
Identification of Codon 146 KRAS Variants in Isolated Epidermal Nevus and Multiple Lesions in Oculoectodermal Syndrome: Confirmation of the Phenotypic Continuum of Mosaic RASopathies

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
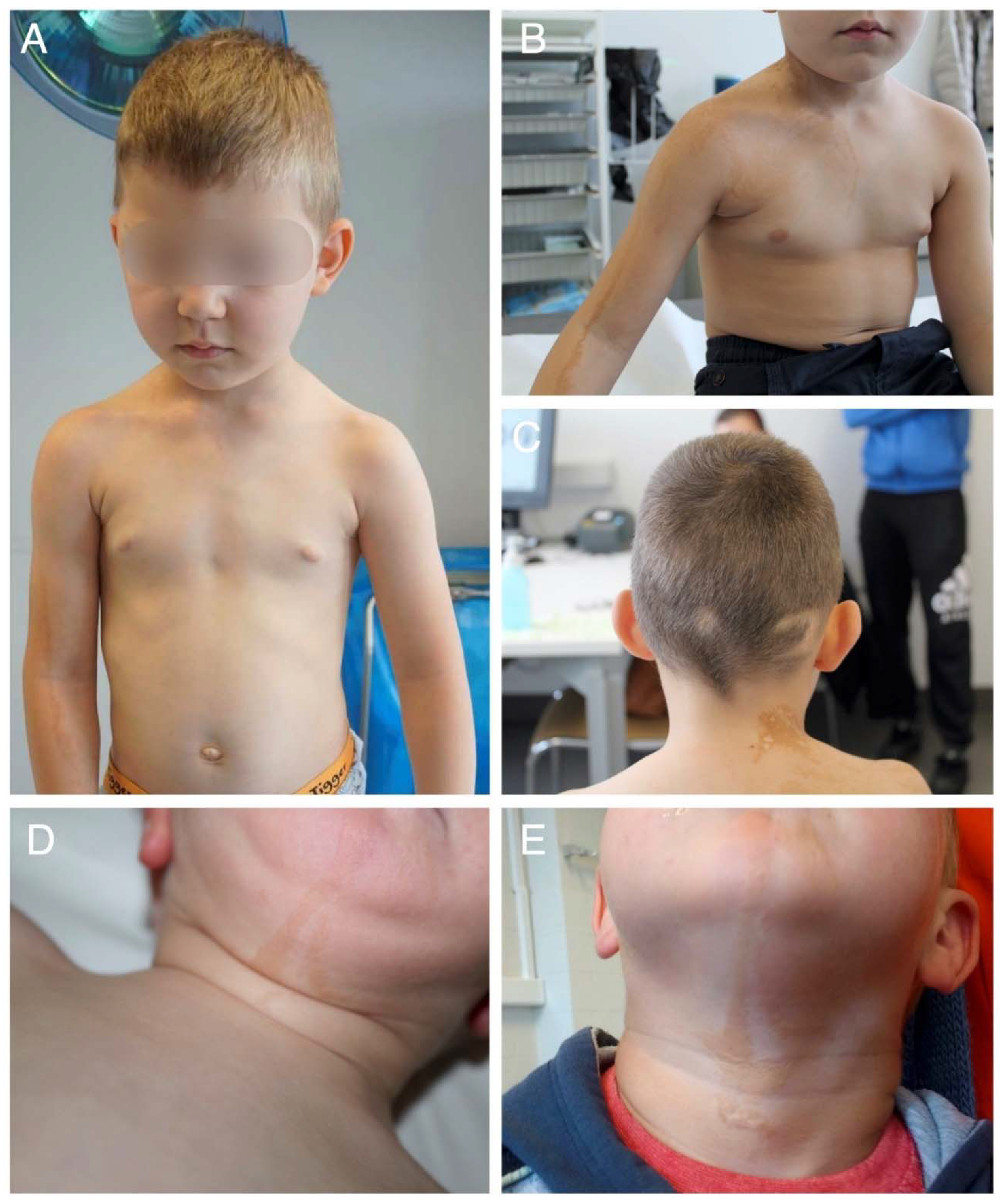
2.1. Clinical Findings
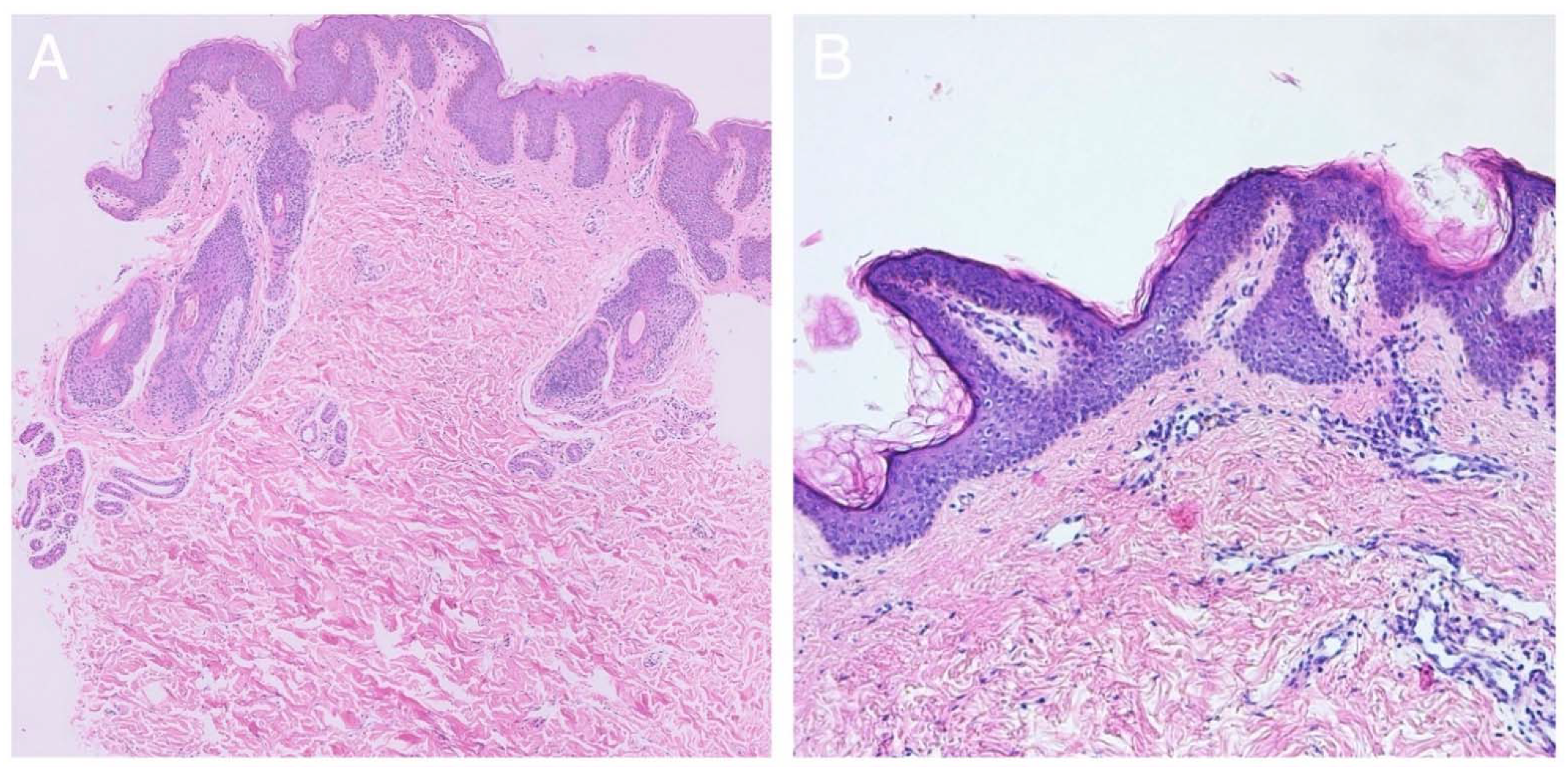
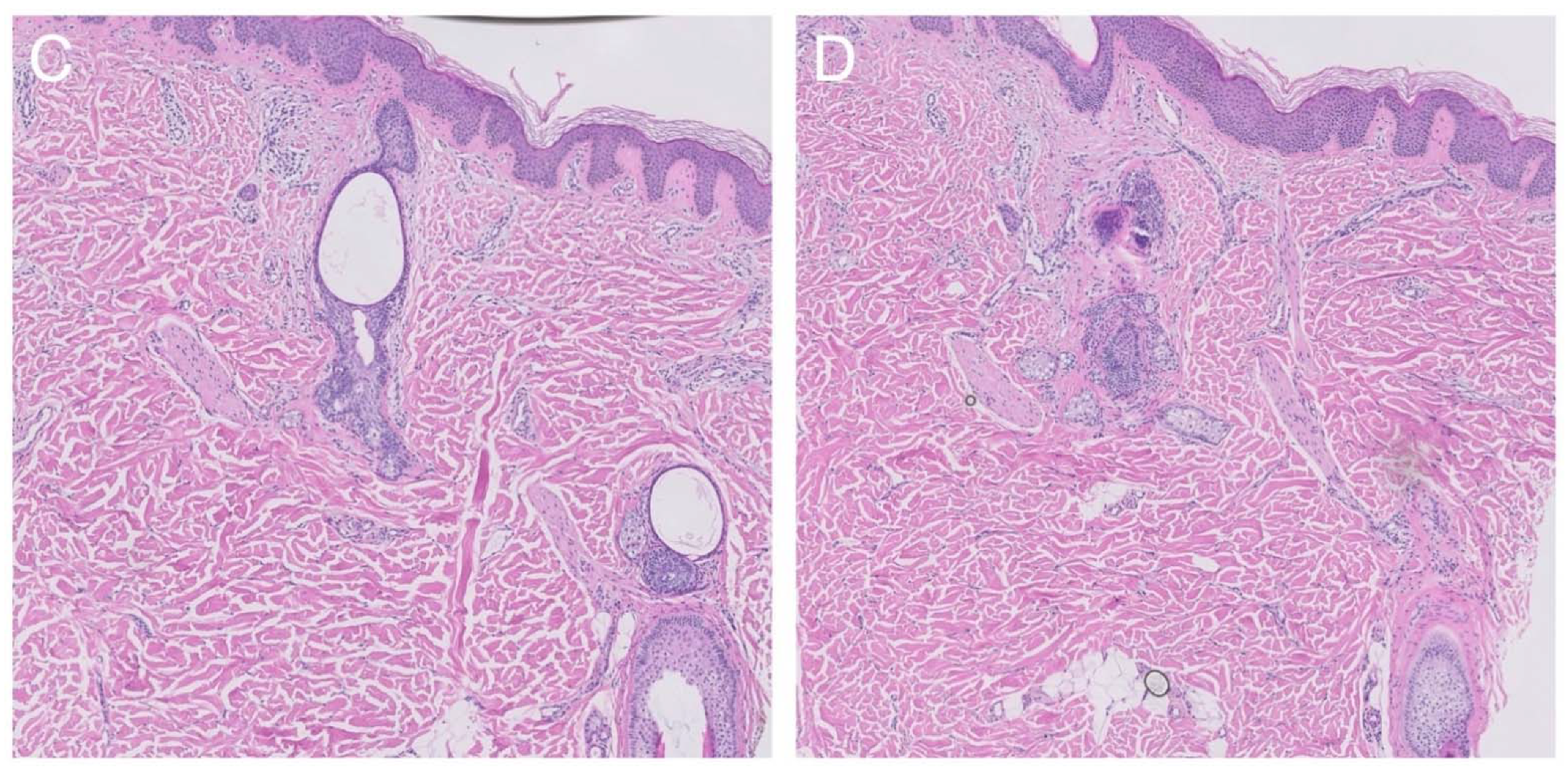
2.2. Histological Findings
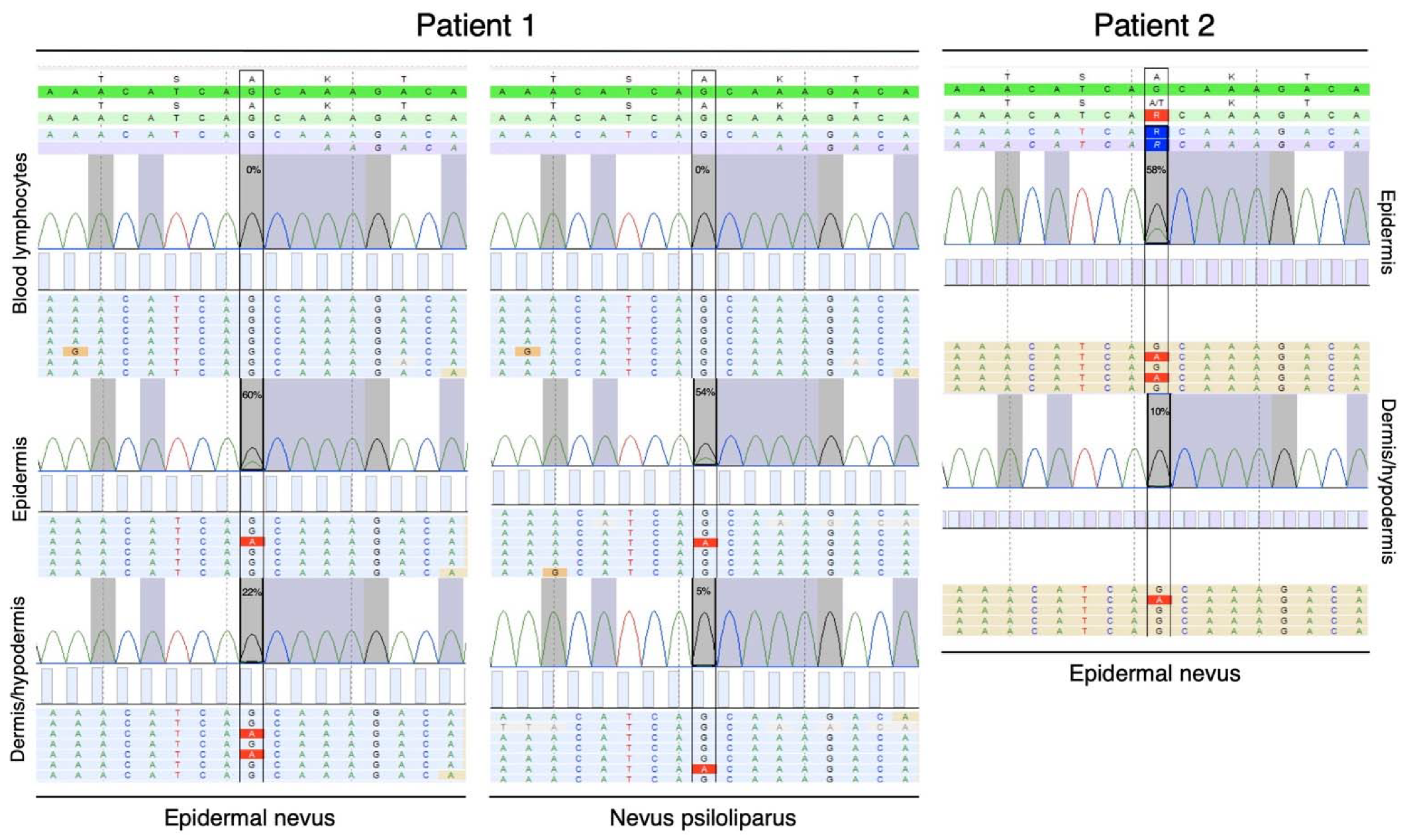
2.3. Molecular Genetic Testing
3. Discussion
4. Materials and Methods
4.1. Consent
4.2. Molecular Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hafner, C.; Groesser, L. Mosaic RASopathies. Cell Cycle 2013, 12, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Toriello, H.V.; Lacassie, Y.; Droste, P.; Higgins, J.V. Provisionally unique syndrome of ocular and ectodermal defects in two unrelated boys. Am. J. Med. Genet. 1993, 45, 764–766. [Google Scholar] [CrossRef]
- Boppudi, S.; Bogershausen, N.; Hove, H.B.; Percin, E.F.; Aslan, D.; Dvorsky, R.; Kayhan, G.; Li, Y.; Cursiefen, C.; Tantcheva-Poor, I.; et al. Specific mosaic KRAS mutations affecting codon 146 cause oculoectodermal syndrome and encephalocraniocutaneous lipomatosis. Clin. Genet. 2016, 90, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Aslan, D.; Akata, R.F.; Schroder, J.; Happle, R.; Moog, U.; Bartsch, O. Oculoectodermal syndrome: Report of a new case with a broad clinical spectrum. Am. J. Med. Genet. Part A 2014, 164, 2947–2951. [Google Scholar] [CrossRef]
- Peacock, J.D.; Dykema, K.J.; Toriello, H.V.; Mooney, M.; Scholten, D.J.; Winn, M.E.; Borgman, A.; Duesbery, N.; Hiemenga, J.A.; Liu, C.; et al. Oculoectodermal syndrome is a mosaic RASopathy associated with KRAS alterations. Am. J. Med. Genet. Part A 2015, 167, 1429–1435. [Google Scholar] [CrossRef]
- Chacon-Camacho, O.F.; Lopez-Moreno, D.; Morales-Sanchez, M.A.; Hofmann, E.; Pacheco-Quito, M.; Wieland, I.; Cortes-Gonzalez, V.; Villanueva-Mendoza, C.; Zenker, M.; Zenteno, J.C. Expansion of the phenotypic spectrum and description of molecular findings in a cohort of patients with oculocutaneous mosaic RASopathies. Mol. Genet. Genom. Med. 2019, 7, e625. [Google Scholar] [CrossRef]
- Asch, S.; Sugarman, J.L. Epidermal nevus syndromes: New insights into whorls and swirls. Pediatr. Dermatol. 2018, 35, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M. Epidermal nevi and the epidermal nevus syndromes: A review of 233 cases. Pediatr. Dermatol. 1992, 9, 342–344. [Google Scholar] [CrossRef]
- Groesser, L.; Herschberger, E.; Ruetten, A.; Ruivenkamp, C.; Lopriore, E.; Zutt, M.; Langmann, T.; Singer, S.; Klingseisen, L.; Schneider-Brachert, W.; et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat. Genet. 2012, 44, 783–787. [Google Scholar] [CrossRef]
- Levinsohn, J.L.; Tian, L.C.; Boyden, L.M.; McNiff, J.M.; Narayan, D.; Loring, E.S.; Yun, D.; Sugarman, J.L.; Overton, J.D.; Mane, S.M.; et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus. J. Investig. Dermatol. 2013, 133, 827–830. [Google Scholar] [CrossRef] [Green Version]
- Froyen, G.; Corbett, M.; Vandewalle, J.; Jarvela, I.; Lawrence, O.; Meldrum, C.; Bauters, M.; Govaerts, K.; Vandeleur, L.; Van Esch, H.; et al. Submicroscopic duplications of the hydroxys-teroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am. J. Hum. Genet. 2008, 82, 432–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, D.H.; Tower, R.; Drolet, B.A. What do mosaic RASopathies tell us about carcinogenesis? Br. J. Dermatol. 2018, 179, 1031–1032. [Google Scholar] [CrossRef] [PubMed]
- Gremer, L.; Merbitz-Zahradnik, T.; Dvorsky, R.; Cirstea, I.C.; Kratz, C.P.; Zenker, M.; Wittinghofer, A.; Ahmadian, M.R. Germline KRAS mutations cause aberrant biochemical and physical properties leading to developmental disorders. Hum. Mutat. 2011, 32, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sol-Church, K.; Stabley, D.L.; Demmer, L.A.; Agbulos, A.; Lin, A.E.; Smoot, L.; Nicholson, L.; Gripp, K.W. Male-to-Male Transmission of Costello Syndrome: G12S HRAS Germline Mutation inherited from a Father With Somatic Mosaicism. Am. J. Med. Genet. Part A 2009, 149, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Niihori, T.; Aoki, Y.; Narumi, Y.; Neri, G.; Cavé, H.; Verloes, A.; Okamoto, N.; Hennekam, R.; Gillessen-Kaesbach, G.; Wieczorek, D.; et al. Germline KRAS and BRAF mutations in car-dio-facio-cutaneous syndrome. Nat. Genet. 2006, 38, 294–296. [Google Scholar] [CrossRef]
- Jongmans, M.C.J.; Van Der Burgt, I.; Hoogerbrugge, P.M.; Noordam, K.; Yntema, H.G.; Nillesen, W.M.; Kuiper, R.P.; Ligtenberg, M.J.; Van Kessel, A.G.; Van Krieken, J.H.J.; et al. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur. J. Hum. Genet. EJHG 2011, 19, 870–874. [Google Scholar] [CrossRef] [Green Version]
- Moog, U. Encephalocraniocutaneous lipomatosis. J. Med. Genet. 2009, 46, 721–729. [Google Scholar] [CrossRef] [Green Version]
- Happle, R.; Kuster, W. Nevus psiloliparus: A distinct fatty tissue nevus. Dermatology 1998, 197, 6–10. [Google Scholar] [CrossRef]
- Happle, R.; Horster, S. Nevus psiloliparus: Report of two nonsyndromic cases. Eur. J. Dermatol. 2004, 14, 314–316. [Google Scholar]
- Happle, R.; Konig, A. Didymosis aplasticosebacea: Coexistence of aplasia cutis congenita and nevus sebaceus may be explained as a twin spot phenomenon. Dermatology 2001, 202, 246–248. [Google Scholar] [CrossRef]
- Happle, R. The group of epidermal nevus syndromes Part I. Well defined phenotypes. J. Am. Acad. Dermatol. 2010, 63, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Happle, R.; Hoffmann, R.; Restano, L.; Caputo, R.; Tadini, G. Phacomatosis pigmentokeratotica: A melanocytic-epidermal twin ne-vus syndrome. Am. J. Med. Genet. 1996, 65, 363–365. [Google Scholar] [CrossRef]
- Prieto-Barrios, M.; Llamas-Martin, R.; Velasco-Tamariz, V.; Calleja-Algarra, A.; Ruano, Y.; Ortiz-Romero, P.L.; Rodriguez-Peralto, J.L. Phacomatosis pigmentokeratotica: A case of HRAS mosaicism causing rhabdomyosarcoma. Br. J. Dermatol. 2018, 179, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Groesser, L.; Herschberger, E.; Sagrera, A.; Shwayder, T.; Flux, K.; Ehmann, L.; Wollenberg, A.; Torrelo, A.; Bagazgoitia, L.; Diaz-Ley, B.; et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J. Investig. Dermatol. 2013, 133, 1998–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waltz, K.M.; Helm, K.F.; Billingsley, E.M. The spectrum of epidermal nevi: A case of verrucous epidermal nevus contiguous with nevus sebaceus. Pediatr. Dermatol. 1999, 16, 211–213. [Google Scholar] [CrossRef]
- Hall, B.E.; Bar-Sagi, D.; Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. USA 2002, 99, 12138–12142. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Feig, L.A.; Cooper, G.M. Relationship among guanine nucleotide exchange, GTP hydrolysis, and transforming potential of mu-tated ras proteins. Mol. Cell. Biol. 1988, 8, 2472–2478. [Google Scholar]
- Neumann, T.E.; Allanson, J.; Kavamura, I.; Kerr, B.; Neri, G.; Noonan, J.; Cordeddu, V.; Gibson, K.; Tzschach, A.; Krueger, G.; et al. Multiple giant cell lesions in patients with Noonan syndrome and cardio-facio-cutaneous syndrome. Eur. J. Hum. Genet. 2009, 17, 420–425. [Google Scholar] [CrossRef] [Green Version]
- Lees, M.; Taylor, D.; Atherton, D.; Reardon, W. Oculo-ectodermal syndrome: Report of two further cases. Am. J. Med. Genet. 2000, 91, 391–395. [Google Scholar] [CrossRef]
- Kratz, C.P.; Rapisuwon, S.; Reed, H.; Hasle, H.; Rosenberg, P.S. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2011, 157, 83–89. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beyens, A.; Dequeker, L.; Brems, H.; Janssens, S.; Syryn, H.; D’Hooghe, A.; De Paepe, P.; Vanwalleghem, L.; Stockman, A.; Vankwikelberge, E.; et al. Identification of Codon 146 KRAS Variants in Isolated Epidermal Nevus and Multiple Lesions in Oculoectodermal Syndrome: Confirmation of the Phenotypic Continuum of Mosaic RASopathies. Int. J. Mol. Sci. 2022, 23, 4036. https://doi.org/10.3390/ijms23074036
Beyens A, Dequeker L, Brems H, Janssens S, Syryn H, D’Hooghe A, De Paepe P, Vanwalleghem L, Stockman A, Vankwikelberge E, et al. Identification of Codon 146 KRAS Variants in Isolated Epidermal Nevus and Multiple Lesions in Oculoectodermal Syndrome: Confirmation of the Phenotypic Continuum of Mosaic RASopathies. International Journal of Molecular Sciences. 2022; 23(7):4036. https://doi.org/10.3390/ijms23074036
Chicago/Turabian StyleBeyens, Aude, Laure Dequeker, Hilde Brems, Sandra Janssens, Hannes Syryn, Anne D’Hooghe, Pascale De Paepe, Lieve Vanwalleghem, Annelies Stockman, Elena Vankwikelberge, and et al. 2022. "Identification of Codon 146 KRAS Variants in Isolated Epidermal Nevus and Multiple Lesions in Oculoectodermal Syndrome: Confirmation of the Phenotypic Continuum of Mosaic RASopathies" International Journal of Molecular Sciences 23, no. 7: 4036. https://doi.org/10.3390/ijms23074036
APA StyleBeyens, A., Dequeker, L., Brems, H., Janssens, S., Syryn, H., D’Hooghe, A., De Paepe, P., Vanwalleghem, L., Stockman, A., Vankwikelberge, E., De Schepper, S., Goeteyn, M., Delbeke, P., & Callewaert, B. (2022). Identification of Codon 146 KRAS Variants in Isolated Epidermal Nevus and Multiple Lesions in Oculoectodermal Syndrome: Confirmation of the Phenotypic Continuum of Mosaic RASopathies. International Journal of Molecular Sciences, 23(7), 4036. https://doi.org/10.3390/ijms23074036