
Implications of Sphingolipid Metabolites in Kidney Diseases


Abstract
:1. Introduction
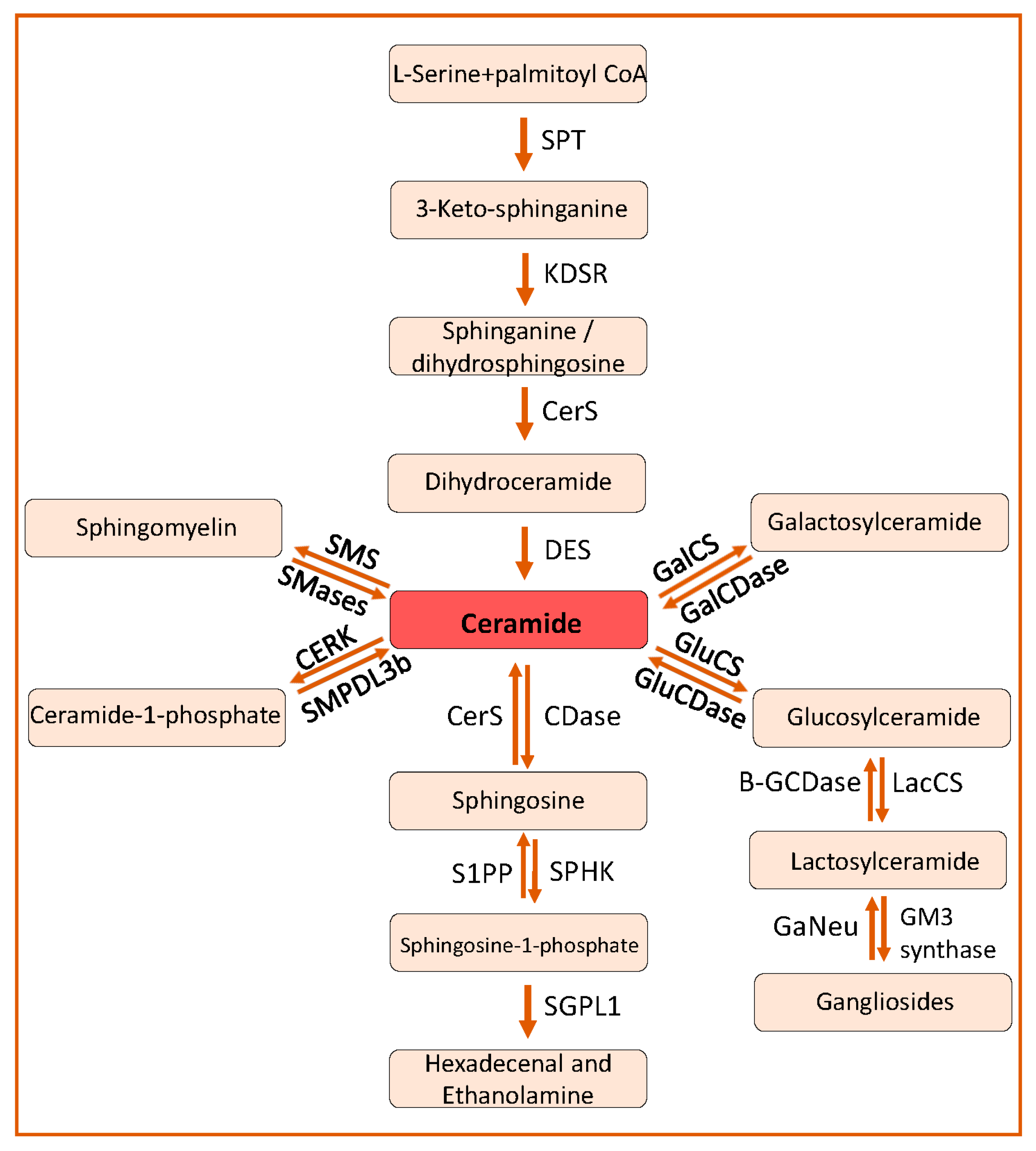
2. Sphingolipid Biosynthesis and Catabolism
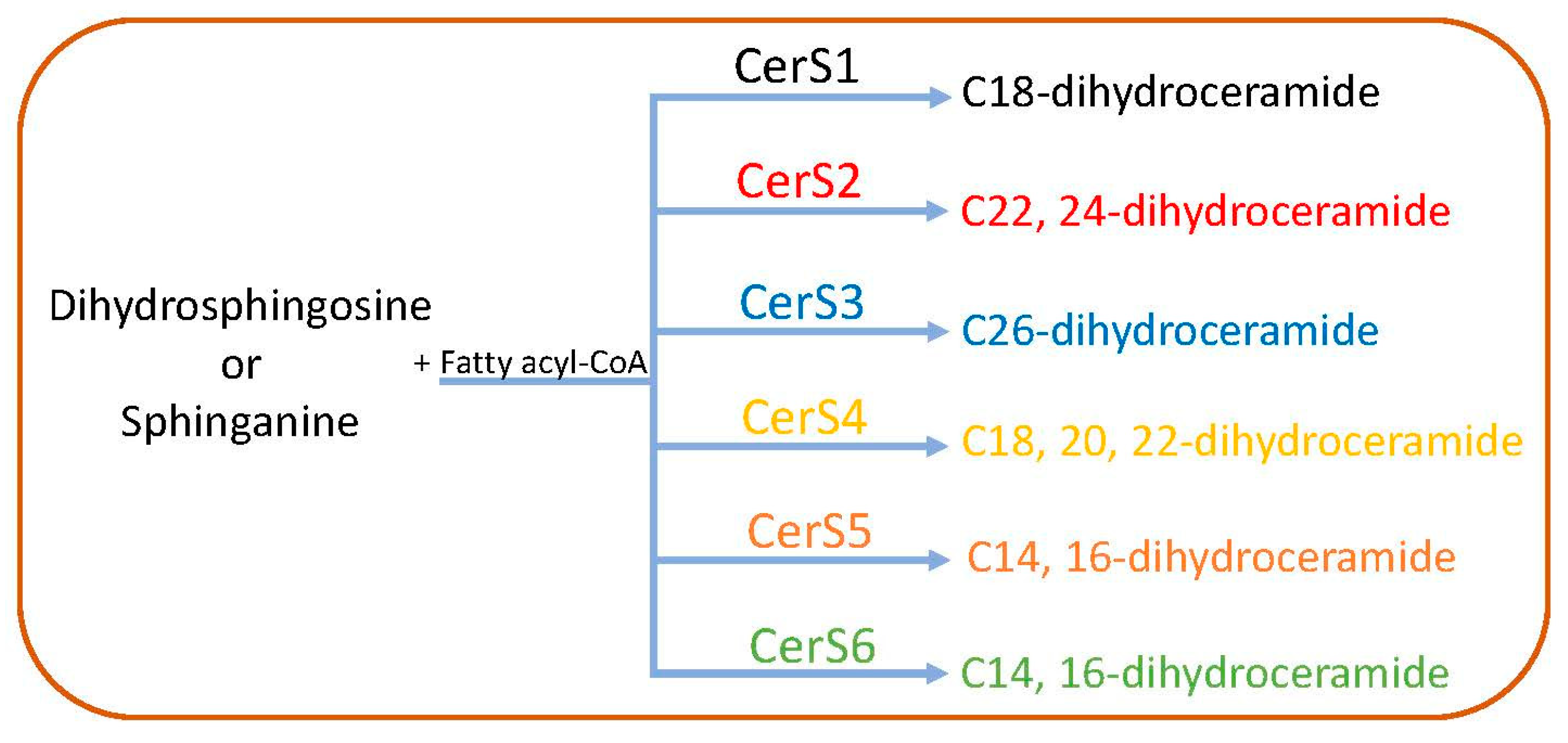
2.1. De Novo Pathway
2.2. The Salvage Pathway
2.3. Catabolism of Ceramides and Synthesis of Other Sphingolipids
3. Sphingolipids in Kidney Diseases
3.1. Role of Sphingolipids in Podocyte Injury in Glomerular Diseases
3.1.1. Diabetic Kidney Disease
Ceramides
Sphingosine-1-Phosphate
Ceramide-1-Phosphate
Glycosphingolipids
3.1.2. Focal Segmental Glomerulosclerosis
3.1.3. Alport Syndrome
3.1.4. IgA Nephropathy
3.1.5. Lupus Nephritis
3.1.6. Fabry’s Disease
3.1.7. COVID-Mediated Kidney Injury
3.1.8. Radiation-Induced Kidney Injury
3.1.9. Treatment Strategies
3.2. Role of Sphingolipids in Tubulointerstitial Fibrosis and Acute Kidney Injury
Treatment Strategies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thudichum, J.L.W. A Treatise on the Chemical Constitution of the Brain: Based Throughout upon Original Researches. Glasgow Med. J. 1884, 22, 363–364. [Google Scholar]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 19, 175–191. [Google Scholar] [CrossRef]
- Takehara, M.; Bandou, H.; Kobayashi, K.; Nagahama, M. Clostridium perfringens α-toxin specifically induces endothelial cell death by promoting ceramide-mediated apoptosis. Anaerobe 2020, 65, 102262. [Google Scholar] [CrossRef]
- Rutherford, C.; Childs, S.; Ohotski, J.; McGlynn, L.; Riddick, M.; MacFarlane, S.; Tasker, D.; Pyne, S.; Pyne, N.; Edwards, J.; et al. Regulation of cell survival by sphingosine-1-phosphate receptor S1P1 via reciprocal ERK-dependent suppression of Bim and PI-3-kinase/protein kinase C-mediated upregulation of Mcl-1. Cell Death Dis. 2013, 4, e927. [Google Scholar] [CrossRef] [Green Version]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, J.S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Shiffman, D.; Pare, G.; Oberbauer, R.; Louie, J.Z.; Rowland, C.M.; Devlin, J.J.; Mann, J.F.; McQueen, M.J. A Gene Variant in CERS2 Is Associated with Rate of Increase in Albuminuria in Patients with Diabetes from ONTARGET and TRANSCEND. PLoS ONE 2014, 9, e106631. [Google Scholar] [CrossRef] [Green Version]
- Summers, S.A.; Garza, L.A.; Zhou, H.; Birnbaum, M.J. Regulation of Insulin-Stimulated Glucose Transporter GLUT4 Translocation and Akt Kinase Activity by Ceramide. Mol. Cell. Biol. 1998, 18, 5457–5464. [Google Scholar] [CrossRef] [Green Version]
- Woo, C.-Y.; Baek, J.Y.; Kim, A.-R.; Hong, C.H.; Yoon, J.E.; Kim, H.S.; Yoo, H.J.; Park, T.-S.; Kc, R.; Lee, K.-U.; et al. Inhibition of Ceramide Accumulation in Podocytes by Myriocin Prevents Diabetic Nephropathy. Diabetes Metab. J. 2020, 44, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Boini, K.M.; Xia, M.; Li, C.; Zhang, C.; Payne, L.P.; Abais, J.M.; Poklis, J.L.; Hylemon, P.B.; Li, P.-L. Acid Sphingomyelinase Gene Deficiency Ameliorates the Hyperhomocysteinemia-Induced Glomerular Injury in Mice. Am. J. Pathol. 2011, 179, 2210–2219. [Google Scholar] [CrossRef]
- Schuchman, E.H. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann–Pick disease. J. Inherit. Metab. Dis. 2007, 30, 654–663. [Google Scholar] [CrossRef]
- Ahmad, A.; Mitrofanova, A.; Bielawski, J.; Yang, Y.; Marples, B.; Fornoni, A.; Zeidan, Y.H. Sphingomyelinase-like phosphodiesterase 3b mediates radiation-induced damage of renal podocytes. FASEB J. 2016, 31, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Zhang, M.; Shi, X.; Wang, K.; Gao, G.; Shen, L.; Sun, T. Kinetic study of Aβ(1-42) amyloidosis in the presence of ganglioside-containing vesicles. Colloids Surf. B Biointerfaces 2019, 185, 110615. [Google Scholar] [CrossRef]
- Orsini, M.; Chateauvieux, S.; Rhim, J.; Gaigneaux, A.; Cheillan, D.; Christov, C.; Dicato, M.; Morceau, F.; Diederich, M. Sphingolipid-mediated inflammatory signaling leading to autophagy inhibition converts erythropoiesis to myelopoiesis in human hematopoietic stem/progenitor cells. Cell Death Differ. 2018, 26, 1796–1812. [Google Scholar] [CrossRef] [Green Version]
- Ponnusamy, S.; Selvam, S.P.; Mehrotra, S.; Kawamori, T.; Snider, A.J.; Obeid, L.; Shao, Y.; Sabbadini, R.; Ogretmen, B. Communication between host organism and cancer cells is transduced by systemic sphingosine kinase 1/sphingosine 1-phosphate signalling to regulate tumour metastasis. EMBO Mol. Med. 2012, 4, 761–775. [Google Scholar] [CrossRef]
- Wang, Y.; Niu, Y.; Zhang, Z.; Gable, K.; Gupta, S.D.; Somashekarappa, N.; Han, G.; Zhao, H.; Myasnikov, A.G.; Kalathur, R.C.; et al. Structural insights into the regulation of human serine palmitoyltransferase complexes. Nat. Struct. Mol. Biol. 2021, 28, 240–248. [Google Scholar] [CrossRef]
- Park, K.-H.; Ye, Z.-W.; Zhang, J.; Hammad, S.M.; Townsend, D.M.; Rockey, D.C.; Kim, S.-H. 3-ketodihydrosphingosine reductase mutation induces steatosis and hepatic injury in zebrafish. Sci. Rep. 2019, 9, 1138. [Google Scholar] [CrossRef] [Green Version]
- Tidhar, R.; Futerman, A.H. The complexity of sphingolipid biosynthesis in the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2511–2518. [Google Scholar] [CrossRef] [Green Version]
- Ailincai, D.; Porzio, W.; Marin, L. Hydrogels based on imino-chitosan amphiphiles as a matrix for drug delivery systems. Polymers 2020, 12, 2687. [Google Scholar] [CrossRef]
- Cingolani, F.; Futerman, A.H.; Casas, J. Ceramide synthases in biomedical research. Chem. Phys. Lipids 2016, 197, 25–32. [Google Scholar] [CrossRef]
- Merrill, A.H. De Novo Sphingolipid Biosynthesis: A Necessary, but Dangerous, Pathway. J. Biol. Chem. 2002, 277, 25843–25846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An Overview of Sphingolipid Metabolism: From Synthesis to Breakdown. In Sphingolipids as Signaling and Regulatory Molecules; Springer Science + Business Media, LLC: New York, NY, USA, 2010; pp. 1–23. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, N.; Hannun, Y.A. Acid and neutral sphingomyelinases: Roles and mechanisms of regulation. Biochem. Cell Biol. 2004, 82, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Duarte, C.; Akkaoui, J.; Yamada, C.; Ho, A.; Mao, C.; Movila, A. Elusive Roles of the Different Ceramidases in Human Health, Pathophysiology, and Tissue Regeneration. Cells 2020, 9, 1379. [Google Scholar] [CrossRef]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell. Signal. 2008, 20, 1010–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandala, S.M.; Thornton, R.; Galve-Roperh, I.; Poulton, S.; Peterson, C.; Olivera, A.; Bergstrom, J.; Kurtz, M.B.; Spiegel, S. Molecular cloning and characterization of a lipid phosphohydrolase that degrades sphingosine-1- phosphate and induces cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 7859–7864. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, C.; Kihara, A.; Gokoh, M.; Igarashi, Y. Identification and Characterization of a Novel Human Sphingosine-1-phosphate Phosphohydrolase, hSPP2. J. Biol. Chem. 2003, 278, 1268–1272. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Kihara, A.; Igarashi, Y. Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5′-phosphate binding domain exposed to the cytosol. Biochem. Biophys. Res. Commun. 2004, 325, 338–343. [Google Scholar] [CrossRef]
- Long, J.; Darroch, P.; Wan, K.F.; Kong, K.C.; Ktistakis, N.; Pyne, N.; Pyne, S. Regulation of cell survival by lipid phosphate phosphatases involves the modulation of intracellular phosphatidic acid and sphingosine 1-phosphate pools. Biochem. J. 2005, 391, 25–32. [Google Scholar] [CrossRef]
- Mitrofanova, A.; Mallela, S.K.; Ducasa, G.M.; Yoo, T.-H.; Rosenfeld-Gur, E.; Zelnik, I.D.; Molina, J.; Santos, J.V.; Ge, M.; Sloan, A.; et al. SMPDL3b modulates insulin receptor signaling in diabetic kidney disease. Nat. Commun. 2019, 10, 2692. [Google Scholar] [CrossRef] [Green Version]
- Mallela, S.K.; Mitrofanova, A.; Merscher, S.; Fornoni, A. Regulation of the amount of ceramide-1-phosphate synthesized in differentiated human podocytes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 158517. [Google Scholar] [CrossRef]
- D’Angelo, G.; Capasso, S.; Sticco, L.; Russo, D. Glycosphingolipids: Synthesis and functions. FEBS J. 2013, 280, 6338–6353. [Google Scholar] [CrossRef]
- Ruotsalainen, V.; Ljungberg, P.; Wartiovaara, J.; Lenkkeri, U.; Kestilä, M.; Jalanko, H.; Holmberg, C.; Tryggvason, K. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 7962–7967. [Google Scholar] [CrossRef] [Green Version]
- Nagata, M. Podocyte injury and its consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef]
- Saran, R.; Robinson, B.; Abbott, K.C.; Bragg-Gresham, J.; Chen, X.; Gipson, D.; Gu, H.; Hirth, R.A.; Hutton, D.; Jin, Y.; et al. US Renal Data System 2019 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. 2020, 75, A6–A7. [Google Scholar] [CrossRef]
- White, K.E.; Bilous, R.W.; Marshall, S.M.; El Nahas, M.; Remuzzi, G.; Piras, G.; De Cosmo, S.; Viberti, G. Podocyte Number in Normotensive Type 1 Diabetic Patients With Albuminuria. Diabetes 2002, 51, 3083–3089. [Google Scholar] [CrossRef] [Green Version]
- Pagtalunan, M.E.; Miller, P.L.; Jumping-Eagle, S.; Nelson, R.G.; Myers, B.D.; Rennke, H.G.; Coplon, N.S.; Sun, L.; Meyer, T.W. Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Investig. 1997, 99, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Emerscher, S.; Fornoni, A. Podocyte Pathology and Nephropathy–Sphingolipids in Glomerular Diseases. Front. Endocrinol. 2014, 5, 127. [Google Scholar] [CrossRef] [Green Version]
- Górska, M.; Dobrzyn, A.; Baranowski, M. Concentrations of sphingosine and sphinganine in plasma of patients with type 2 diabetes. Med. Sci. Monit. 2005, 11, CR35–CR38. [Google Scholar]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; DeFronzo, R.A.; Kirwan, J.P. Plasma Ceramides Are Elevated in Obese Subjects With Type 2 Diabetes and Correlate With the Severity of Insulin Resistance. Diabetes 2009, 58, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Kremer, G.J.; Atzpodien, W.; Schnellbacher, E. Plasma glycosphingolipids in diabetics and normals. Klin. Wochenschr. 1975, 53, 637–638. [Google Scholar] [CrossRef] [PubMed]
- Tofte, N.; Suvitaival, T.; Ahonen, L.; Winther, S.A.; Theilade, S.; Frimodt-Møller, M.; Ahluwalia, T.S.; Rossing, P. Lipidomic analysis reveals sphingomyelin and phosphatidylcholine species associated with renal impairment and all-cause mortality in type 1 diabetes. Sci. Rep. 2019, 9, 16398. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.; Sun, L.; Wu, Q.; Zong, G.; Qi, Q.; Li, H.; Zheng, H.; Zeng, R.; Liang, L.; Lin, X. Associations among circulating sphingolipids, β-cell function, and risk of developing type 2 diabetes: A population-based cohort study in China. PLoS Med. 2020, 17, e1003451. [Google Scholar] [CrossRef]
- Chew, W.S.; Torta, F.; Ji, S.; Choi, H.; Begum, H.; Sim, X.; Khoo, C.M.; Khoo, E.Y.H.; Ong, W.-Y.; Van Dam, R.M.; et al. Large-scale lipidomics identifies associations between plasma sphingolipids and T2DM incidence. JCI Insight 2019, 4, e126925. [Google Scholar] [CrossRef] [PubMed]
- Nair, K.M.S.V.; Kayampilly, J.B.P.; Saha, H.Z.J.; Sas, K.; Byun, J.; Zhang, H.; Iii, F.C.B.; Kretzler, M.; Pennathur, S. Targeted Lipidomic and Transcriptomic Analysis Identifies Dysregulated Renal Ceramide Metabolism in a Mouse Model of Diabetic Kidney Disease. J. Proteom. Bioinform. 2015, 8. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-J.; Ghosh, S.; Kovalik, J.-P.; Ching, J.; Choi, H.W.; Tavintharan, S.; Ong, C.N.; Sum, C.F.; Summers, S.A.; Tai, E.S.; et al. Profiling of Plasma Metabolites Suggests Altered Mitochondrial Fuel Usage and Remodeling of Sphingolipid Metabolism in Individuals With Type 2 Diabetes and Kidney Disease. Kidney Int. Rep. 2016, 2, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Morita, Y.; Kurano, M.; Sakai, E.; Nishikawa, T.; Nishikawa, M.; Sawabe, M.; Aoki, J.; Yatomi, Y. Analysis of urinary sphingolipids using liquid chromatography-tandem mass spectrometry in diabetic nephropathy. J. Diabetes Investig. 2019, 11, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Klein, R.L.; Hammad, S.M.; Baker, N.L.; Hunt, K.J.; Al Gadban, M.M.; Cleary, P.A.; Virella, G.; Lopes-Virella, M.F. Decreased plasma levels of select very long chain ceramide species Are associated with the development of nephropathy in type 1 diabetes. Metabolism 2014, 63, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Yoo, T.-H.; Pedigo, C.E.; Guzman, J.; Correa-Medina, M.; Wei, C.; Villarreal, R.; Mitrofanova, A.; Leclercq, F.; Faul, C.; Li, J.; et al. Sphingomyelinase-Like Phosphodiesterase 3b Expression Levels Determine Podocyte Injury Phenotypes in Glomerular Disease. J. Am. Soc. Nephrol. 2014, 26, 133–147. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Babelova, A.; Moreth, K.; Xin, C.; Eberhardt, W.; Doller, A.; Pavenstädt, H.; Schaefer, L.; Pfeilschifter, J.; Huwiler, A. Transforming growth factor-β2 upregulates sphingosine kinase-1 activity, which in turn attenuates the fibrotic response to TGF-β2 by impeding CTGF expression. Kidney Int. 2009, 76, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Lan, T.; Shen, X.; Liu, P.; Liu, W.; Xu, S.; Xie, X.; Jiang, Q.; Li, W.; Huang, H. Berberine ameliorates renal injury in diabetic C57BL/6 mice: Involvement of suppression of SphK–S1P signaling pathway. Arch. Biochem. Biophys. 2010, 502, 112–120. [Google Scholar] [CrossRef]
- Geoffroy, K.; Troncy, L.; Wiernsperger, N.; Lagarde, M.; El Bawab, S. Glomerular proliferation during early stages of diabetic nephropathy is associated with local increase of sphingosine-1-phosphate levels. FEBS Lett. 2005, 579, 1249–1254. [Google Scholar] [CrossRef] [Green Version]
- Nojiri, T.; Kurano, M.; Tokuhara, Y.; Ohkubo, S.; Hara, M.; Ikeda, H.; Tsukamoto, K.; Yatomi, Y. Modulation of sphingosine-1-phosphate and apolipoprotein M levels in the plasma, liver and kidneys in streptozotocin-induced diabetic mice. J. Diabetes Investig. 2014, 5, 639–648. [Google Scholar] [CrossRef]
- Imeri, F.; Tanturovska, B.S.; Schwalm, S.; Saha, S.; Zeng-Brouwers, J.; Pavenstädt, H.; Pfeilschifter, J.; Schaefer, L.; Huwiler, A. Loss of sphingosine kinase 2 enhances Wilm’s tumor suppressor gene 1 and nephrin expression in podocytes and protects from streptozotocin-induced podocytopathy and albuminuria in mice. Matrix Biol. 2021, 98, 32–48. [Google Scholar] [CrossRef]
- Sui, J.; He, M.; Wang, Y.; Zhao, X.; He, Y.; Shi, B. Sphingolipid metabolism in type 2 diabetes and associated cardiovascular complications. Exp. Ther. Med. 2019, 18, 3603–3614. [Google Scholar] [CrossRef] [Green Version]
- Awad, A.S.; Ye, H.; Huang, L.; Li, L.; Foss, F.W.; Macdonald, T.L.; Lynch, K.R.; Okusa, M.D. Selective sphingosine 1-phosphate 1 receptor activation reduces ischemia-reperfusion injury in mouse kidney. Am. J. Physiol. Physiol. 2006, 290, F1516–F1524. [Google Scholar] [CrossRef] [Green Version]
- Awad, A.S.; Rouse, M.D.; Khutsishvili, K.; Huang, L.; Bolton, W.K.; Lynch, K.R.; Okusa, M.D. Chronic sphingosine 1-phosphate 1 receptor activation attenuates early-stage diabetic nephropathy independent of lymphocytes. Kidney Int. 2011, 79, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Imasawa, T.; Kitamura, H.; Ohkawa, R.; Satoh, Y.; Miyashita, A.; Yatomi, Y. Unbalanced expression of sphingosine 1-phosphate receptors in diabetic nephropathy. Exp. Toxicol. Pathol. 2010, 62, 53–60. [Google Scholar] [CrossRef]
- Su, K.; Zeng, P.; Liang, W.; Luo, Z.; Wang, Y.; Lv, X.; Han, Q.; Yan, M.; Chen, C. FTY720 Attenuates Angiotensin II-Induced Podocyte Damage via Inhibiting Inflammatory Cytokines. Mediat. Inflamm. 2017, 2017, 3701385. [Google Scholar] [CrossRef]
- Schümann, J.; Grevot, A.; Ledieu, D.; Wolf, A.; Schubart, A.; Piaia, A.; Sutter, E.; Côté, S.; Beerli, C.; Pognan, F.; et al. Reduced Activity of Sphingosine-1-Phosphate Lyase Induces Podocyte-related Glomerular Proteinuria, Skin Irritation, and Platelet Activation. Toxicol. Pathol. 2015, 43, 694–703. [Google Scholar] [CrossRef] [Green Version]
- Lovric, S.S.; Goncalves, S.; Gee, H.Y.; Oskouian, B.; Srinivas, H.; Choi, W.-I.; Shril, S.; Ashraf, S.; Tan, W.; Rao, J.; et al. Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J. Clin. Investig. 2017, 127, 912–928. [Google Scholar] [CrossRef] [Green Version]
- Linhares, N.; Arantes, R.R.; Araujo, S.A.; Pena, S.D.J. Nephrotic syndrome and adrenal insufficiency caused by a variant in SGPL1. Clin. Kidney J. 2017, 11, 462–467. [Google Scholar] [CrossRef]
- Pastukhov, O.; Schwalm, S.; Römer, I.; Zangemeister-Wittke, U.; Pfeilschifter, J.; Huwiler, A. Ceramide Kinase Contributes to Proliferation but not to Prostaglandin E2 Formation in Renal Mesangial Cells and Fibroblasts. Cell. Physiol. Biochem. 2014, 34, 119–133. [Google Scholar] [CrossRef] [Green Version]
- Zador, I.Z.; Deshmukh, G.D.; Kunkel, R.; Johnson, K.; Radin, N.S.; Shayman, J.A. A role for glycosphingolipid accumulation in the renal hypertrophy of streptozotocin-induced diabetes mellitus. J. Clin. Investig. 1993, 91, 797–803. [Google Scholar] [CrossRef] [Green Version]
- Subathra, M.; Korrapati, M.; Howell, L.A.; Arthur, J.M.; Shayman, J.A.; Schnellmann, R.G.; Siskind, L.J. Kidney glycosphingolipids are elevated early in diabetic nephropathy and mediate hypertrophy of mesangial cells. Am. J. Physiol. Physiol. 2015, 309, F204–F215. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Virella, M.F.; Baker, N.L.; Hunt, K.J.; Hammad, S.; Arthur, J.; Virella, G.; Klein, R.L. Glycosylated sphingolipids and progression to kidney dysfunction in type 1 diabetes. J. Clin. Lipidol. 2019, 13, 481–491.e1. [Google Scholar] [CrossRef]
- Hou, B.; He, P.; Ma, P.; Yang, X.; Xu, C.; Lam, S.M.; Shui, G.; Yang, X.; Zhang, L.; Qiang, G.; et al. Comprehensive Lipidome Profiling of the Kidney in Early-Stage Diabetic Nephropathy. Front. Endocrinol. 2020, 11, 359. [Google Scholar] [CrossRef]
- Galeano, B.; Klootwijk, R.; Manoli, I.; Sun, M.; Ciccone, C.; Darvish, D.; Starost, M.F.; Zerfas, P.M.; Hoffmann, V.J.; Hoogstraten-Miller, S.; et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J. Clin. Investig. 2007, 117, 1585–1594. [Google Scholar] [CrossRef] [Green Version]
- Hoon, D.S.; Okun, E.; Neuwirth, H.; Morton, D.L.; Irie, R.F. Aberrant Expression of Gangliosides in Human Renal Cell Carcinomas. J. Urol. 1993, 150, 2013–2018. [Google Scholar] [CrossRef]
- Vukovic, I.; Ljubicic, S.; Kurir, T.T.; Bozic, J.; Markotic, A. The Missing Link—Likely Pathogenetic Role of GM3 and Other Gangliosides in the Development of Diabetic Nephropathy. Kidney Blood Press. Res. 2015, 40, 306–314. [Google Scholar] [CrossRef]
- Ene, C.D.; Penescu, M.; Anghel, A.; Neagu, M.; Budu, V.; Nicolae, I. Monitoring Diabetic Nephropathy by Circulating Gangliosides. J. Immunoass. Immunochem. 2015, 37, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Alroy, J.; Sabnis, S.; Kopp, J. Renal Pathology in Fabry Disease. J. Am. Soc. Nephrol. 2002, 13, S134–S138. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Hadjidemetriou, I.; Maharaj, A.; Meimaridou, E.; Buonocore, F.; Saleem, M.; Hurcombe, J.; Bierzynska, A.; Barbagelata, E.; Bergadá, I.; et al. Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. J. Clin. Investig. 2017, 127, 942–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemper, M.J.; Lemke, A. Treatment of Genetic Forms of Nephrotic Syndrome. Front. Pediatr. 2018, 6, 72. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-Y.; Park, S.; Lee, S.-W.; Lee, J.-H.; Lee, E.; Kim, M.; Kim, Y.; Kang, J.; Chung, C.; Moon, J.-S.; et al. RIPK3 Contributes to Lyso-Gb3-Induced Podocyte Death. Cells 2021, 10, 245. [Google Scholar] [CrossRef]
- Kitiyakara, C.; Eggers, P.; Kopp, J.B. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am. J. Kidney Dis. 2004, 44, 815–825. [Google Scholar] [CrossRef]
- Choi, S.R.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Kim, Y.; Choi, B.S.; Kim, Y.-S.; Kim, H.W.; Lim, K.-M.; Park, C.W. Adiponectin receptor agonist AdipoRon decreased ceramide, and lipotoxicity, and ameliorated diabetic nephropathy. Metabolism 2018, 85, 348–360. [Google Scholar] [CrossRef]
- Li, G.; Kidd, J.; Kaspar, C.; Dempsey, S.; Bhat, O.M.; Camus, S.; Ritter, J.K.; Gehr, T.W.; Gulbins, E.; Li, P.-L. Podocytopathy and Nephrotic Syndrome in Mice with Podocyte-Specific Deletion of the Asah1 Gene. Am. J. Pathol. 2020, 190, 1211–1223. [Google Scholar] [CrossRef]
- Voyer, L.E.; Alvarado, C.; Cuttica, R.J.; Balestracci, A.; Zardini, M.; Lago, N. Nephrotic syndrome due to immunoglobulin M mesangial glomerulonephritis preceding juvenile idiopathic arthritis. Iran. J. Kidney Dis. 2013, 7, 231–234. [Google Scholar]
- Hasbal, N.B.; Caglayan, F.B.; Sakaci, T.; Ahbap, E.; Koc, Y.; Sevinc, M.; Ucar, Z.A.; Unsal, A.; Basturk, T. Unexpectedly High Prevalence of Low Alpha-Galactosidase A Enzyme Activity in Patients with Focal Segmental Glomerulosclerosis. Clinics 2020, 75. [Google Scholar] [CrossRef]
- Ding, F.; Wickman, L.; Wang, S.Q.; Zhang, Y.; Wang, F.; Afshinnia, F.; Hodgin, J.; Ding, J.; Wiggins, R.C. Accelerated podocyte detachment and progressive podocyte loss from glomeruli with age in Alport Syndrome. Kidney Int. 2017, 92, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Hu, R.; Kamijo, Y.; Nakajima, T.; Aoyama, T.; Ehara, T.; Shigematsu, H.; Kannagi, R.; Kyogashima, M.; Hara, A. Kidney dysfunction induced by protein overload nephropathy reduces serum sulfatide levels in mice. Nephrology 2009, 14, 658–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Suzuki, T. Role of sulfatide in normal and pathological cells and tissues. J. Lipid Res. 2012, 53, 1437–1450. [Google Scholar] [CrossRef] [Green Version]
- Gessel, M.M.; Spraggins, J.M.; Voziyan, P.A.; Abrahamson, D.R.; Caprioli, R.M.; Hudson, B.G. Two Specific Sulfatide Species Are Dysregulated during Renal Development in a Mouse Model of Alport Syndrome. Lipids 2019, 54, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Coppo, R. Clinical and histological risk factors for progression of IgA nephropathy: An update in children, young and adult patients. J. Nephrol. 2016, 30, 339–346. [Google Scholar] [CrossRef]
- Al Hussain, T.; Hussein, M.H.; Al Mana, H.; Akhtar, M. Pathophysiology of IgA Nephropathy. Adv. Anat. Pathol. 2017, 24, 56–62. [Google Scholar] [CrossRef]
- Fogo, A.B.; Lusco, M.A.; Najafian, B.; Alpers, C.E. AJKD Atlas of Renal Pathology: IgA Nephropathy. Am. J. Kidney Dis. 2015, 66, e33–e34. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.N.; Leung, J.C.K.; Chan, L.Y.Y.; Saleem, M.A.; Mathieson, P.W.; Lai, F.M.; Tang, S.C.W. Activation of podocytes by mesangial-derived TNF-α: Glomerulo-podocytic communication in IgA nephropathy. Am. J. Physiol. Physiol. 2008, 294, F945–F955. [Google Scholar] [CrossRef] [Green Version]
- Asao, R.; Asanuma, K.; Kodama, F.; Akiba-Takagi, M.; Nagai-Hosoe, Y.; Seki, T.; Takeda, Y.; Ohsawa, I.; Mano, S.; Matsuoka, K.; et al. Relationships between Levels of Urinary Podocalyxin, Number of Urinary Podocytes, and Histologic Injury in Adult Patients with IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2012, 7, 1385–1393. [Google Scholar] [CrossRef]
- Tewari, R.; Nada, R.; Rayat, C.S.; Boruah, D.; Dudeja, P.; Joshi, K.; Sakhuja, V. Correlation of Proteinuria with Podocyte Foot Process Effacement in IgA Nephropathy: An Ultrastructural Study. Ultrastruct. Pathol. 2014, 39, 147–151. [Google Scholar] [CrossRef]
- Li, Q.; Zhu, L.; Shi, S.; Xu, D.; Lv, J.; Zhang, H. Case Report: A Pathogenic Missense Variant of WT1 Cosegregates With Proteinuria in a Six-Generation Chinese Family With IgA Nephropathy. Front. Med. 2022, 8, 810940. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, S.; Muso, E.; Takeuchi, E.; Matsushima, H.; Shibata, Y.; Sasayama, S.; Yoshida, H. Selective Breeding for High Serum IgA Levels from Noninbred ddY Mice: Isolation of a Strain with an Early Onset of Glomerular IgA Deposition. Nephron Exp. Nephrol. 1997, 76, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Kurano, M.; Tsuneyama, K.; Morimoto, Y.; Nishikawa, M.; Yatomi, Y. Apolipoprotein M suppresses the phenotypes of IgA nephropathy in hyper-IgA mice. FASEB J. 2019, 33, 5181–5195. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, H.; Hirata, D.; Kamimura, T.; Sato, H.; Iwamoto, M.; Yoshio, T.; Masuyama, J.; Fujimura, A.; Kobayashi, E.; Kano, S.; et al. Effects of FTY720 in MRL-lpr/lpr mice: Therapeutic potential in systemic lupus erythematosus. J. Rheumatol. 2002, 29, 707–716. [Google Scholar]
- Davidson, A. What is damaging the kidney in lupus nephritis? Nat. Rev. Rheumatol. 2015, 12, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Fogo, A.B.; Lusco, M.A.; Najafian, B.; Alpers, C.E. AJKD Atlas of Renal Pathology: Focal and Diffuse Lupus Nephritis (ISN/RPS Class III and IV). Am. J. Kidney Dis. 2017, 70, e9–e11. [Google Scholar] [CrossRef]
- Hajji, M.; Harzallah, A.; Kaaroud, H.; Barbouch, S.; Ben Hamida, F.; Ben Abdallah, T. Factors associated with relapse of lupus nephritis: A single center study of 249 cases. Saudi J. Kidney Dis. Transplant. 2017, 28, 1349–1355. [Google Scholar] [CrossRef]
- Doria, A.; Gatto, M.; Zen, M.; Iaccarino, L.; Punzi, L. Optimizing outcome in SLE: Treating-to-target and definition of treatment goals. Autoimmun. Rev. 2014, 13, 770–777. [Google Scholar] [CrossRef]
- Matsui, I.; Hamano, T.; Tomida, K.; Inoue, K.; Takabatake, Y.; Nagasawa, Y.; Kawada, N.; Ito, T.; Kawachi, H.; Rakugi, H.; et al. Active vitamin D and its analogue, 22-oxacalcitriol, ameliorate puromycin aminonucleoside-induced nephrosis in rats. Nephrol. Dial. Transplant. 2009, 24, 2354–2361. [Google Scholar] [CrossRef]
- Nagase, M.; Yoshida, S.; Shibata, S.; Nagase, T.; Gotoda, T.; Ando, K.; Fujita, T. Enhanced Aldosterone Signaling in the Early Nephropathy of Rats with Metabolic Syndrome: Possible Contribution of Fat-Derived Factors. J. Am. Soc. Nephrol. 2006, 17, 3438–3446. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Guo, H.; Miao, X.; Xu, J.; Yang, R.; Zhao, L.; Liu, J.; Yang, L.; Gao, F.; Zhang, W.; et al. Nestin protects podocyte from injury in lupus nephritis by mitophagy and oxidative stress. Cell Death Dis. 2020, 11, 319. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhu, C.; Yu, N.; Yang, L. Tim-1 alleviates lupus nephritis-induced podocyte injury via regulating autophagy. Central Eur. J. Immunol. 2021, 46, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Checa, A.; Idborg, H.; Zandian, A.; Sar, D.G.; Surowiec, I.; Trygg, J.; Svenungsson, E.; Jakobsson, P.-J.; Nilsson, P.; Gunnarsson, I.; et al. Dysregulations in circulating sphingolipids associate with disease activity indices in female patients with systemic lupus erythematosus: A cross-sectional study. Lupus 2017, 26, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xie, X.-W.; Zhou, H.; Wang, B.; Zhang, M.-J.; Tang, F.-Y. Metabolic profiling reveals new serum biomarkers of lupus nephritis. Lupus 2017, 26, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Patyna, S.; Büttner, S.; Eckes, T.; Obermüller, N.; Bartel, C.; Braner, A.; Trautmann, S.; Thomas, D.; Geiger, H.; Pfeilschifter, J.; et al. Blood ceramides as novel markers for renal impairment in systemic lupus erythematosus. Prostaglandins Other Lipid Mediat. 2019, 144, 106348. [Google Scholar] [CrossRef]
- Nowling, T.K.; Mather, A.R.; Thiyagarajan, T.; Hernández-Corbacho, M.J.; Powers, T.W.; Jones, E.E.; Snider, A.J.; Oates, J.C.; Drake, R.R.; Siskind, L.J. Renal Glycosphingolipid Metabolism Is Dysfunctional in Lupus Nephritis. J. Am. Soc. Nephrol. 2014, 26, 1402–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fall, B.; Scott, C.R.; Mauer, M.; Shankland, S.; Pippin, J.; Jefferson, J.A.; Wallace, E.; Warnock, D.; Najafian, B. Urinary Podocyte Loss Is Increased in Patients with Fabry Disease and Correlates with Clinical Severity of Fabry Nephropathy. PLoS ONE 2016, 11, e0168346. [Google Scholar] [CrossRef] [Green Version]
- Askari, H.; Kaneski, C.R.; Semino-Mora, C.; Desai, P.; Ang, A.; Kleiner, D.E.; Perlee, L.T.; Quezado, M.; Spollen, L.E.; Wustman, B.A.; et al. Cellular and tissue localization of globotriaosylceramide in Fabry disease. Virchows Arch. 2007, 451, 823–834. [Google Scholar] [CrossRef]
- Liebau, M.C.; Braun, F.; Höpker, K.; Weitbrecht, C.; Bartels, V.; Müller, R.-U.; Brodesser, S.; Saleem, M.A.; Benzing, T.; Schermer, B.; et al. Dysregulated Autophagy Contributes to Podocyte Damage in Fabry’s Disease. PLoS ONE 2013, 8, e63506. [Google Scholar] [CrossRef] [Green Version]
- Katsuta, H.; Tsuboi, K.; Yamamoto, H.; Goto, H. Correlations Between Serum Cholesterol and Vascular Lesions in Fabry Disease Patients. Circ. J. 2018, 82, 3058–3063. [Google Scholar] [CrossRef] [Green Version]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Fan, J.; Hu, L.-F.; Zhang, Y.; Ooi, J.D.; Meng, T.; Jin, P.; Ding, X.; Peng, L.-K.; Song, L.; et al. Single-cell analysis of angiotensin-converting enzyme II expression in human kidneys and bladders reveals a potential route of 2019 novel coronavirus infection. Chin. Med. J. 2021, 134, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Luo, R.; Wang, K.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Yao, Y.; Ge, S.; Xu, G. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020, 97, 829–838. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Zhang, J.; Yang, H.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat. Commun. 2021, 12, 2506. [Google Scholar] [CrossRef]
- Ahmadian, E.; Khatibi, S.M.H.; Soofiyani, S.R.; Abediazar, S.; Shoja, M.M.; Ardalan, M.; Vahed, S.Z. COVID-19 and kidney injury: Pathophysiology and molecular mechanisms. Rev. Med. Virol. 2020, 31, e2176. [Google Scholar] [CrossRef]
- Larsen, C.P.; Bourne, T.D.; Wilson, J.D.; Saqqa, O.; Sharshir, M.A. Collapsing Glomerulopathy in a Patient With COVID-19. Kidney Int. Rep. 2020, 5, 935–939. [Google Scholar] [CrossRef]
- Fanelli, V.; Fiorentino, M.; Cantaluppi, V.; Gesualdo, L.; Stallone, G.; Ronco, C.; Castellano, G. Acute kidney injury in SARS-CoV-2 infected patients. Crit. Care 2020, 24, 155. [Google Scholar] [CrossRef] [Green Version]
- Shetty, A.A.; Tawhari, I.; Safar-Boueri, L.; Seif, N.; Alahmadi, A.; Gargiulo, R.; Aggarwal, V.; Usman, I.; Kisselev, S.; Gharavi, A.G.; et al. COVID-19–Associated Glomerular Disease. J. Am. Soc. Nephrol. 2020, 32, 33–40. [Google Scholar] [CrossRef]
- Fornoni, A.; Sageshima, J.; Wei, C.; Merscher-Gomez, S.; Aguillon-Prada, R.; Jauregui, A.N.; Li, J.; Mattiazzi, A.; Ciancio, G.; Chen, L.; et al. Rituximab Targets Podocytes in Recurrent Focal Segmental Glomerulosclerosis. Sci. Transl. Med. 2011, 3, 85ra46. [Google Scholar] [CrossRef] [Green Version]
- Torretta, E.; Garziano, M.; Poliseno, M.; Capitanio, D.; Biasin, M.; Santantonio, T.A.; Clerici, M.; Caputo, S.L.; Trabattoni, D.; Gelfi, C. Severity of COVID-19 Patients Predicted by Serum Sphingolipids Signature. Int. J. Mol. Sci. 2021, 22, 10198. [Google Scholar] [CrossRef] [PubMed]
- Dawson, L.A.; Kavanagh, B.D.; Paulino, A.C.; Das, S.K.; Miften, M.; Li, X.A.; Pan, C.; Haken, R.K.T.; Schultheiss, T.E. Radiation-Associated Kidney Injury. Int. J. Radiat. Oncol. 2010, 76, S108–S115. [Google Scholar] [CrossRef] [PubMed]
- Cortes, L.K.; Vainauskas, S.; Dai, N.; McClung, C.M.; Shah, M.; Benner, J.S.; Corrêa, I.R., Jr.; VerBerkmoes, N.C.; Taron, C.H. Proteomic identification of mammalian cell surface derived glycosylphosphatidylinositol-anchored proteins through selective glycan enrichment. Proteomics 2014, 14, 2471–2484. [Google Scholar] [CrossRef] [Green Version]
- Garrouste, C.; Canaud, G.; Büchler, M.; Rivalan, J.; Colosio, C.; Martinez, F.; Aniort, J.; Dudreuilh, C.; Pereira, B.; Caillard, S.; et al. Rituximab for Recurrence of Primary Focal Segmental Glomerulosclerosis After Kidney Transplantation. Transplantation 2017, 101, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, T.; Krochmal, M.; Cisek, K.; Fernandes, M.; Husi, H.; Stevens, R.; Bascands, J.-L.; Schanstra, J.P.; Klein, J. Omics databases on kidney disease: Where they can be found and how to benefit from them. Clin. Kidney J. 2016, 9, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalhorn, T.; Zager, R.A. Renal cortical ceramide patterns during ischemic and toxic injury: Assessments by HPLC-mass spectrometry. Am. J. Physiol. Content 1999, 277, F723–F733. [Google Scholar] [CrossRef] [PubMed]
- Alasfar, S.; Matar, D.; Montgomery, R.A.; Desai, N.; Lonze, B.; Vujjini, V.; Estrella, M.M.; Dieck, J.M.; Khneizer, G.; Sever, S.; et al. Rituximab and Therapeutic Plasma Exchange in Recurrent Focal Segmental Glomerulosclerosis Postkidney Transplantation. Transplantation 2018, 102, e115–e120. [Google Scholar] [CrossRef]
- Lanaret, C.; Anglicheau, D.; Audard, V.; Büchler, M.; Caillard, S.; Couzi, L.; Malvezzi, P.; Mesnard, L.; Bertrand, D.; Martinez, F.; et al. Rituximab for recurrence of primary focal segmental glomerulosclerosis after kidney transplantation: Results of a nationwide study. Am. J. Transplant. 2021, 21, 3021–3033. [Google Scholar] [CrossRef]
- Takahashi, Y.; Ikezumi, Y.; Saitoh, A. Rituximab protects podocytes and exerts anti-proteinuric effects in rat adriamycin-induced nephropathy independent of B-lymphocytes. Nephrology 2016, 22, 49–57. [Google Scholar] [CrossRef]
- Huang, K.; Liu, W.; Lan, T.; Xie, X.; Peng, J.; Huang, J.; Wang, S.; Shen, X.; Liu, P.; Huang, H. Berberine Reduces Fibronectin Expression by Suppressing the S1P-S1P2 Receptor Pathway in Experimental Diabetic Nephropathy Models. PLoS ONE 2012, 7, e43874. [Google Scholar] [CrossRef]
- Ortiz, A.; Oliveira, J.P.; Waldek, S.; Warnock, D.G.; Cianciaruso, B.; Wanner, C.; Registry, O.B.O.T.F. Nephropathy in males and females with Fabry disease: Cross-sectional description of patients before treatment with enzyme replacement therapy. Nephrol. Dial. Transplant. 2008, 23, 1600–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurberg, B.L.; Rennke, H.; Colvin, R.B.; Dikman, S.; Gordon, R.E.; Collins, A.B.; Desnick, R.J.; O’Callaghan, M. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002, 62, 1933–1946. [Google Scholar] [CrossRef] [Green Version]
- Tøndel, C.; Bostad, L.; Larsen, K.K.; Hirth, A.; Vikse, B.E.; Houge, G.; Svarstad, E. Agalsidase Benefits Renal Histology in Young Patients with Fabry Disease. J. Am. Soc. Nephrol. 2012, 24, 137–148. [Google Scholar] [CrossRef]
- Hodgin, J.B.; Nair, V.; Zhang, H.; Randolph, A.; Harris, R.C.; Nelson, R.G.; Weil, E.J.; Cavalcoli, J.D.; Patel, J.M.; Brosius, F.C.; et al. Identification of Cross-Species Shared Transcriptional Networks of Diabetic Nephropathy in Human and Mouse Glomeruli. Diabetes 2013, 62, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Sha, Q.; Lyu, J.; Zhao, M.; Li, H.; Guo, M.; Sun, Q. Multi-Omics Analysis of Diabetic Nephropathy Reveals Potential New Mechanisms and Drug Targets. Front. Genet. 2020, 11, 616435. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.; Mariani, L.H.; Kretzler, M. Integrated multi-omics approaches to improve classification of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Zager, R.A.; Conrad, S.; Lochhead, K.; Sweeney, E.A.; Igarashi, Y.; Burkhart, K. Altered sphingomyelinase and ceramide expression in the setting of ischemic and nephrotoxic acute renal failure. Kidney Int. 1998, 53, 573–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupre, T.; Doll, M.A.; Shah, P.P.; Sharp, C.N.; Siow, D.; Megyesi, J.; Shayman, J.; Bielawska, A.; Bielawski, J.; Beverly, L.J.; et al. Inhibiting glucosylceramide synthase exacerbates cisplatin-induced acute kidney injury. J. Lipid Res. 2017, 58, 1439–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zager, R.A.; Conrad, D.S.; Burkhart, K. Ceramide accumulation during oxidant renal tubular injury: Mechanisms and potential consequences. J. Am. Soc. Nephrol. 1998, 9, 1670–1680. [Google Scholar] [CrossRef]
- Ueda, N.; Camargo, S.; Hong, X.; Basnakian, A.G.; Walker, P.D.; Shah, S.V. Role of Ceramide Synthase in Oxidant Injury to Renal Tubular Epithelial Cells. J. Am. Soc. Nephrol. 2001, 12, 2384–2391. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Herrington, J.; Zager, R.A. Sphingosine: A mediator of acute renal tubular injury and subsequent cytoresistance. Proc. Natl. Acad. Sci. USA 1995, 92, 8970–8974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuvillier, O. Sphingosine in apoptosis signaling. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2002, 1585, 153–162. [Google Scholar] [CrossRef]
- Woodcock, J. Sphingosine and ceramide signalling in apoptosis. IUBMB Life 2006, 58, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing Effects of ERK and JNK-p38 MAP Kinases on Apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef]
- Basnakian, A.G.; Ueda, N.; Hong, X.; Galitovsky, V.E.; Yin, X.; Shah, S.V. Ceramide synthase is essential for endonuclease-mediated death of renal tubular epithelial cells induced by hypoxia-reoxygenation. Am. J. Physiol. Physiol. 2005, 288, F308–F314. [Google Scholar] [CrossRef]
- Siskind, L.J.; Mullen, T.D.; Rosales, K.R.; Clarke, C.J.; Hernandez-Corbacho, M.J.; Edinger, A.L.; Obeid, L. The BCL-2 Protein BAK Is Required for Long-chain Ceramide Generation during Apoptosis. J. Biol. Chem. 2010, 285, 11818–11826. [Google Scholar] [CrossRef] [Green Version]
- Parra, V.; Eisner, V.; Chiong, M.; Criollo, A.; Moraga, F.; Garcia, A.; Härtel, S.; Jaimovich, E.; Zorzano, A.; Hidalgo, C.; et al. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc. Res. 2007, 77, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef]
- Siskind, L.J.; Feinstein, L.; Yu, T.; Davis, J.S.; Jones, D.; Choi, J.; Zuckerman, J.E.; Tan, W.; Hill, R.B.; Hardwick, J.M.; et al. Anti-apoptotic Bcl-2 Family Proteins Disassemble Ceramide Channels. J. Biol. Chem. 2008, 283, 6622–6630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kepp, O.; Rajalingam, K.; Kimmig, S.; Rudel, T. Bak and Bax are non-redundant during infection- and DNA damage-induced apoptosis. EMBO J. 2007, 26, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Zager, R.A.; Burkhart, K.M.; Johnson, A. Sphingomyelinase and Membrane Sphingomyelin Content. J. Am. Soc. Nephrol. 2000, 11, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Liu, W.; Xie, X.; Xu, S.; Huang, K.; Peng, J.; Shen, X.; Liu, P.; Wang, L.; Xia, P.; et al. Sphingosine Kinase-1 Pathway Mediates High Glucose-Induced Fibronectin Expression in Glomerular Mesangial Cells. Mol. Endocrinol. 2011, 25, 2094–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizawa, S.; Takahashi-Fujigasaki, J.; Kanazawa, Y.; Matoba, K.; Kawanami, D.; Yokota, T.; Iwamoto, T.; Tajima, N.; Manome, Y.; Utsunomiya, K. Sphingosine-1-phosphate induces differentiation of cultured renal tubular epithelial cells under Rho kinase activation via the S1P2 receptor. Clin. Exp. Nephrol. 2014, 18, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.-K.; Bajwa, A.; Ye, H.; Vergis, A.L.; Awad, A.S.; Kharel, Y.; Lynch, K.R.; Okusa, M.D. Divergent roles of sphingosine kinases in kidney ischemia–reperfusion injury. Kidney Int. 2009, 75, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Kim, M.; Kim, M.; D’Agati, V.D.; Lee, H.T. Sphingosine kinase 1 protects against renal ischemia–reperfusion injury in mice by sphingosine-1-phosphate1 receptor activation. Kidney Int. 2011, 80, 1315–1327. [Google Scholar] [CrossRef] [Green Version]
- Vidyasagar, A.; Wilson, N.A.; Djamali, A. Heat shock protein 27 (HSP27): Biomarker of disease and therapeutic target. Fibrogenesis Tissue Repair 2012, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Kim, M.; Kim, N.; D’Agati, V.D.; Sr, C.W.E.; Lee, H.T. Isoflurane mediates protection from renal ischemia-reperfusion injury via sphingosine kinase and sphingosine-1-phosphate-dependent pathways. Am. J. Physiol. Physiol. 2007, 293, F1827–F1835. [Google Scholar] [CrossRef] [Green Version]
- Bajwa, A.; Huang, L.; Kurmaeva, E.; Ye, H.; Dondeti, K.R.; Chroscicki, P.; Foley, L.S.; Balogun, Z.A.; Alexander, K.J.; Park, H.; et al. Sphingosine Kinase 2 Deficiency Attenuates Kidney Fibrosis via IFN-γ. J. Am. Soc. Nephrol. 2016, 28, 1145–1161. [Google Scholar] [CrossRef] [Green Version]
- Yaghobian, D.; Don, A.; Yaghobian, S.; Chen, X.; Pollock, C.A.; Saad, S. Increased sphingosine 1-phosphate mediates inflammation and fibrosis in tubular injury in diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 2015, 43, 56–66. [Google Scholar] [CrossRef]
- Park, S.W.; Kim, M.; Kim, J.Y.; Brown, K.M.; Haase, V.H.; D’Agati, V.D.; Lee, H.T. Proximal tubule sphingosine kinase-1 has a critical role in A1 adenosine receptor-mediated renal protection from ischemia. Kidney Int. 2012, 82, 878–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, H.M.; Huang, L.; Ye, H.; Liu, C.; Sung, S.-S.J.; Lynch, K.R.; Rosin, D.L.; Bajwa, A.; Okusa, M.D. Endothelial Sphingosine 1-Phosphate Receptor-1 Mediates Protection and Recovery from Acute Kidney Injury. J. Am. Soc. Nephrol. 2016, 27, 3383–3393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajwa, A.; Rosin, D.L.; Chroscicki, P.; Lee, S.; Dondeti, K.; Ye, H.; Kinsey, G.R.; Stevens, B.K.; Jobin, K.; Kenwood, B.M.; et al. Sphingosine 1-Phosphate Receptor-1 Enhances Mitochondrial Function and Reduces Cisplatin-Induced Tubule Injury. J. Am. Soc. Nephrol. 2014, 26, 908–925. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, A.; Jo, S.-K.; Ye, H.; Huang, L.; Dondeti, K.R.; Rosin, D.L.; Haase, V.H.; Macdonald, T.L.; Lynch, K.R.; Okusa, M.D. Activation of Sphingosine-1-Phosphate 1 Receptor in the Proximal Tubule Protects Against Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2010, 21, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Lai, L.-W.; Yong, K.-C.; Igarashi, S.; Lien, Y.-H. A sphingosine-1-phosphate type 1 receptor agonist inhibits the early T-cell transient following renal ischemia–reperfusion injury. Kidney Int. 2007, 71, 1223–1231. [Google Scholar] [CrossRef] [Green Version]
- Lien, Y.-H.; Yong, K.-C.; Cho, C.; Igarashi, S.; Lai, L.-W. S1P1-selective agonist, SEW2871, ameliorates ischemic acute renal failure. Kidney Int. 2006, 69, 1601–1608. [Google Scholar] [CrossRef] [Green Version]
- Kearney, P.M.; Whelton, M.; Reynolds, K.; Muntner, P.; Whelton, P.K.; He, J. Global burden of hypertension: Analysis of worldwide data. Lancet 2005, 365, 217–223. [Google Scholar] [CrossRef]
- Vanhoutte, P.M. Endothelial dysfunction in hypertension. J. Hypertens Suppl. 1996, 14, S83–S93. [Google Scholar]
- Taddei, S.; Salvetti, A. Pathogenetic Factors in Hypertension Endothelial Factors. Clin. Exp. Hypertens. 1996, 18, 323–335. [Google Scholar] [CrossRef]
- Di Pietro, P.; Carrizzo, A.; Sommella, E.; Oliveti, M.; Iacoviello, L.; Di Castelnuovo, A.; Acernese, F.; Damato, A.; De Lucia, M.; Merciai, F.; et al. Targeting the ASMase/S1P pathway protects from sortilin-evoked vascular damage in hypertension. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Pepe, G.; Cotugno, M.; Marracino, F.; Giova, S.; Capocci, L.; Forte, M.; Stanzione, R.; Bianchi, F.; Marchitti, S.; Di Pardo, A.; et al. Differential Expression of Sphingolipid Metabolizing Enzymes in Spontaneously Hypertensive Rats: A Possible Substrate for Susceptibility to Brain and Kidney Damage. Int. J. Mol. Sci. 2021, 22, 3796. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, A.; Zhang, Y.; Kothiya, M.; Galvani, S.; Obinata, H.; Bucci, M.; Giordano, F.J.; Jiang, X.-C.; Hla, T.; Di Lorenzo, A. Nogo-B regulates endothelial sphingolipid homeostasis to control vascular function and blood pressure. Nat. Med. 2015, 21, 1028–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, B.; Obinata, H.; Galvani, S.; Mendelson, K.; Ding, B.-S.; Skoura, A.; Kinzel, B.; Brinkmann, V.; Rafii, S.; Evans, T.; et al. Flow-Regulated Endothelial S1P Receptor-1 Signaling Sustains Vascular Development. Dev. Cell 2012, 23, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Morace, I.; Pilz, R.; Federico, G.; Jennemann, R.; Krunic, D.; Nordström, V.; von Gerichten, J.; Marsching, C.; Schießl, I.M.; Müthing, J.; et al. Renal globotriaosylceramide facilitates tubular albumin absorption and its inhibition protects against acute kidney injury. Kidney Int. 2019, 96, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Jeon, Y.J.; Jung, N.; Park, J.-W.; Park, H.-Y.; Jung, S.-C. Epithelial–Mesenchymal Transition in Kidney Tubular Epithelial Cells Induced by Globotriaosylsphingosine and Globotriaosylceramide. PLoS ONE 2015, 10, e0136442. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, M.; Ohkita, M.; Kobayashi, Y.; Yuba, M.; Matsumura, Y. Protective Effect Of alpha-LIPOIC Acid Against Ischaemic Acute Renal Failure In Rats. Clin. Exp. Pharmacol. Physiol. 2002, 29, 189–194. [Google Scholar] [CrossRef]
- Ghorbani, A. Renal protective effect of selenium on cisplatin-induced nephrotoxicity. J. Ren. Inj. Prev. 2012, 1, 31–32. [Google Scholar] [CrossRef]
- Feng, Y.; Bai, T.; Ma, H.; Wang, J.-K. Propofol Attenuates Human Proximal Renal Tubular Epithelial Cell Injury Induced by Anoxia-Reoxygenation. Lab. Med. 2015, 39, 356–360. [Google Scholar] [CrossRef] [Green Version]
- Peters, E.; Geraci, S.; Heemskerk, S.; Wilmer, M.J.; Bilos, A.; Kraenzlin, B.; Gretz, N.; Pickkers, P.; Masereeuw, R. Alkaline phosphatase protects against renal inflammation through dephosphorylation of lipopolysaccharide and adenosine triphosphate. J. Cereb. Blood Flow Metab. 2015, 172, 4932–4945. [Google Scholar] [CrossRef]
- Okusa, M.D.; Linden, J.; Macdonald, T.; Huang, L. Selective A2A adenosine receptor activation reduces ischemia-reperfusion injury in rat kidney. Am. J. Physiol. Content 1999, 277, F404–F412. [Google Scholar] [CrossRef]
- Grange, C.; Tapparo, M.; Bruno, S.; Chatterjee, D.; Quesenberry, P.J.; Tetta, C.; Camussi, G. Biodistribution of mesenchymal stem cell-derived extracellular vesicles in a model of acute kidney injury monitored by optical imaging. Int. J. Mol. Med. 2014, 33, 1055–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Kidney Disease | Sphingolipids Altered | Potential Drug Targets | References |
|---|---|---|---|
| Diabetic kidney disease | Sphingosine, Ceramides, S1P, C1P, Glycosphingolipids | CerS, ASAH1, SPHKs, S1PR1/S1PR2, SMPDL3b | [25,44,45,54,76,103,108] |
| Focal segmental glomerulosclerosis | Ceramides, S1P | ASAH1, SGPL1 | [57,76,77] |
| Alport syndrome | Sulfohexosyl ceramides | CerS | [78] |
| IgA nephropathy | S1P | SPHKs, S1PR2 | [56,82] |
| Lupus nephritis | Sphingosine, Ceramides, Lactosylceramides | CerS, NEU1 | [86,88,89] |
| Fabrys disease | Gb3 | α-galactosidase A | [90,91,92] |
| COVID mediated kidney injury | Sphingosine, Ceramides | SMase, SPT | [100] |
| Radiation induced kidney injury | S1P | SMPDL3b, SPHKs | [45] |
| Acute kidney injury | Sphingosine, Ceramides, S1P | CerS, SPHKs, S1PR1/S1PR2 | [53,120,124,125,127,128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mallela, S.k.; Merscher, S.; Fornoni, A. Implications of Sphingolipid Metabolites in Kidney Diseases. Int. J. Mol. Sci. 2022, 23, 4244. https://doi.org/10.3390/ijms23084244
Mallela Sk, Merscher S, Fornoni A. Implications of Sphingolipid Metabolites in Kidney Diseases. International Journal of Molecular Sciences. 2022; 23(8):4244. https://doi.org/10.3390/ijms23084244
Chicago/Turabian StyleMallela, Shamroop kumar, Sandra Merscher, and Alessia Fornoni. 2022. "Implications of Sphingolipid Metabolites in Kidney Diseases" International Journal of Molecular Sciences 23, no. 8: 4244. https://doi.org/10.3390/ijms23084244
APA StyleMallela, S. k., Merscher, S., & Fornoni, A. (2022). Implications of Sphingolipid Metabolites in Kidney Diseases. International Journal of Molecular Sciences, 23(8), 4244. https://doi.org/10.3390/ijms23084244