
Hepatic PTEN Signaling Regulates Systemic Metabolic Homeostasis through Hepatokines-Mediated Liver-to-Peripheral Organs Crosstalk


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
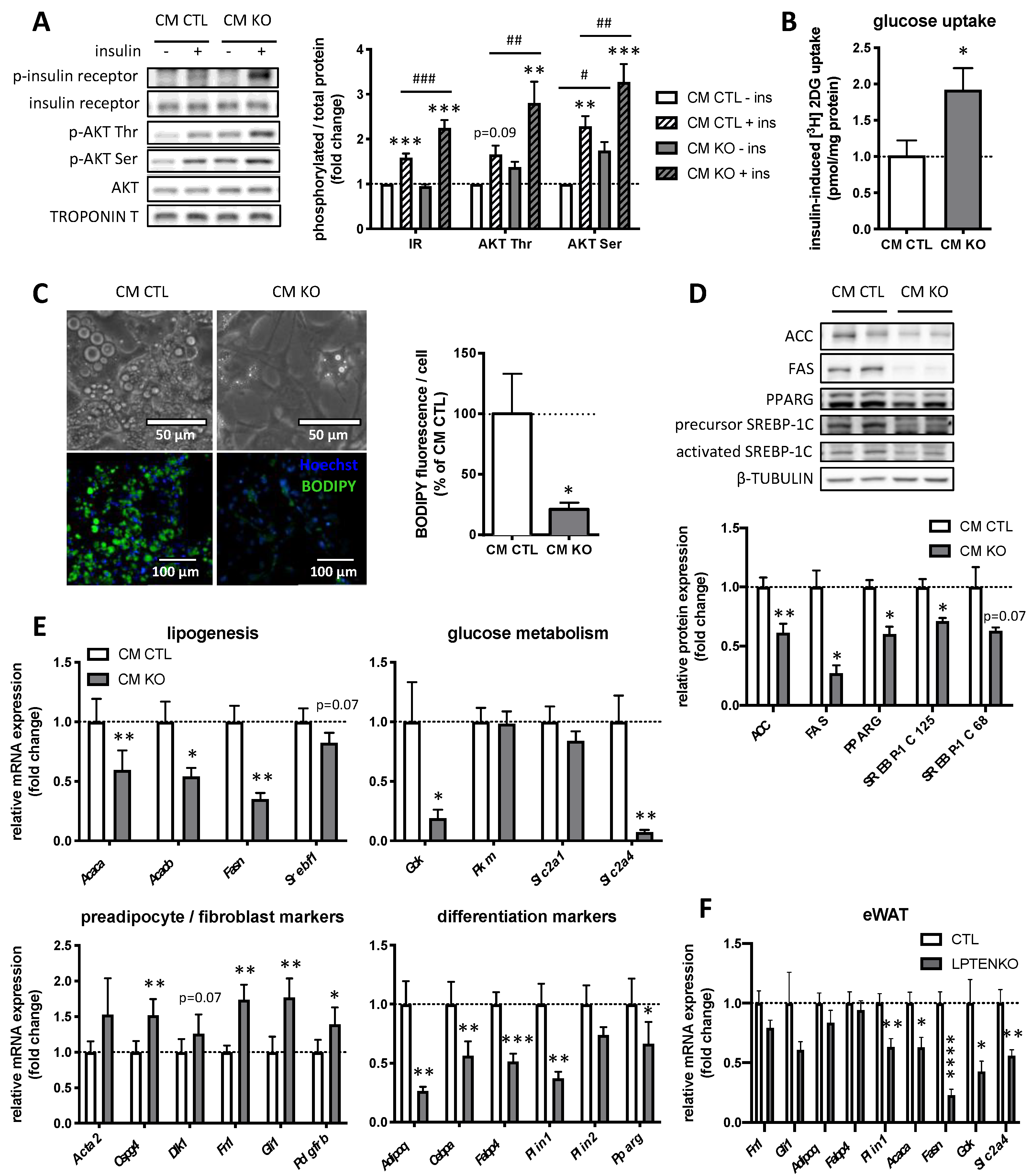
2.1. Conditioned Medium from LPTENKO Primary Hepatocytes Improves Insulin Sensitivity in Muscle Cells and Inhibits Lipogenesis in Adipocytes
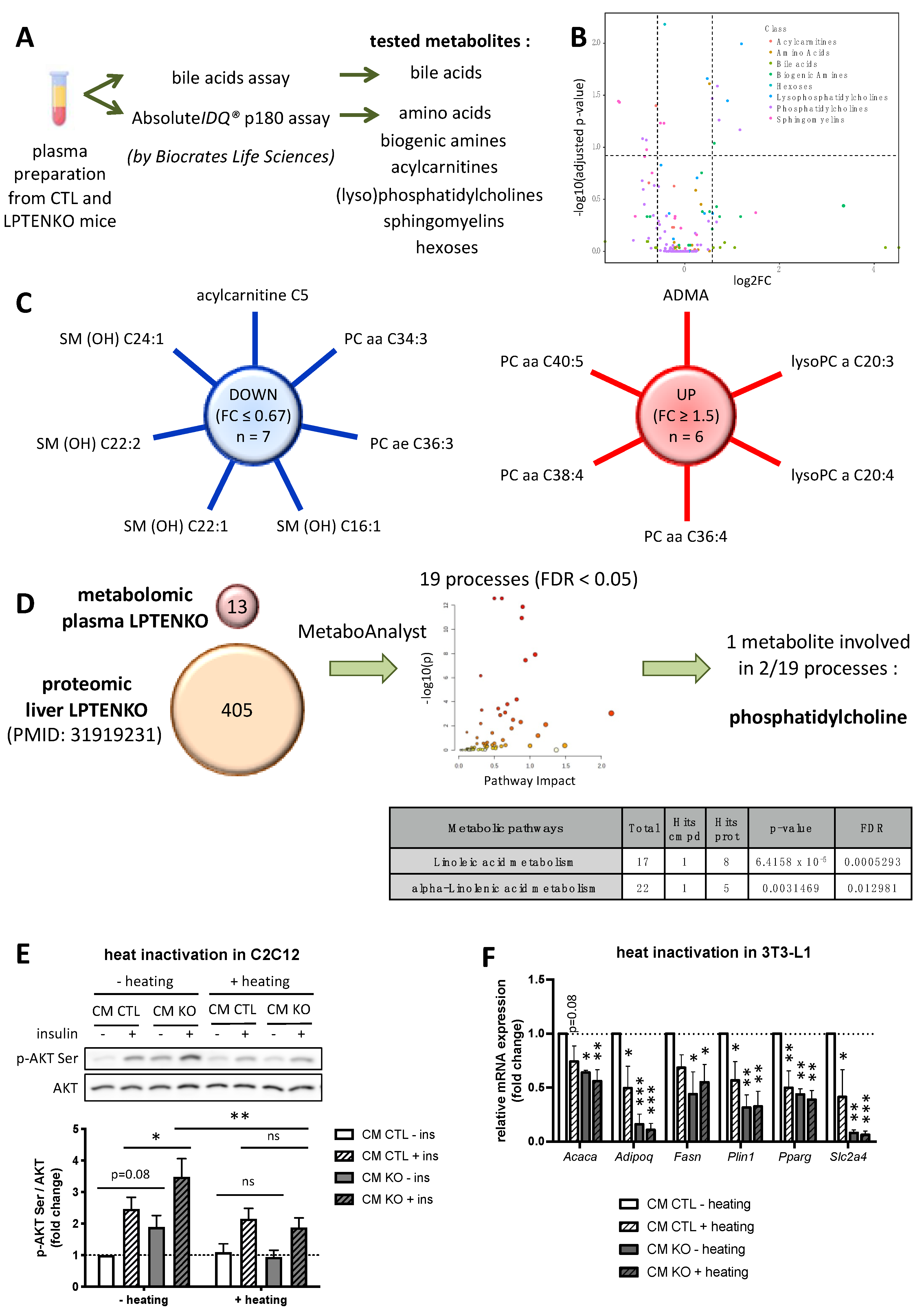
2.2. Protein Factors, but Not Metabolites, Secreted by PTEN-Deficient Hepatocytes Predominantly Trigger an Increased Insulin Sensitivity in Muscle Cells and Pre-Adipocytes Differentiation
2.3. PTEN Deficiency in Hepatocytes Alters the Expression of a Whole Network of Hepatokines
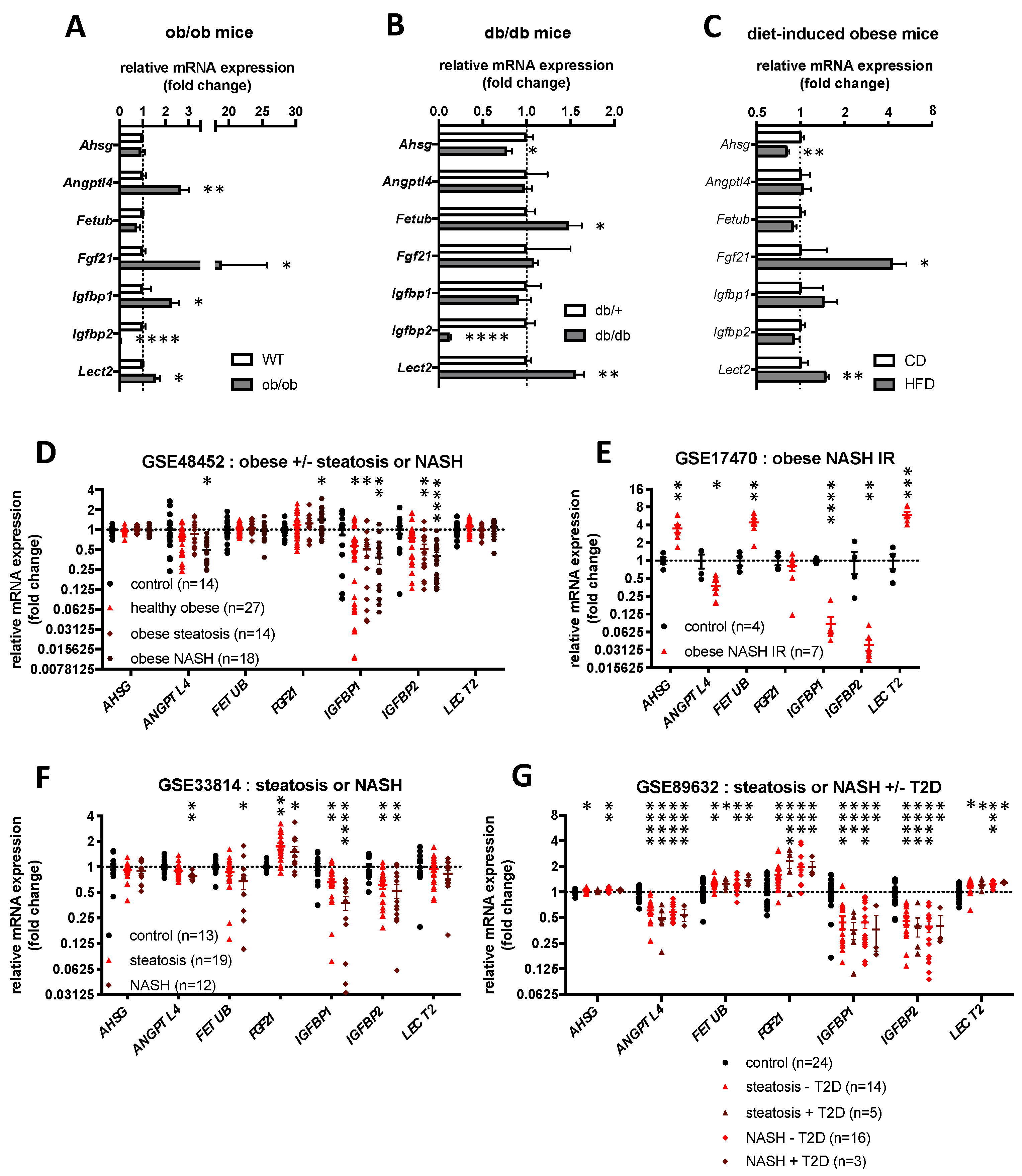
2.4. Hepatokines Expression in Human and Mouse Models of Obesity, Hepatic Steatosis and Insulin Resistance
2.5. FGF21 Secreted by Primary Hepatocytes from LPTENKO Mice Triggers Insulin Hypersensitivity in C2C12 Myotubes
2.6. AHSG, ANGPTL4 and LECT2 Are Required for 3T3-L1 Cells Differentiation and Lipogenesis
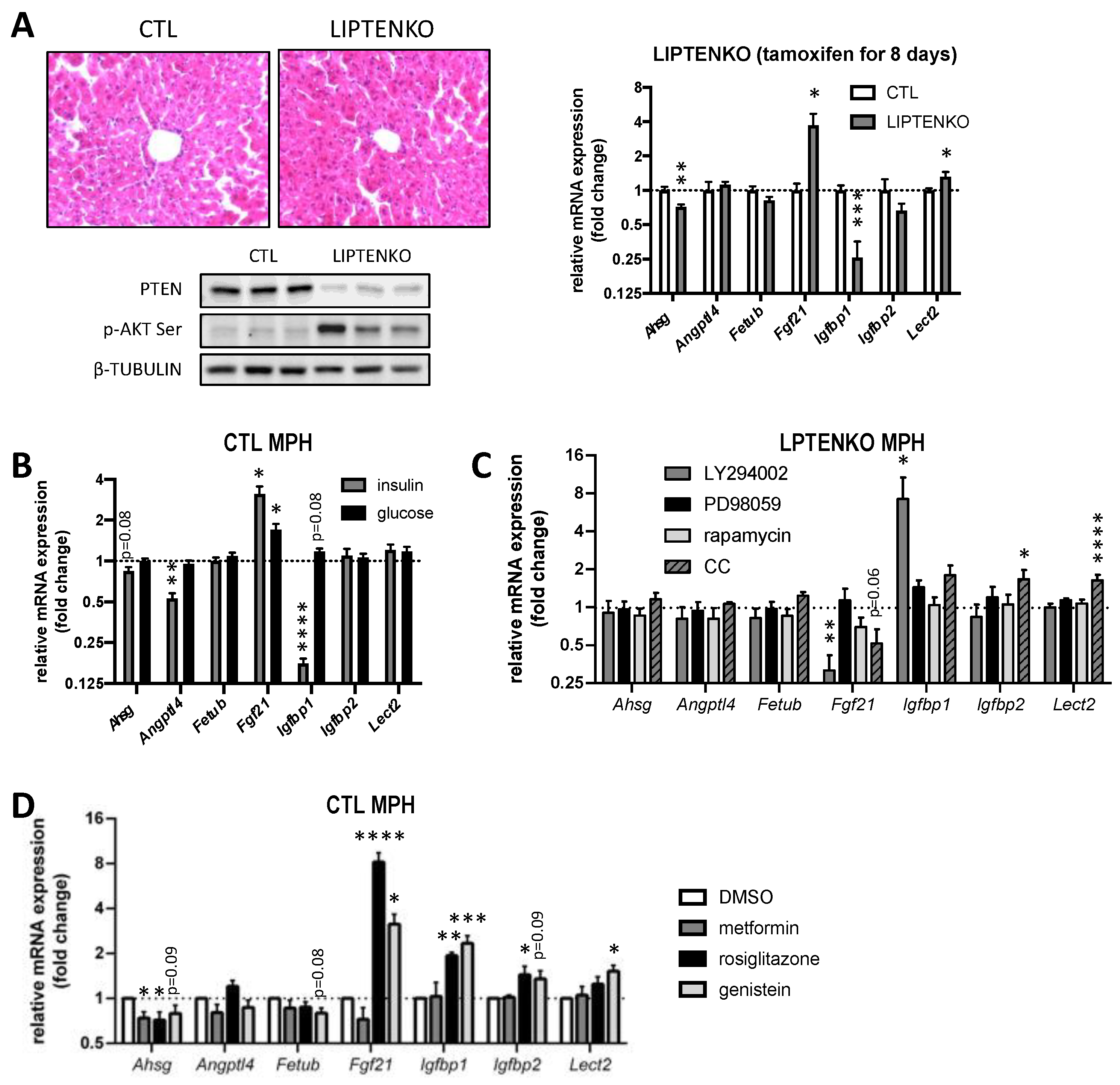
2.7. Hepatokine Expression/Secretion in Hepatocytes Is Regulated by PTEN-Dependent Mechanisms and/or the Hepatic Metabolic Status
3. Discussion
4. Materials and Methods
4.1. Cells, Antibodies, siRNAs, Primers, Kits, Reagents and Diets
4.2. Animals
4.3. Metabolomic Analysis
4.4. Preparation of Conditioned Media from Primary Hepatocytes
4.5. Proteomic Analysis of Conditioned Media by LC-MS/MS
4.6. Protein Extraction and Western Blot
4.7. mRNA Extraction and RT-qPCR
4.8. Bioinformatics Analysis
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Abranches, M.V.; Oliveira, F.C.; Conceicao, L.L.; Peluzio, M.D. Obesity and diabetes: The link between adipose tissue dysfunction and glucose homeostasis. Nutr. Res. Rev. 2015, 28, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J. Mechanisms of insulin resistance in obesity. Front. Med. 2013, 7, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.M. The Impact of Organokines on Insulin Resistance, Inflammation, and Atherosclerosis. Endocrinol. Metab. 2016, 31, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Droujinine, I.A.; Perrimon, N. Defining the interorgan communication network: Systemic coordination of organismal cellular processes under homeostasis and localized stress. Front. Cell Infect. Microbiol. 2013, 3, 82. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Haring, H.U. The role of hepatokines in metabolism. Nat. Rev. Endocrinol 2013, 9, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Choi, K.M. Hepatokines as a Link between Obesity and Cardiovascular Diseases. Diabetes Metab. J. 2015, 39, 10–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefan, N.; Hennige, A.M.; Staiger, H.; Machann, J.; Schick, F.; Krober, S.M.; Machicao, F.; Fritsche, A.; Haring, H.U. Alpha2-Heremans-Schmid glycoprotein/fetuin-A is associated with insulin resistance and fat accumulation in the liver in humans. Diabetes Care 2006, 29, 853–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, S.T.; Chellam, N.; Srinivas, P.R.; Cintron, V.J.; Leon, M.A.; Goustin, A.S.; Grunberger, G. Alpha2-HSG, a specific inhibitor of insulin receptor autophosphorylation, interacts with the insulin receptor. Mol. Cell Endocrinol. 2000, 164, 87–98. [Google Scholar] [CrossRef]
- Mathews, S.T.; Singh, G.P.; Ranalletta, M.; Cintron, V.J.; Qiang, X.; Goustin, A.S.; Jen, K.L.; Charron, M.J.; Jahnen-Dechent, W.; Grunberger, G. Improved insulin sensitivity and resistance to weight gain in mice null for the Ahsg gene. Diabetes 2002, 51, 2450–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennige, A.M.; Staiger, H.; Wicke, C.; Machicao, F.; Fritsche, A.; Haring, H.U.; Stefan, N. Fetuin-A induces cytokine expression and suppresses adiponectin production. PLoS ONE 2008, 3, e1765. [Google Scholar] [CrossRef] [Green Version]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef]
- Jung, T.W.; Chung, Y.H.; Kim, H.C.; Abd El-Aty, A.M.; Jeong, J.H. LECT2 promotes inflammation and insulin resistance in adipocytes via P38 pathways. J. Mol. Endocrinol. 2018, 61, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Misu, H.; Chikamoto, K.; Takayama, H.; Kikuchi, A.; Mohri, K.; Takata, N.; Hayashi, H.; Matsuzawa-Nagata, N.; Takeshita, Y.; et al. LECT2 functions as a hepatokine that links obesity to skeletal muscle insulin resistance. Diabetes 2014, 63, 1649–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 2010, 12, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.T.; Ou, H.Y.; Hung, H.C.; Su, Y.C.; Lu, F.H.; Wu, J.S.; Yang, Y.C.; Wu, C.L.; Chang, C.J. A novel hepatokine, HFREP1, plays a crucial role in the development of insulin resistance and type 2 diabetes. Diabetologia 2016, 59, 1732–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Sharma, V.P.; Shen, H.; Xiao, Y.; Zhu, Q.; Xiong, X.; Guo, L.; Jiang, L.; Ohta, K.; Li, S.; et al. The hepatokine Tsukushi gates energy expenditure via brown fat sympathetic innervation. Nat. Metab. 2019, 1, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, M.; Camire, E.; Aldow, M.; Caron, A.; Jubinville, E.; Turcotte, L.; Kaci, I.; Beaulieu, M.J.; Roy, C.; Labbe, S.M.; et al. The hepatokine Tsukushi is released in response to NAFLD and impacts cholesterol homeostasis. JCI Insight 2019, 4, e129492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouchiroud, M.; Camire, E.; Aldow, M.; Caron, A.; Jubinville, E.; Turcotte, L.; Kaci, I.; Beaulieu, M.J.; Roy, C.; Labbe, S.M.; et al. The Hepatokine TSK does not affect brown fat thermogenic capacity, body weight gain, and glucose homeostasis. Mol. Metab. 2019, 30, 184–191. [Google Scholar] [CrossRef]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.S.; Lindberg, R.A.; et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camporez, J.P.; Jornayvaz, F.R.; Petersen, M.C.; Pesta, D.; Guigni, B.A.; Serr, J.; Zhang, D.; Kahn, M.; Samuel, V.T.; Jurczak, M.J.; et al. Cellular mechanisms by which FGF21 improves insulin sensitivity in male mice. Endocrinology 2013, 154, 3099–3109. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Tian, H.; Lam, K.S.; Lin, S.; Hoo, R.C.; Konishi, M.; Itoh, N.; Wang, Y.; Bornstein, S.R.; Xu, A.; et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013, 17, 779–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashili, F.L.; Austin, R.L.; Deshmukh, A.S.; Fritz, T.; Caidahl, K.; Bergdahl, K.; Zierath, J.R.; Chibalin, A.V.; Moller, D.E.; Kharitonenkov, A.; et al. Direct effects of FGF21 on glucose uptake in human skeletal muscle: Implications for type 2 diabetes and obesity. Diabetes Metab. Res. Rev. 2011, 27, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emanuelli, B.; Vienberg, S.G.; Smyth, G.; Cheng, C.; Stanford, K.I.; Arumugam, M.; Michael, M.D.; Adams, A.C.; Kharitonenkov, A.; Kahn, C.R. Interplay between FGF21 and insulin action in the liver regulates metabolism. J. Clin. Investig. 2014, 124, 515–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, J.M.; Grunberg, J.R.; Church, C.; Elias, I.; Palsdottir, V.; Jansson, J.O.; Bosch, F.; Hammarstedt, A.; Hedjazifar, S.; Smith, U. BMP4 Gene Therapy in Mature Mice Reduces BAT Activation but Protects from Obesity by Browning Subcutaneous Adipose Tissue. Cell Rep. 2017, 20, 1038–1049. [Google Scholar] [CrossRef] [PubMed]
- Elsen, M.; Raschke, S.; Tennagels, N.; Schwahn, U.; Jelenik, T.; Roden, M.; Romacho, T.; Eckel, J. BMP4 and BMP7 induce the white-to-brown transition of primary human adipose stem cells. Am. J. Physiol. Cell Physiol. 2014, 306, C431–C440. [Google Scholar] [CrossRef] [Green Version]
- Schulz, T.J.; Huang, T.L.; Tran, T.T.; Zhang, H.; Townsend, K.L.; Shadrach, J.L.; Cerletti, M.; McDougall, L.E.; Giorgadze, N.; Tchkonia, T.; et al. Identification of inducible brown adipocyte progenitors residing in skeletal muscle and white fat. Proc. Natl. Acad. Sci. USA 2011, 108, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; McMillan, R.P.; Zhu, Q.; Lopaschuk, G.D.; Hulver, M.W.; Butler, A.A. Therapeutic effects of adropin on glucose tolerance and substrate utilization in diet-induced obese mice with insulin resistance. Mol. Metab. 2015, 4, 310–324. [Google Scholar] [CrossRef]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Chen, X.; Chen, Y.; Zhao, Q. Decreased irisin secretion contributes to muscle insulin resistance in high-fat diet mice. Int. J. Clin. Exp. Pathol. 2015, 8, 6490–6497. [Google Scholar] [PubMed]
- Seo, J.A.; Kang, M.C.; Yang, W.M.; Hwang, W.M.; Kim, S.S.; Hong, S.H.; Heo, J.I.; Vijyakumar, A.; Pereira de Moura, L.; Uner, A.; et al. Apolipoprotein J is a hepatokine regulating muscle glucose metabolism and insulin sensitivity. Nat. Commun. 2020, 11, 2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyrou, M.; Bourgoin, L.; Poher, A.L.; Altirriba, J.; Maeder, C.; Caillon, A.; Fournier, M.; Montet, X.; Rohner-Jeanrenaud, F.; Foti, M. Hepatic PTEN deficiency improves muscle insulin sensitivity and decreases adiposity in mice. J. Hepatol. 2015, 62, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobolewski, C.; Abegg, D.; Berthou, F.; Dolicka, D.; Calo, N.; Sempoux, C.; Fournier, M.; Maeder, C.; Ay, A.S.; Clavien, P.A.; et al. S100A11/ANXA2 belongs to a tumour suppressor/oncogene network deregulated early with steatosis and involved in inflammation and hepatocellular carcinoma development. Gut 2020, 69, 1841–1854. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.; Hoy, A.J.; Morris, A.; Brown, R.D.; Lo, J.C.; Burke, M.; Goode, R.J.; Kingwell, B.A.; Kraakman, M.J.; Febbraio, M.A.; et al. Fetuin B Is a Secreted Hepatocyte Factor Linking Steatosis to Impaired Glucose Metabolism. Cell Metab. 2015, 22, 1078–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, A.; Kovarova, M.; Staiger, H.; Machann, J.; Schick, F.; Konigsrainer, A.; Konigsrainer, I.; Schleicher, E.; Fritsche, A.; Haring, H.U.; et al. The hepatokines fetuin-A and fetuin-B are upregulated in the state of hepatic steatosis and may differently impact on glucose homeostasis in humans. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E266–E273. [Google Scholar] [CrossRef]
- Kachaylo, E.; Tschuor, C.; Calo, N.; Borgeaud, N.; Ungethum, U.; Limani, P.; Piguet, A.C.; Dufour, J.F.; Foti, M.; Graf, R.; et al. PTEN Down-Regulation Promotes beta-Oxidation to Fuel Hypertrophic Liver Growth After Hepatectomy in Mice. Hepatology 2017, 66, 908–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinciguerra, M.; Veyrat-Durebex, C.; Moukil, M.A.; Rubbia-Brandt, L.; Rohner-Jeanrenaud, F.; Foti, M. PTEN down-regulation by unsaturated fatty acids triggers hepatic steatosis via an NF-kappaBp65/mTOR-dependent mechanism. Gastroenterology 2008, 134, 268–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Pareja, A.; Clement, S.; Peyrou, M.; Spahr, L.; Negro, F.; Rubbia-Brandt, L.; Foti, M. Phosphatase and tensin homolog is a differential diagnostic marker between nonalcoholic and alcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 3735–3745. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.S.; Bril, F.; Cusi, K.; Newsome, P.N. Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 70, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Calo, N.; Ramadori, P.; Sobolewski, C.; Romero, Y.; Maeder, C.; Fournier, M.; Rantakari, P.; Zhang, F.P.; Poutanen, M.; Dufour, J.F.; et al. Stress-activated miR-21/miR-21* in hepatocytes promotes lipid and glucose metabolic disorders associated with high-fat diet consumption. Gut 2016, 65, 1871–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, F.M.; Chui, P.C.; Antonellis, P.J.; Bina, H.A.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E. Obesity is a fibroblast growth factor 21 (FGF21)-resistant state. Diabetes 2010, 59, 2781–2789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehead, A.; Krause, F.N.; Moran, A.; MacCannell, A.D.V.; Scragg, J.L.; McNally, B.D.; Boateng, E.; Murfitt, S.A.; Virtue, S.; Wright, J.; et al. Brown and beige adipose tissue regulate systemic metabolism through a metabolite interorgan signaling axis. Nat. Commun. 2021, 12, 1905. [Google Scholar] [CrossRef] [PubMed]
- Gancheva, S.; Jelenik, T.; Alvarez-Hernandez, E.; Roden, M. Interorgan Metabolic Crosstalk in Human Insulin Resistance. Physiol. Rev. 2018, 98, 1371–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Vijayakumar, A.; Kahn, B.B. Metabolites as regulators of insulin sensitivity and metabolism. Nat. Rev. Mol. Cell Biol. 2018, 19, 654–672. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Lee, Y.K.; Mamrosh, J.L.; Busby, S.A.; Griffin, P.R.; Pathak, M.C.; Ortlund, E.A.; Moore, D.D. A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature 2011, 474, 506–510. [Google Scholar] [CrossRef]
- Liu, S.; Brown, J.D.; Stanya, K.J.; Homan, E.; Leidl, M.; Inouye, K.; Bhargava, P.; Gangl, M.R.; Dai, L.; Hatano, B.; et al. A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature 2013, 502, 550–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Sundaram, K.; Mu, J.; Dryden, G.W.; Sriwastva, M.K.; Lei, C.; Zhang, L.; Qiu, X.; Xu, F.; Yan, J.; et al. High-fat diet-induced upregulation of exosomal phosphatidylcholine contributes to insulin resistance. Nat. Commun. 2021, 12, 213. [Google Scholar] [CrossRef]
- van der Veen, J.N.; Lingrell, S.; McCloskey, N.; LeBlond, N.D.; Galleguillos, D.; Zhao, Y.Y.; Curtis, J.M.; Sipione, S.; Fullerton, M.D.; Vance, D.E.; et al. A role for phosphatidylcholine and phosphatidylethanolamine in hepatic insulin signaling. FASEB J. 2019, 33, 5045–5057. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Lu, W.; Lin, T.; You, P.; Ye, M.; Huang, Y.; Jiang, X.; Wang, C.; Wang, F.; Lee, M.H.; et al. Activation of Liver FGF21 in hepatocarcinogenesis and during hepatic stress. BMC Gastroenterol. 2013, 13, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, N. FGF21 as a Hepatokine, Adipokine, and Myokine in Metabolism and Diseases. Front. Endocrinol. 2014, 5, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.S.; Choi, S.E.; Ha, E.S.; An, S.Y.; Kim, T.H.; Han, S.J.; Kim, H.J.; Kim, D.J.; Kang, Y.; Lee, K.W. Fibroblast growth factor-21 protects human skeletal muscle myotubes from palmitate-induced insulin resistance by inhibiting stress kinase and NF-kappaB. Metabolism 2012, 61, 1142–1151. [Google Scholar] [CrossRef]
- Holland, W.L.; Adams, A.C.; Brozinick, J.T.; Bui, H.H.; Miyauchi, Y.; Kusminski, C.M.; Bauer, S.M.; Wade, M.; Singhal, E.; Cheng, C.C.; et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 2013, 17, 790–797. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Kurosu, H.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Kuro-o, M. BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proc. Natl. Acad. Sci. USA 2007, 104, 7432–7437. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Kinoshita, S.; Shiraishi, N.; Nakagawa, S.; Sekine, S.; Fujimori, T.; Nabeshima, Y.I. Molecular cloning and expression analyses of mouse betaklotho, which encodes a novel Klotho family protein. Mech. Dev. 2000, 98, 115–119. [Google Scholar] [CrossRef]
- Tomiyama, K.; Maeda, R.; Urakawa, I.; Yamazaki, Y.; Tanaka, T.; Ito, S.; Nabeshima, Y.; Tomita, T.; Odori, S.; Hosoda, K.; et al. Relevant use of Klotho in FGF19 subfamily signaling system in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 1666–1671. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.Y.; Choi, S.E.; Ha, E.S.; Kim, T.H.; Jung, J.G.; Han, S.J.; Kim, H.J.; Kim, D.J.; Kang, Y.; Lee, K.W. Association between insulin resistance and impairment of FGF21 signal transduction in skeletal muscles. Endocrine 2016, 53, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Zheng, J.; Lv, J.; Xu, J.; Ji, X.; Luo, Y.B.; Li, W.; Zhao, Y.; Yan, C. Skeletal muscle increases FGF21 expression in mitochondrial disorders to compensate for energy metabolic insufficiency by activating the mTOR-YY1-PGC1alpha pathway. Free Radic Biol Med. 2015, 84, 161–170. [Google Scholar] [CrossRef]
- Bonen, A. PGC-1alpha-induced improvements in skeletal muscle metabolism and insulin sensitivity. Appl. Physiol. Nutr. Metab. 2009, 34, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Xiao, D.; Gao, Q.; Zeng, L. PGC-1alpha activation: A therapeutic target for type 2 diabetes? Eat. Weight Disor. 2019, 24, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Hale, C.; Chen, M.M.; Stanislaus, S.; Chinookoswong, N.; Hager, T.; Wang, M.; Veniant, M.M.; Xu, J. Lack of overt FGF21 resistance in two mouse models of obesity and insulin resistance. Endocrinology 2012, 153, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markan, K.R. Defining “FGF21 Resistance” during obesity: Controversy, criteria and unresolved questions. F1000Research 2018, 7, 289. [Google Scholar] [CrossRef] [PubMed]
- Ambele, M.A.; Dhanraj, P.; Giles, R.; Pepper, M.S. Adipogenesis: A Complex Interplay of Multiple Molecular Determinants and Pathways. Int. J. Mol. Sci. 2020, 21, 4283. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Kajimura, S. Metabolic adaptation and maladaptation in adipose tissue. Nat. Metab. 2019, 1, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Chen, Z.; Friedman, J. Transcriptional regulation of adipogenesis by KLF4. Cell Metab. 2008, 7, 339–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chang, A.; Li, Y.; Gao, Y.; Wang, H.; Ma, Z.; Li, X.; Wang, B. miR-140-5p regulates adipocyte differentiation by targeting transforming growth factor-beta signaling. Sci. Rep. 2015, 5, 18118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brix, J.M.; Stingl, H.; Hollerl, F.; Schernthaner, G.H.; Kopp, H.P.; Schernthaner, G. Elevated Fetuin-A concentrations in morbid obesity decrease after dramatic weight loss. J. Clin. Endocrinol. Metab. 2010, 95, 4877–4881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pampanini, V.; Inzaghi, E.; Germani, D.; Alterio, A.; Puglianiello, A.; Alisi, A.; Nobili, V.; Cianfarani, S. Serum Fetuin-A levels in obese children with biopsy proven nonalcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 71–76. [Google Scholar] [CrossRef]
- Schinzari, F.; Vizioli, G.; Campia, U.; Tesauro, M.; Cardillo, C. Variable Changes of Circulating ANGPTL3 and ANGPTL4 in Different Obese Phenotypes: Relationship with Vasodilator Dysfunction. Biomedicines 2021, 9, 1037. [Google Scholar] [CrossRef]
- Barja-Fernandez, S.; Moreno-Navarrete, J.M.; Folgueira, C.; Xifra, G.; Sabater, M.; Castelao, C.; Fern, O.J.; Leis, R.; Dieguez, C.; Casanueva, F.F.; et al. Plasma ANGPTL-4 is Associated with Obesity and Glucose Tolerance: Cross-Sectional and Longitudinal Findings. Mol. Nutr. Food Res. 2018, 62, e1800060. [Google Scholar] [CrossRef] [PubMed]
- Okumura, A.; Unoki-Kubota, H.; Matsushita, Y.; Shiga, T.; Moriyoshi, Y.; Yamagoe, S.; Kaburagi, Y. Increased serum leukocyte cell-derived chemotaxin 2 (LECT2) levels in obesity and fatty liver. Biosci. Trends 2013, 7, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.J.; Hwang, S.Y.; Choi, J.H.; Lee, H.J.; Chung, H.S.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; Choi, D.S.; et al. Association of leukocyte cell-derived chemotaxin 2 (LECT2) with NAFLD, metabolic syndrome, and atherosclerosis. PLoS ONE 2017, 12, e0174717. [Google Scholar] [CrossRef]
- Perez-Sotelo, D.; Roca-Rivada, A.; Larrosa-Garcia, M.; Castelao, C.; Baamonde, I.; Baltar, J.; Crujeiras, A.B.; Seoane, L.M.; Casanueva, F.F.; Pardo, M. Visceral and subcutaneous adipose tissue express and secrete functional alpha2hsglycoprotein (fetuin a) especially in obesity. Endocrine 2017, 55, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Dutton, S.; Trayhurn, P. Regulation of angiopoietin-like protein 4/fasting-induced adipose factor (Angptl4/FIAF) expression in mouse white adipose tissue and 3T3-L1 adipocytes. Br. J. Nutr. 2008, 100, 18–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barchetta, I.; Chiappetta, C.; Ceccarelli, V.; Cimini, F.A.; Bertoccini, L.; Gaggini, M.; Cristofano, C.D.; Silecchia, G.; Lenzi, A.; Leonetti, F.; et al. Angiopoietin-Like Protein 4 Overexpression in Visceral Adipose Tissue from Obese Subjects with Impaired Glucose Metabolism and Relationship with Lipoprotein Lipase. Int. J. Mol. Sci. 2020, 21, 7197. [Google Scholar] [CrossRef] [PubMed]
- Nagai, H.; Hamada, T.; Uchida, T.; Yamagoe, S.; Suzuki, K. Systemic expression of a newly recognized protein, LECT2, in the human body. Pathol. Int. 1998, 48, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, D.; Ailhaud, G.; Negrel, R. Fetuin modulates growth and differentiation of Ob17 preadipose cells in serum-free hormone-supplemented medium. Biochim. Biophys. Acta 1985, 846, 185–191. [Google Scholar] [CrossRef]
- Strieder-Barboza, C.; Contreras, G.A. Fetuin-A modulates lipid mobilization in bovine adipose tissue by enhancing lipogenic activity of adipocytes. J. Dairy Sci. 2019, 102, 4628–4638. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Chattopadhyay, M.; Mukherjee, S.; Dasgupta, S.; Mukhopadhyay, S.; Bhattacharya, S. Fetuin-A downregulates adiponectin through Wnt-PPARgamma pathway in lipid induced inflamed adipocyte. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Chattopadhyay, D.; Chatterjee, S.K.; Mondal, S.A.; Majumdar, S.S.; Mukhopadhyay, S.; Saha, N.; Velayutham, R.; Bhattacharya, S.; Mukherjee, S. Increase in PPARgamma inhibitory phosphorylation by Fetuin-A through the activation of Ras-MEK-ERK pathway causes insulin resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166050. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.W.; Youn, B.S.; Choi, H.Y.; Lee, S.Y.; Hong, H.C.; Yang, S.J.; Yoo, H.J.; Kim, B.H.; Baik, S.H.; Choi, K.M. Salsalate and adiponectin ameliorate hepatic steatosis by inhibition of the hepatokine fetuin-A. Biochem. Pharmacol. 2013, 86, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Seal, S.; Mukherjee, S.; Kundu, R.; Mukherjee, S.; Ray, S.; Mukhopadhyay, S.; Majumdar, S.S.; Bhattacharya, S. Adipocyte fetuin-A contributes to macrophage migration into adipose tissue and polarization of macrophages. J. Biol Chem. 2013, 288, 28324–28330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Shimizugawa, T.; Ono, M.; Furukawa, H. Angiopoietin-like protein 4 is a potent hyperlipidemia-inducing factor in mice and inhibitor of lipoprotein lipase. J. Lipid Res. 2002, 43, 1770–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukonina, V.; Lookene, A.; Olivecrona, T.; Olivecrona, G. Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc. Natl. Acad. Sci. USA 2006, 103, 17450–17455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryal, B.; Singh, A.K.; Zhang, X.; Varela, L.; Rotllan, N.; Goedeke, L.; Chaube, B.; Camporez, J.P.; Vatner, D.F.; Horvath, T.L.; et al. Absence of ANGPTL4 in adipose tissue improves glucose tolerance and attenuates atherogenesis. JCI Insight 2018, 3, e97918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Aryal, B.; Chaube, B.; Rotllan, N.; Varela, L.; Horvath, T.L.; Suarez, Y.; Fernandez-Hernando, C. Brown adipose tissue derived ANGPTL4 controls glucose and lipid metabolism and regulates thermogenesis. Mol. Metab. 2018, 11, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Youn, B.S.; Shin, M.S.; Namkoong, C.; Park, K.H.; Baik, J.H.; Kim, J.B.; Park, J.Y.; Lee, K.U.; Kim, Y.B.; et al. Hypothalamic Angptl4/Fiaf is a novel regulator of food intake and body weight. Diabetes 2010, 59, 2772–2780. [Google Scholar] [CrossRef] [Green Version]
- Janssen, A.W.F.; Katiraei, S.; Bartosinska, B.; Eberhard, D.; Willems van Dijk, K.; Kersten, S. Loss of angiopoietin-like 4 (ANGPTL4) in mice with diet-induced obesity uncouples visceral obesity from glucose intolerance partly via the gut microbiota. Diabetologia 2018, 61, 1447–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandard, S.; Zandbergen, F.; van Straten, E.; Wahli, W.; Kuipers, F.; Muller, M.; Kersten, S. The fasting-induced adipose factor/angiopoietin-like protein 4 is physically associated with lipoproteins and governs plasma lipid levels and adiposity. J. Biol. Chem. 2006, 281, 934–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, N.E.; Lam, L.N.; Yang, K.; Zhou, A.Y.; Koliwad, S.; Wang, J.C. Angiopoietin-like 4 (Angptl4) protein is a physiological mediator of intracellular lipolysis in murine adipocytes. J. Biol. Chem. 2012, 287, 8444–8456. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.C.; Chickering, T.W.; Rosen, E.D.; Dussault, B.; Qin, Y.; Soukas, A.; Friedman, J.M.; Holmes, W.E.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma target gene encoding a novel angiopoietin-related protein associated with adipose differentiation. Mol. Cell Biol. 2000, 20, 5343–5349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Xiao, Y.; Zhang, W.; Xu, E.; Liu, W.; Yi, X.; Chang, M. DNA microarray analysis of genes differentially expressed in adipocyte differentiation. J. Biosci. 2014, 39, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Cheng, G.; Zhu, H.; Guan, G. A study of genes involved in adipocyte differentiation. J. Pediatr. Endocrinol. Metab. 2015, 28, 93–99. [Google Scholar] [CrossRef]
- Singh, A.K.; Chaube, B.; Zhang, X.; Sun, J.; Citrin, K.M.; Canfran-Duque, A.; Aryal, B.; Rotllan, N.; Varela, L.; Lee, R.G.; et al. Hepatocyte-specific suppression of ANGPTL4 improves obesity-associated diabetes and mitigates atherosclerosis in mice. J. Clin. Investig. 2021, 131, e140989. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Lee, R.A.; Tsai, S.L.; Kanamaluru, D.; Gray, N.E.; Yiv, N.; Cheang, R.T.; Tan, J.H.; Lee, J.Y.; Fitch, M.D.; et al. An ANGPTL4-ceramide-protein kinase Czeta axis mediates chronic glucocorticoid exposure-induced hepatic steatosis and hypertriglyceridemia in mice. J. Biol. Chem. 2019, 294, 9213–9224. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, Y.; Pan, R.; Wu, C.; Chen, S.; Li, L.; Li, Y.; Yu, C.; Meng, Z.X.; Xu, C. Leukocyte cell-derived chemotaxin 2 promotes the development of nonalcoholic fatty liver disease through STAT-1 pathway in mice. Liver Int. 2021, 41, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Takata, N.; Ishii, K.A.; Takayama, H.; Nagashimada, M.; Kamoshita, K.; Tanaka, T.; Kikuchi, A.; Takeshita, Y.; Matsumoto, Y.; Ota, T.; et al. LECT2 as a hepatokine links liver steatosis to inflammation via activating tissue macrophages in NASH. Sci. Rep. 2021, 11, 555. [Google Scholar] [CrossRef]
- Xu, M.; Xu, H.H.; Lin, Y.; Sun, X.; Wang, L.J.; Fang, Z.P.; Su, X.H.; Liang, X.J.; Hu, Y.; Liu, Z.M.; et al. LECT2, a Ligand for Tie1, Plays a Crucial Role in Liver Fibrogenesis. Cell 2019, 178, 1478–1492.e20. [Google Scholar] [CrossRef]
- Esfahani, M.; Baranchi, M.; Goodarzi, M.T. The implication of hepatokines in metabolic syndrome. Diabetes Metab. Syndr. 2019, 13, 2477–2480. [Google Scholar] [CrossRef]
- Ong, H.T.; Tan, P.K.; Wang, S.M.; Hian Low, D.T.; Ooi, L.L.; Hui, K.M. The tumor suppressor function of LECT2 in human hepatocellular carcinoma makes it a potential therapeutic target. Cancer Gene Ther. 2011, 18, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K.; Yang, C.Y.; Hua, K.T.; Ho, M.C.; Johansson, G.; Jeng, Y.M.; Chen, C.N.; Chen, M.W.; Lee, W.J.; Su, J.L.; et al. Leukocyte cell-derived chemotaxin 2 antagonizes MET receptor activation to suppress hepatocellular carcinoma vascular invasion by protein tyrosine phosphatase 1B recruitment. Hepatology 2014, 59, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.J.; Jung, T.W.; Kim, B.H.; Hong, H.C.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Choi, K.M.; Choi, D.S.; Baik, S.H.; et al. A dipeptidyl peptidase-IV inhibitor improves hepatic steatosis and insulin resistance by AMPK-dependent and JNK-dependent inhibition of LECT2 expression. Biochem. Pharmacol. 2015, 98, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Stiles, B.; Wang, Y.; Stahl, A.; Bassilian, S.; Lee, W.P.; Kim, Y.J.; Sherwin, R.; Devaskar, S.; Lesche, R.; Magnuson, M.A.; et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity. Proc. Natl. Acad. Sci. USA 2004, 101, 2082–2087. [Google Scholar] [CrossRef] [Green Version]
- Hong, X.; Song, R.; Song, H.; Zheng, T.; Wang, J.; Liang, Y.; Qi, S.; Lu, Z.; Song, X.; Jiang, H.; et al. PTEN antagonises Tcl1/hnRNPK-mediated G6PD pre-mRNA splicing which contributes to hepatocarcinogenesis. Gut 2014, 63, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, B.; Liu, E.; Zhang, Z. Loss of PTEN expression is associated with PI3K pathway-dependent metabolic reprogramming in hepatocellular carcinoma. Cell Commun. Signal. 2020, 18, 131. [Google Scholar] [CrossRef] [PubMed]
- Blouin, M.J.; Zhao, Y.; Zakikhani, M.; Algire, C.; Piura, E.; Pollak, M. Loss of function of PTEN alters the relationship between glucose concentration and cell proliferation, increases glycolysis, and sensitizes cells to 2-deoxyglucose. Cancer Lett. 2010, 289, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.R.; Foti, M. Non-genomic loss of PTEN function in cancer: Not in my genes. Trends Pharmacol. Sci. 2011, 32, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Chetram, M.A.; Hinton, C.V. PTEN regulation of ERK1/2 signaling in cancer. J. Recept. Signal. Transduct. Res. 2012, 32, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumiya, Y.; Bina, H.A.; Ouchi, N.; Akasaki, Y.; Kharitonenkov, A.; Walsh, K. FGF21 is an Akt-regulated myokine. FEBS Lett. 2008, 582, 3805–3810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, T.; Ozaki, N.; Kato, Y.; Miura, Y.; Oiso, Y. Insulin downregulates angiopoietin-like protein 4 mRNA in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2006, 347, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Mehrian-Shai, R.; Chen, C.D.; Shi, T.; Horvath, S.; Nelson, S.F.; Reichardt, J.K.; Sawyers, C.L. Insulin growth factor-binding protein 2 is a candidate biomarker for PTEN status and PI3K/Akt pathway activation in glioblastoma and prostate cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 5563–5568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, F.; Kassner, F.; Schmid, G.; Kratzsch, J.; Laner, A.; Wabitsch, M.; Korner, A.; Kiess, W.; Garten, A. Phosphatidylinositol 3-kinase (PI3K) signalling regulates insulin-like-growth factor binding protein-2 (IGFBP-2) production in human adipocytes. Growth Horm. IGF Res. 2015, 25, 115–120. [Google Scholar] [CrossRef]
- Mireuta, M.; Darnel, A.; Pollak, M. IGFBP-2 expression in MCF-7 cells is regulated by the PI3K/AKT/mTOR pathway through Sp1-induced increase in transcription. Growth Factors 2010, 28, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Haukeland, J.W.; Dahl, T.B.; Yndestad, A.; Gladhaug, I.P.; Loberg, E.M.; Haaland, T.; Konopski, Z.; Wium, C.; Aasheim, E.T.; Johansen, O.E.; et al. Fetuin A in nonalcoholic fatty liver disease: In vivo and in vitro studies. Eur. J. Endocrinol. 2012, 166, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Esteghamati, A.; Afarideh, M.; Feyzi, S.; Noshad, S.; Nakhjavani, M. Comparative effects of metformin and pioglitazone on fetuin-A and osteoprotegerin concentrations in patients with newly diagnosed diabetes: A randomized clinical trial. Diabetes Metab. Syndr. 2015, 9, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Ismail, T.A.; Soliman, M.M.; Nassan, M.A. Molecular and immunohistochemical effects of metformin in a rat model of type 2 diabetes mellitus. Exp. Ther. Med. 2015, 9, 1921–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Emoto, M.; Araki, T.; Yokoyama, H.; Lee, E.; Teramura, M.; Koyama, H.; Shoji, T.; Inaba, M.; Nishizawa, Y. Effects of pioglitazone on serum fetuin-A levels in patients with type 2 diabetes mellitus. Metabolism 2008, 57, 1248–1252. [Google Scholar] [CrossRef]
- Ochi, A.; Mori, K.; Emoto, M.; Nakatani, S.; Morioka, T.; Motoyama, K.; Fukumoto, S.; Imanishi, Y.; Koyama, H.; Ishimura, E.; et al. Direct inhibitory effects of pioglitazone on hepatic fetuin-A expression. PLoS ONE 2014, 9, e88704. [Google Scholar] [CrossRef] [Green Version]
- Paolini, P.; Pick, D.; Lapira, J.; Sannino, G.; Pasqualini, L.; Ludka, C.; Sprague, L.J.; Zhang, X.; Bartolotta, E.A.; Vazquez-Hidalgo, E.; et al. Developmental and extracellular matrix-remodeling processes in rosiglitazone-exposed neonatal rat cardiomyocytes. Pharmacogenomics 2014, 15, 759–774. [Google Scholar] [CrossRef]
- Gealekman, O.; Burkart, A.; Chouinard, M.; Nicoloro, S.M.; Straubhaar, J.; Corvera, S. Enhanced angiogenesis in obesity and in response to PPARgamma activators through adipocyte VEGF and ANGPTL4 production. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1056–E1064. [Google Scholar] [CrossRef] [PubMed]
- Mandard, S.; Zandbergen, F.; Tan, N.S.; Escher, P.; Patsouris, D.; Koenig, W.; Kleemann, R.; Bakker, A.; Veenman, F.; Wahli, W.; et al. The direct peroxisome proliferator-activated receptor target fasting-induced adipose factor (FIAF/PGAR/ANGPTL4) is present in blood plasma as a truncated protein that is increased by fenofibrate treatment. J. Biol. Chem. 2004, 279, 34411–34420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhshandehroo, M.; Stienstra, R.; de Wit, N.J.; Bragt, M.C.; Haluzik, M.; Mensink, R.P.; Muller, M.; Kersten, S. Plasma mannose-binding lectin is stimulated by PPARalpha in humans. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E595–E602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frenzel, E.; Wrenger, S.; Immenschuh, S.; Koczulla, R.; Mahadeva, R.; Deeg, H.J.; Dinarello, C.A.; Welte, T.; Marcondes, A.M.; Janciauskiene, S. Acute-phase protein alpha1-antitrypsin--a novel regulator of angiopoietin-like protein 4 transcription and secretion. J. Immunol. 2014, 192, 5354–5362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, K.; Tomita, T. Thiazolidinediones are potent inducers of fibroblast growth factor 21 expression in the liver. Biol. Pharm. Bull. 2011, 34, 1120–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.S.; Imran, K.M.; Hossain, M.; Lee, T.J.; Kim, Y.S. Biochanin A induces a brown-fat phenotype via improvement of mitochondrial biogenesis and activation of AMPK signaling in murine C3H10T1/2 mesenchymal stem cells. Phytother. Res. 2021, 35, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, E.B.; Vienberg, S.G.; Orskov, C.; Hansen, H.S.; Andersen, B. Metformin stimulates FGF21 expression in primary hepatocytes. Exp. Diabetes Res. 2012, 2012, 465282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.K.; Lee, S.H.; Jhun, J.Y.; Byun, J.K.; Jeong, J.H.; Lee, S.Y.; Kim, J.K.; Choi, J.Y.; Cho, M.L. Metformin Prevents Fatty Liver and Improves Balance of White/Brown Adipose in an Obesity Mouse Model by Inducing FGF21. Mediat. Inflamm. 2016, 2016, 5813030. [Google Scholar] [CrossRef] [PubMed]
- Coskun, T.; Bina, H.A.; Schneider, M.A.; Dunbar, J.D.; Hu, C.C.; Chen, Y.; Moller, D.E.; Kharitonenkov, A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 2008, 149, 6018–6027. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.D.; Li, C.Y.; Bina, H.A.; Lynes, S.E.; Michael, M.D.; Shanafelt, A.B.; Kharitonenkov, A.; Wasserman, D.H. Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology 2009, 150, 4084–4093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, Q.; Huang, Z.; Pang, S.; Yang, X.; Li, J.; Yu, B.; Tang, L.; Li, X.; Wang, X. Two-hundred-liter scale fermentation, purification of recombinant human fibroblast growth factor-21, and its anti-diabetic effects on ob/ob mice. Appl. Microbiol. Biotechnol. 2019, 103, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Qi, J.; Yu, D.; Wu, Y.; Zhu, S.; Li, S.; Wu, Q.; Ren, G.; Li, D. Pharmacological efficacy of FGF21 analogue, liraglutide and insulin glargine in treatment of type 2 diabetes. J. Diabetes Complicat. 2017, 31, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Lee, J.E.; Cha, J.J.; Hyun, Y.Y.; Kim, J.E.; Lee, M.H.; Song, H.K.; Nam, D.H.; Han, J.Y.; Han, S.Y.; et al. Fibroblast growth factor 21 improves insulin resistance and ameliorates renal injury in db/db mice. Endocrinology 2013, 154, 3366–3376. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Qi, J.; Wu, Y.; Yu, D.; Xu, P.; Li, S.; Zhu, S.; Wu, Q.; Ren, G.; Li, D. Comparison of PEGylated FGF-21 with insulin glargine for long-lasting hypoglycaemic effect in db/db mice. Diabetes Metab. 2015, 41, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Veniant, M.M.; Komorowski, R.; Chen, P.; Stanislaus, S.; Winters, K.; Hager, T.; Zhou, L.; Wada, R.; Hecht, R.; Xu, J. Long-acting FGF21 has enhanced efficacy in diet-induced obese mice and in obese rhesus monkeys. Endocrinology 2012, 153, 4192–4203. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Stanislaus, S.; Chinookoswong, N.; Lau, Y.Y.; Hager, T.; Patel, J.; Ge, H.; Weiszmann, J.; Lu, S.C.; Graham, M.; et al. Acute glucose-lowering and insulin-sensitizing action of FGF21 in insulin-resistant mouse models--association with liver and adipose tissue effects. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1105–E1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaich, G.; Chien, J.Y.; Fu, H.; Glass, L.C.; Deeg, M.A.; Holland, W.L.; Kharitonenkov, A.; Bumol, T.; Schilske, H.K.; Moller, D.E. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013, 18, 333–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukdar, S.; Zhou, Y.; Li, D.; Rossulek, M.; Dong, J.; Somayaji, V.; Weng, Y.; Clark, R.; Lanba, A.; Owen, B.M.; et al. A Long-Acting FGF21 Molecule, PF-05231023, Decreases Body Weight and Improves Lipid Profile in Non-human Primates and Type 2 Diabetic Subjects. Cell Metab. 2016, 23, 427–440. [Google Scholar] [CrossRef] [Green Version]
- Charles, E.D.; Neuschwander-Tetri, B.A.; Pablo Frias, J.; Kundu, S.; Luo, Y.; Tirucherai, G.S.; Christian, R. Pegbelfermin (BMS-986036), PEGylated FGF21, in Patients with Obesity and Type 2 Diabetes: Results from a Randomized Phase 2 Study. Obesity 2019, 27, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Pan, Q.; Wu, G.; Qian, L.; Zhang, J.; Zhang, L.; Fang, Q.; Zang, G.; Wang, Y.; Lau, G.; et al. Diverse Changes of Circulating Fibroblast Growth Factor 21 Levels in Hepatitis B Virus-Related Diseases. Sci. Rep. 2017, 7, 16482. [Google Scholar] [CrossRef] [Green Version]
- Singhal, G.; Kumar, G.; Chan, S.; Fisher, F.M.; Ma, Y.; Vardeh, H.G.; Nasser, I.A.; Flier, J.S.; Maratos-Flier, E. Deficiency of fibroblast growth factor 21 (FGF21) promotes hepatocellular carcinoma (HCC) in mice on a long term obesogenic diet. Mol. Metab. 2018, 13, 56–66. [Google Scholar] [CrossRef]
- Zheng, Q.; Martin, R.C.; Shi, X.; Pandit, H.; Yu, Y.; Liu, X.; Guo, W.; Tan, M.; Bai, O.; Meng, X.; et al. Lack of FGF21 promotes NASH-HCC transition via hepatocyte-TLR4-IL-17A signaling. Theranostics 2020, 10, 9923–9936. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Hu, W.; Zhang, L.; Jiang, F.; Mao, X.; Yang, G.; Li, L. FGF21 facilitates autophagy in prostate cancer cells by inhibiting the PI3K-Akt-mTOR signaling pathway. Cell Death Dis. 2021, 12, 303. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yang, Y.; Liu, M.; Wang, D.; Wang, F.; Bi, Y.; Ji, J.; Li, S.; Liu, Y.; Chen, R.; et al. Oncogenic KRAS Reduces Expression of FGF21 in Acinar Cells to Promote Pancreatic Tumorigenesis in Mice on a High-Fat Diet. Gastroenterology 2019, 157, 1413–1428 e11. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.E.; Kim, J.T.; Lim, M.A.; Oh, C.; Liu, L.; Jung, S.N.; Won, H.R.; Lee, K.; Chang, J.W.; Yi, H.S.; et al. Association between Circulating Fibroblast Growth Factor 21 and Aggressiveness in Thyroid Cancer. Cancers 2019, 11, 1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Li, Y.; Jiang, G.; Fang, J.; You, Z.; Shao, G.; Zhang, Z.; Jiao, A.; Peng, X. FGF21 promotes non-small cell lung cancer progression by SIRT1/PI3K/AKT signaling. Life Sci. 2021, 269, 118875. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.; Harris, R.B.; Klimentidis, Y.C.; Kohler, L.N.; Jurutka, P.W.; Jacobs, E.T. Circulating Fibroblast Growth Factor-21 and Risk of Metachronous Colorectal Adenoma. J. Gastrointest. Cancer 2021, 52, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gu, X.; Fang, M.; Ji, J.; Yi, C.; Gao, C. The diagnostic value of serum fucosylated fetuin A in hepatitis B virus-related liver diseases. Clin. Chem. Lab. Med. 2016, 54, 693–701. [Google Scholar] [CrossRef]
- Aleksandrova, K.; Boeing, H.; Nothlings, U.; Jenab, M.; Fedirko, V.; Kaaks, R.; Lukanova, A.; Trichopoulou, A.; Trichopoulos, D.; Boffetta, P.; et al. Inflammatory and metabolic biomarkers and risk of liver and biliary tract cancer. Hepatology 2014, 60, 858–871. [Google Scholar] [CrossRef]
- Xiang, Y.; Liu, Y.; Yang, Y.; Hu, H.; Hu, P.; Ren, H.; Zhang, D. A secretomic study on human hepatocellular carcinoma multiple drug-resistant cell lines. Oncol. Rep. 2015, 34, 1249–1260. [Google Scholar] [CrossRef]
- Kondoh, N.; Wakatsuki, T.; Ryo, A.; Hada, A.; Aihara, T.; Horiuchi, S.; Goseki, N.; Matsubara, O.; Takenaka, K.; Shichita, M.; et al. Identification and characterization of genes associated with human hepatocellular carcinogenesis. Cancer Res. 1999, 59, 4990–4996. [Google Scholar]
- Dechassa, M.L.; Tryndyak, V.; de Conti, A.; Xiao, W.; Beland, F.A.; Pogribny, I.P. Identification of chromatin-accessible domains in non-alcoholic steatohepatitis-derived hepatocellular carcinoma. Mol. Carcinog. 2018, 57, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.L.; Huang, S.P.; Lan, W.S.; Lee, P.D. Elevated insulin, proinsulin and insulin-like growth factor-binding protein-1 in liver disease. Growth Horm. IGF Res. 2003, 13, 316–321. [Google Scholar] [CrossRef]
- Ranke, M.B.; Maier, K.P.; Schweizer, R.; Stadler, B.; Schleicher, S.; Elmlinger, M.W.; Flehmig, B. Pilot study of elevated levels of insulin-like growth factor-binding protein-2 as indicators of hepatocellular carcinoma. Horm. Res. 2003, 60, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Mao, Y.Q.; Jiang, W.D.; Chen, Y.R.; Huang, R.Y.; Zhou, X.B.; Wang, Y.F.; Shi, Z.; Wang, Z.S.; Huang, R.P. Development of IGF signaling antibody arrays for the identification of hepatocellular carcinoma biomarkers. PLoS ONE 2012, 7, e46851. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Z.; Huang, R.; Yang, Z.; Lv, Z.; Chen, X.; Xu, F.; Yi, Y.H.; Wu, J.; Huang, R.P. Quantitative screening of serum protein biomarkers by reverse phase protein arrays. Oncotarget 2018, 9, 32624–32641. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Nagai, H.; Gotoh, K.; Kanagawa, H.; Kouyama, H.; Kawanishi, T.; Mima, S.; Yamagoe, S.; Suzuki, K. Expression pattern of a newly recognized protein, LECT2, in hepatocellular carcinoma and its premalignant lesion. Pathol. Int. 1999, 49, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Ovejero, C.; Cavard, C.; Perianin, A.; Hakvoort, T.; Vermeulen, J.; Godard, C.; Fabre, M.; Chafey, P.; Suzuki, K.; Romagnolo, B.; et al. Identification of the leukocyte cell-derived chemotaxin 2 as a direct target gene of beta-catenin in the liver. Hepatology 2004, 40, 167–176. [Google Scholar] [CrossRef] [PubMed]
- L’Hermitte, A.; Pham, S.; Cadoux, M.; Couchy, G.; Caruso, S.; Anson, M.; Crain-Denoyelle, A.M.; Celton-Morizur, S.; Yamagoe, S.; Zucman-Rossi, J.; et al. Lect2 Controls Inflammatory Monocytes to Constrain the Growth and Progression of Hepatocellular Carcinoma. Hepatology 2019, 69, 160–178. [Google Scholar] [CrossRef] [Green Version]
- El-Shal, A.S.; Zidan, H.E.; Rashad, N.M.; Wadea, F.M. Angiopoietin-like protein 3 and 4 expression 4 and their serum levels in hepatocellular carcinoma. Cytokine 2017, 96, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ge, C.; Zhao, F.; Yan, M.; Hu, C.; Jia, D.; Tian, H.; Zhu, M.; Chen, T.; Jiang, G.; et al. Hypoxia-inducible factor 1 alpha-activated angiopoietin-like protein 4 contributes to tumor metastasis via vascular cell adhesion molecule-1/integrin beta1 signaling in human hepatocellular carcinoma. Hepatology 2011, 54, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wei, S.; Ning, S.; Jie, Y.; Ru, Y.; Gu, Y. Evaluation of TGFbeta, XPO4, elF5A2 and ANGPTL4 as biomarkers in HCC. Exp. Ther. Med. 2013, 5, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, K.T.; Xu, A.; Cheng, Q.; Guo, D.Y.; Lim, Z.X.; Sun, C.K.; Fung, J.H.; Poon, R.T.; Fan, S.T.; Lo, C.M.; et al. Clinical relevance and therapeutic potential of angiopoietin-like protein 4 in hepatocellular carcinoma. Mol. Cancer 2014, 13, 196. [Google Scholar] [CrossRef] [Green Version]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Investig. 2004, 113, 1774–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar] [CrossRef]
- Alessi, D.R.; Cuenda, A.; Cohen, P.; Dudley, D.T.; Saltiel, A.R. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995, 270, 27489–27494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berthou, F.; Sobolewski, C.; Abegg, D.; Fournier, M.; Maeder, C.; Dolicka, D.; Correia de Sousa, M.; Adibekian, A.; Foti, M. Hepatic PTEN Signaling Regulates Systemic Metabolic Homeostasis through Hepatokines-Mediated Liver-to-Peripheral Organs Crosstalk. Int. J. Mol. Sci. 2022, 23, 3959. https://doi.org/10.3390/ijms23073959
Berthou F, Sobolewski C, Abegg D, Fournier M, Maeder C, Dolicka D, Correia de Sousa M, Adibekian A, Foti M. Hepatic PTEN Signaling Regulates Systemic Metabolic Homeostasis through Hepatokines-Mediated Liver-to-Peripheral Organs Crosstalk. International Journal of Molecular Sciences. 2022; 23(7):3959. https://doi.org/10.3390/ijms23073959
Chicago/Turabian StyleBerthou, Flavien, Cyril Sobolewski, Daniel Abegg, Margot Fournier, Christine Maeder, Dobrochna Dolicka, Marta Correia de Sousa, Alexander Adibekian, and Michelangelo Foti. 2022. "Hepatic PTEN Signaling Regulates Systemic Metabolic Homeostasis through Hepatokines-Mediated Liver-to-Peripheral Organs Crosstalk" International Journal of Molecular Sciences 23, no. 7: 3959. https://doi.org/10.3390/ijms23073959
APA StyleBerthou, F., Sobolewski, C., Abegg, D., Fournier, M., Maeder, C., Dolicka, D., Correia de Sousa, M., Adibekian, A., & Foti, M. (2022). Hepatic PTEN Signaling Regulates Systemic Metabolic Homeostasis through Hepatokines-Mediated Liver-to-Peripheral Organs Crosstalk. International Journal of Molecular Sciences, 23(7), 3959. https://doi.org/10.3390/ijms23073959