, C.; Niranjan, T.; Kahler, S.G.; Chiurazzi, P.; Steindl, K.; Van Der Spek, P.J.; Swagemakers, S.; et al. ZC4H2, an XLID gene, is required for the generation of a specific subset of CNS interneurons. Hum. Mol. Genet. 2015, 24, 4848–4861. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.G.; Kim, H.T.; Leach, N.T.; Lan, F.; Ullmann, R.; Silahtaroglu, A.; Kurth, I.; Nowka, A.; Seong, I.S.; Shen, Y.; et al. Translocations disrupting PHF21A in the Potocki-Shaffer-syndrome region are associated with intellectual disability and craniofacial anomalies. Am. J. Hum. Genet. 2012, 91, 56–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaro, F.P.; Alvizi, L.; Zechi-Ceide, R.M.; Bertola, D.; Felix, T.M.; de Souza, J.; Raskin, S.; Twigg, S.R.; Weiner, A.M.; Armas, P.; et al. A noncoding expansion in EIF4A3 causes Richieri-Costa-Pereira syndrome, a craniofacial disorder associated with limb defects. Am. J. Hum. Genet. 2014, 94, 120–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keefe, M.D.; Soderholm, H.E.; Shih, H.Y.; Stevenson, T.J.; Glaittli, K.A.; Bowles, D.M.; Scholl, E.; Colby, S.; Merchant, S.; Hsu, E.W.; et al. Vanishing white matter disease expression of truncated EIF2B5 activates induced stress response. eLife. 2020, 9, e56319. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Kim, S.H.; Ben-Mahmoud, A.; Kim, O.H.; Choi, T.I.; Lee, K.H.; Ku, B.; Eum, J.; Kee, Y.; Lee, S.; et al. Eif2b3 mutants recapitulate phenotypes of vanishing white matter disease and validate novel disease alleles in zebrafish. Hum. Mol. Genet. 2021, 30, 331–342. [Google Scholar] [CrossRef]
- Choi, J.H.; Jeong, Y.M.; Kim, S.; Lee, B.; Ariyasiri, K.; Kim, H.T.; Jung, S.H.; Hwang, K.S.; Choi, T.I.; Park, C.O.; et al. Targeted knockout of a chemokine-like gene increases anxiety and fear responses. Proc. Natl. Acad. Sci. USA 2018, 115, E1041–E1050. [Google Scholar] [CrossRef] [Green Version]
- Bishop, K.M.; Garel, S.; Nakagawa, Y.; Rubenstein, J.L.; O’Leary, D.D. Emx1 and Emx2 cooperate to regulate cortical size, lamination, neuronal differentiation, development of cortical efferents, and thalamocortical pathfinding. J. Comp. Neurol. 2003, 457, 345–360. [Google Scholar] [CrossRef]
- Wu, Y.Q.; Badano, J.L.; McCaskill, C.; Vogel, H.; Potocki, L.; Shaffer, L.G. Haploinsufficiency of ALX4 as a potential cause of parietal foramina in the 11p11.2 contiguous gene-deletion syndrome. Am. J. Hum. Genet. 2000, 67, 1327–1332. [Google Scholar] [CrossRef] [Green Version]
- Wakui, K.; Gregato, G.; Ballif, B.C.; Glotzbach, C.D.; Bailey, K.A.; Kuo, P.L.; Sue, W.C.; Sheffield, L.J.; Irons, M.; Gomez, E.G.; et al. Construction of a natural panel of 11p11.2 deletions and further delineation of the critical region involved in Potocki-Shaffer syndrome. Eur. J. Hum. Genet. 2005, 13, 528–540. [Google Scholar] [CrossRef] [Green Version]
- Wuyts, W.; Waeber, G.; Meinecke, P.; Schüler, H.; Goecke, T.O.; Van Hul, W.; Bartsch, O. Proximal 11p deletion syndrome (P11pDS): Additional evaluation of the clinical and molecular aspects. Eur. J. Hum. Genet. 2004, 12, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Wuyts, W.; Di Gennaro, G.; Bianco, F.; Wauters, J.; Morocutti, C.; Pierelli, F.; Bossuyt, P.; Van Hul, W.; Casali, C. Molecular and clinical examination of an Italian DEFECT11 family. Eur. J. Hum. Genet. 1999, 7, 579–584. [Google Scholar] [CrossRef]
- Bartsch, O.; Wuyts, W.; Van Hul, W.; Hecht, J.T.; Meinecke, P.; Hogue, D.; Werner, W.; Zabel, B.; Hinkel, G.K.; Powell, C.M.; et al. Delineation of a contiguous gene syndrome with multiple exostoses, enlarged parietal foramina, craniofacial dysostosis, and mental retardation, caused by deletions in the short arm of chromosome 11. Am. J. Hum. Genet. 1996, 58, 734–742. [Google Scholar] [PubMed]
- Yamamoto, T.; Akaboshi, S.; Ninomiya, H.; Nanba, E. DEFECT 11 syndrome associated with agenesis of the corpus callosum. J. Med. Genet. 2001, 38, E5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stickens, D.; Clines, G.; Burbee, D.; Ramos, P.; Thomas, S.; Hogue, D.; Hecht, J.T.; Lovett, M.; Evans, G.A. The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nat. Genet. 1996, 14, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Mavrogiannis, L.A.; Antonopoulou, I.; Baxová, A.; Kutílek, S.; Kim, C.A.; Sugayama, S.M.; Salamanca, A.; Wall, S.A.; Morriss-Kay, G.M.; Wilkie, A.O. Haploinsufficiency of the human homeobox gene ALX4 causes skull ossification defects. Nat. Genet. 2001, 27, 17–18. [Google Scholar] [CrossRef]
- Kim, H.G.; Rosenfeld, J.A.; Scott, D.A.; Bénédicte, G.; Labonne, J.D.; Brown, J.; McGuire, M.; Mahida, S.; Naidu, S.; Gutierrez, J.; et al. Disruption of PHF21A causes syndromic intellectual disability with craniofacial anomalies, epilepsy, hypotonia, and neurobehavioral problems including autism. Mol. Autism 2019, 10, 35. [Google Scholar] [CrossRef]
- Miles, J.H.; Carpenter, N.J. Unique X-linked mental retardation syndrome with fingertip arches and contractures linked to Xq21.31. Am. J. Med. Genet. 1991, 38, 215–223. [Google Scholar] [CrossRef]
- Neri, G.; Schwartz, C.E.; Lubs, H.A.; Stevenson, R.E. X-linked intellectual disability update 2017. Am. J. Med. Genet. A 2018, 176, 1375–1388. [Google Scholar] [CrossRef] [Green Version]
- Labonne, J.D.; Lee, K.H.; Iwase, S.; Kong, I.K.; Diamond, M.P.; Layman, L.C.; Kim, C.H.; Kim, H.G. An atypical 12q24.31 microdeletion implicates six genes including a histone demethylase KDM2B and a histone methyltransferase SETD1B in syndromic intellectual disability. Hum. Genet. 2016, 135, 757–771. [Google Scholar] [CrossRef]
- Fukuda, T.; Tokunaga, A.; Sakamoto, R.; Yoshida, N. Fbxl10/Kdm2b deficiency accelerates neural progenitor cell death and leads to exencephaly. Mol. Cell. Neurosci. 2011, 46, 614–624. [Google Scholar] [CrossRef]
- Soppa, U.; Schumacher, J.; Florencio Ortiz, V.; Pasqualon, T.; Tejedor, F.J.; Becker, W. The Down syndrome-related protein kinase DYRK1A phosphorylates p27(Kip1) and Cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle 2014, 13, 2084–2100. [Google Scholar] [CrossRef] [Green Version]
- van Bon, B.W.; Hoischen, A.; Hehir-Kwa, J.; de Brouwer, A.P.; Ruivenkamp, C.; Gijsbers, A.C.; Marcelis, C.L.; de Leeuw, N.; Veltman, J.A.; Brunner, H.G.; et al. Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clin. Genet. 2011, 79, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Ruaud, L.; Mignot, C.; Guët, A.; Ohl, C.; Nava, C.; Héron, D.; Keren, B.; Depienne, C.; Benoit, V.; Maystadt, I.; et al. DYRK1A mutations in two unrelated patients. Eur. J. Med. Genet. 2015, 58, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Bronicki, L.M.; Redin, C.; Drunat, S.; Piton, A.; Lyons, M.; Passemard, S.; Baumann, C.; Faivre, L.; Thevenon, J.; Rivière, J.B.; et al. Ten new cases further delineate the syndromic intellectual disability phenotype caused by mutations in DYRK1A. Eur. J. Med. Genet. 2015, 23, 1482–1487. [Google Scholar]
- van Bon, B.W.; Coe, B.P.; Bernier, R.; Green, C.; Gerdts, J.; Witherspoon, K.; Kleefstra, T.; Willemsen, M.H.; Kumar, R.; Bosco, P.; et al. Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol. Psychiatry 2016, 21, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Gerlai, R. Zebrafish antipredatory responses: A future for translational research? Behav. Brain Res. 2010, 207, 223–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, K.J.; Hawkins, T.A.; Yáñez, J.; Anadón, R.; Wilson, S.W.; Folgueira, M. Afferent Connectivity of the Zebrafish Habenulae. Front. Neural Circuits 2016, 10, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.B.; Yao, Y.Y.; Zhang, H.F.; Kawakami, K.; Du, J.L. Left Habenula Mediates Light-Preference Behavior in Zebrafish via an Asymmetrical Visual Pathway. Neuron 2017, 93, 914–928.e4. [Google Scholar] [CrossRef] [Green Version]
- Hikosaka, O. The habenula: From stress evasion to value-based decision-making. Nat. Rev. Neurosci. 2010, 11, 503–513. [Google Scholar] [CrossRef]
- Amo, R.; Aizawa, H.; Takahoko, M.; Kobayashi, M.; Takahashi, R.; Aoki, T.; Okamoto, H. Identification of the zebrafish ventral habenula as a homolog of the mammalian lateral habenula. J. Neurosci. 2010, 30, 1566–1574. [Google Scholar] [CrossRef]
- Morton, N.E.; Rao, D.C.; Lang-Brown, H.; Maclean, C.J.; Bart, R.D.; Lew, R. Colchester revisited: A genetic study of mental defect. J. Med. Genet. 1977, 14, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Armfield, K.; Nelson, R.; Lubs, H.A.; Häne, B.; Schroer, R.J.; Arena, F.; Schwartz, C.E.; Stevenson, R.E. X-linked mental retardation syndrome with short stature, small hands and feet, seizures, cleft palate, and glaucoma is linked to Xq28. Am. J. Med. Genet. 1999, 85, 236–242. [Google Scholar] [CrossRef]
- Lei, L.; Yan, S.Y.; Yang, R.; Chen, J.Y.; Li, Y.; Bu, Y.; Chang, N.; Zhou, Q.; Zhu, X.; Li, C.Y.; et al. Spliceosomal protein eftud2 mutation leads to p53-dependent apoptosis in zebrafish neural progenitors. Nucleic Acids Res. 2017, 45, 3422–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Knaap, M.S.; Breiter, S.N.; Naidu, S.; Hart, A.A.; Valk, J. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology 1999, 213, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Anzil, A.P.; Gessaga, E. Late-life cavitating dystrophy of the cerebral and cerebellar white matter. A form of sudanophil leucodystrophy. Eur. Neurol. 1972, 7, 79–94. [Google Scholar] [CrossRef]
- Deisenhammer, E.; Jellinger, K. Höhlenbildende Neutralfett-Leukodystrophie mit Schubverlauf. Neuropadiatrie 1976, 7, 111–121. [Google Scholar] [CrossRef]
- Gautier, J.C.; Gray, F.; Awada, A.; Escourolle, R. Leucodystrophie orthochromatique cavitaire de l’adulte. Prolifération et inclusions oligodendrogliales. Rev. Neurol. 1984, 140, 493–501. [Google Scholar]
- Girard, P.F.; Tommasi, M.; Rochet, M.; Boucher, M. Leuco-encéphalopathie avec cavitations massives, bilatérales et symétriques. Syndrome de décortication post-traumatique. Presse Med. 1968, 76, 163–166. [Google Scholar]
- Graveleau, P.; Gray, F.; Plas, J.; Graveleau, J.; Brion, S. Leucodystrophie orthochromatique cavitaire avec modifications oligodendrogliales. Un cas sporadique adulte [Cavitary orthochromatic leukodystrophy with oligodendroglial changes. A sporadic adult case]. Rev. Neurol. 1985, 141, 713–718. [Google Scholar]
- Macchi, G.; Taramelli, M.; Borri, P.F. Su di un caso di leucopatia spongio-cavitaria. Acta Neurol. 1969, 24, 565–571. [Google Scholar]
- Watanabe, I.; Muller, J. Cavitating “diffuse sclerosis”. J. Neuropathol. Exp. Neurol. 1967, 26, 437–455. [Google Scholar] [CrossRef]
- De Jonghe, P.; Timmerman, V.; Nelis, E.; Martin, J.J.; Van Broeckhoven, C. Charcot-Marie-Tooth disease and related peripheral neuropathies. J. Peripher. Nerv. Syst. 1997, 2, 370–387. [Google Scholar] [PubMed]
- Ramchandren, S. Charcot-Marie-Tooth Disease and Other Genetic Polyneuropathies. Continuum (Minneap Minn). 2017, 23, 1360–1377. [Google Scholar] [CrossRef] [PubMed]
- Saporta, A.S.; Sottile, S.L.; Miller, L.J.; Feely, S.M.; Siskind, C.E.; Shy, M.E. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann. Neurol. 2011, 69, 22–33. [Google Scholar] [CrossRef]
- Jones, E.A.; Brewer, M.H.; Srinivasan, R.; Krueger, C.; Sun, G.; Charney, K.N.; Keles, S.; Antonellis, A.; Svaren, J. Distal enhancers upstream of the Charcot-Marie-Tooth type 1A disease gene PMP22. Hum. Mol. Genet. 2012, 21, 1581–1591. [Google Scholar] [CrossRef] [Green Version]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Gitcho, M.A.; Baloh, R.H.; Chakraverty, S.; Mayo, K.; Norton, J.B.; Levitch, D.; Hatanpaa, K.J.; White, C.L., 3rd; Bigio, E.H.; Caselli, R.; et al. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 2008, 63, 535–538. [Google Scholar] [CrossRef] [Green Version]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Rademakers, R. The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Opin. Neurol. 2008, 21, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Valdmanis, P.N.; Rouleau, G.A. Genetics of familial amyotrophic lateral sclerosis. Neurology 2008, 70, 144–152. [Google Scholar] [CrossRef]
- Elshafey, A.; Lanyon, W.G.; Connor, J.M. Identification of a new missense point mutation in exon 4 of the Cu/Zn superoxide dismutase (SOD-1) gene in a family with amyotrophic lateral sclerosis. Hum. Mol. Genet. 1994, 3, 363–364. [Google Scholar] [CrossRef]
- Turner, B.J.; Talbot, K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 2008, 85, 94–134. [Google Scholar] [CrossRef]
- Buratti, E.; Baralle, F.E. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci. 2008, 13, 867–878. [Google Scholar] [CrossRef] [Green Version]
- Lagier-Tourenne, C.; Cleveland, D.W. Rethinking ALS: The FUS about TDP-43. Cell 2009, 136, 1001–1004. [Google Scholar] [CrossRef] [Green Version]
- Zinszner, H.; Sok, J.; Immanuel, D.; Yin, Y.; Ron, D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci. 1997, 110, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 2008, 121, 3778–3785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rademakers, R.; Stewart, H.; Dejesus-Hernandez, M.; Krieger, C.; Graff-Radford, N.; Fabros, M.; Briemberg, H.; Cashman, N.; Eisen, A.; Mackenzie, I.R. Fus gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve 2010, 42, 170–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: Summary and update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef] [PubMed]
- Hicks, G.G.; Singh, N.; Nashabi, A.; Mai, S.; Bozek, G.; Klewes, L.; Arapovic, D.; White, E.K.; Koury, M.J.; Oltz, E.M.; et al. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat. Genet. 2000, 24, 175–179. [Google Scholar] [CrossRef]
- Frickenhaus, M.; Wagner, M.; Mallik, M.; Catinozzi, M.; Storkebaum, E. Highly efficient cell-type-specific gene inactivation reveals a key function for the Drosophila FUS homolog cabeza in neurons. Sci. Rep. 2015, 5, 9107. [Google Scholar] [CrossRef] [Green Version]
- Lissouba, A.; Liao, M.; Kabashi, E.; Drapeau, P. Transcriptomic Analysis of Zebrafish TDP-43 Transgenic Lines. Front. Mol. Neurosci. 2018, 11, 463. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Program/Network Name | Goals | Homepage Address |
|---|---|---|
| Genetic and Rare Diseases Information Center | Providing the general public with the latest information on various rare diseases in an easy-to-understand manner. | https://rarediseases.info.nih.gov/diseases |
| International rare disease research consortium | Contributing to the development of new treatments for rare diseases and methods to uncover the genetic causes of rare diseases. | https://irdirc.org/ |
| National Organization for Rare Disorders | Raising awareness of rare diseases and improve access to treatment and medical services for patients and their families. | https://rarediseases.org |
| Orphanet (global network) | Providing international reference knowledge base for rare diseases and orphan drugs. | https://www.orpha.net/ |
| Providing the scientific datasets (research, clinical trials, drugs, etc.) related to rare diseases and orphan drugs. | http://www.orphadata.org/ | |
| Rare Diseases Clinical Research Network | Providing support for clinical studies and facilitating collaboration, study enrollment, and data sharing. | https://ncats.nih.gov/rdcrn |
| Undiagnosed Diseases Network | Accelerating identification of genetic causes of rare diseases by validating candidate genes, using model organisms. | https://undiagnosed.hms.harvard.edu/research |
| Gene Name | Related Disease | Zebrafish Phenotype | Publication |
|---|---|---|---|
| sod1 | Amyotrophic Lateral Sclerosis | Motor neuron loss Muscle atrophy | [36] |
| fus | Shortened motor neuron length Decreased neuromuscular junction Impaired motor behavior Decreased life span Increase of the smallest tau transcripts | [37] | |
| tardbp | Axonopathy of the motor neurons Premature of axonal branch | [38] | |
| c9orf72 | Impaired motor behavior Cognitive impairment Muscle atrophy Motor neuron loss | [39] | |
| fam50a | Armfield XLID syndrome | Abnormal neurogenesis Abnormal craniofacial patterning | [40] |
| dyrk1a | Down Syndrome and Autism | Decreased brain size Increased anxiolytic behavior Impaired social interaction/cohesion | [41] |
| wdr11 | Idiopathic Hypogonadotropic Hypogonadism Kallmann Syndrome | Delayed puberty Impaired sense of smell | [42] |
| eftud2 | Mandibulofacial Dysostosis, Guion–Almeida Type | Decreased brain size Small eyes Curved body Early embryonic lethality | [43] |
| zc4h2 | Miles–Carpenter Syndrome | Abnormal swimming Increased twitching Motor hyperactivity Eye movement deficits Pectoral fin contractures | [44] |
| phf21a | Potocki–Shaffer Syndrome | Abnormal head and jaw size Change of head and face shape | [45] |
| eif4a3 | Richieri–Costa–Pereira Syndrome | Underdevelopment of craniofacial cartilage and bone structures | [46] |
| eif2b5 | Vanishing White Matter Disease | Early embryonic lethality Loss of oligodendrocyte precursor cells Impaired motor behavior | [47] |
| eif2b3 | Defected myelin gene expression Defected glial cell differentiation | [48] | |
| sam2 | 12q14.1 Deletion Syndrome | Increased of fear, anxiety-related behaviors, and autism | [49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Son, M.; Kim, D.Y.; Kim, C.-H. Disease Modeling of Rare Neurological Disorders in Zebrafish. Int. J. Mol. Sci. 2022, 23, 3946. https://doi.org/10.3390/ijms23073946
Son M, Kim DY, Kim C-H. Disease Modeling of Rare Neurological Disorders in Zebrafish. International Journal of Molecular Sciences. 2022; 23(7):3946. https://doi.org/10.3390/ijms23073946
Chicago/Turabian StyleSon, Myeongjoo, Dae Yu Kim, and Cheol-Hee Kim. 2022. "Disease Modeling of Rare Neurological Disorders in Zebrafish" International Journal of Molecular Sciences 23, no. 7: 3946. https://doi.org/10.3390/ijms23073946
APA StyleSon, M., Kim, D. Y., & Kim, C. -H. (2022). Disease Modeling of Rare Neurological Disorders in Zebrafish. International Journal of Molecular Sciences, 23(7), 3946. https://doi.org/10.3390/ijms23073946