
Serrated Colorectal Lesions: An Up-to-Date Review from Histological Pattern to Molecular Pathogenesis


 ,
, 
 , and
, and
Abstract
:1. Introduction
2. The WHO Classification of Colorectal Serrated Lesions
- Hyperplastic polyps (HPs)
- Sessile serrated lesions (SSLs)
- Traditional serrated adenoma (TSAs)
- Serrated adenomas, unclassified
3. Colorectal Serrated Lesions: Histologic and Endoscopic Features
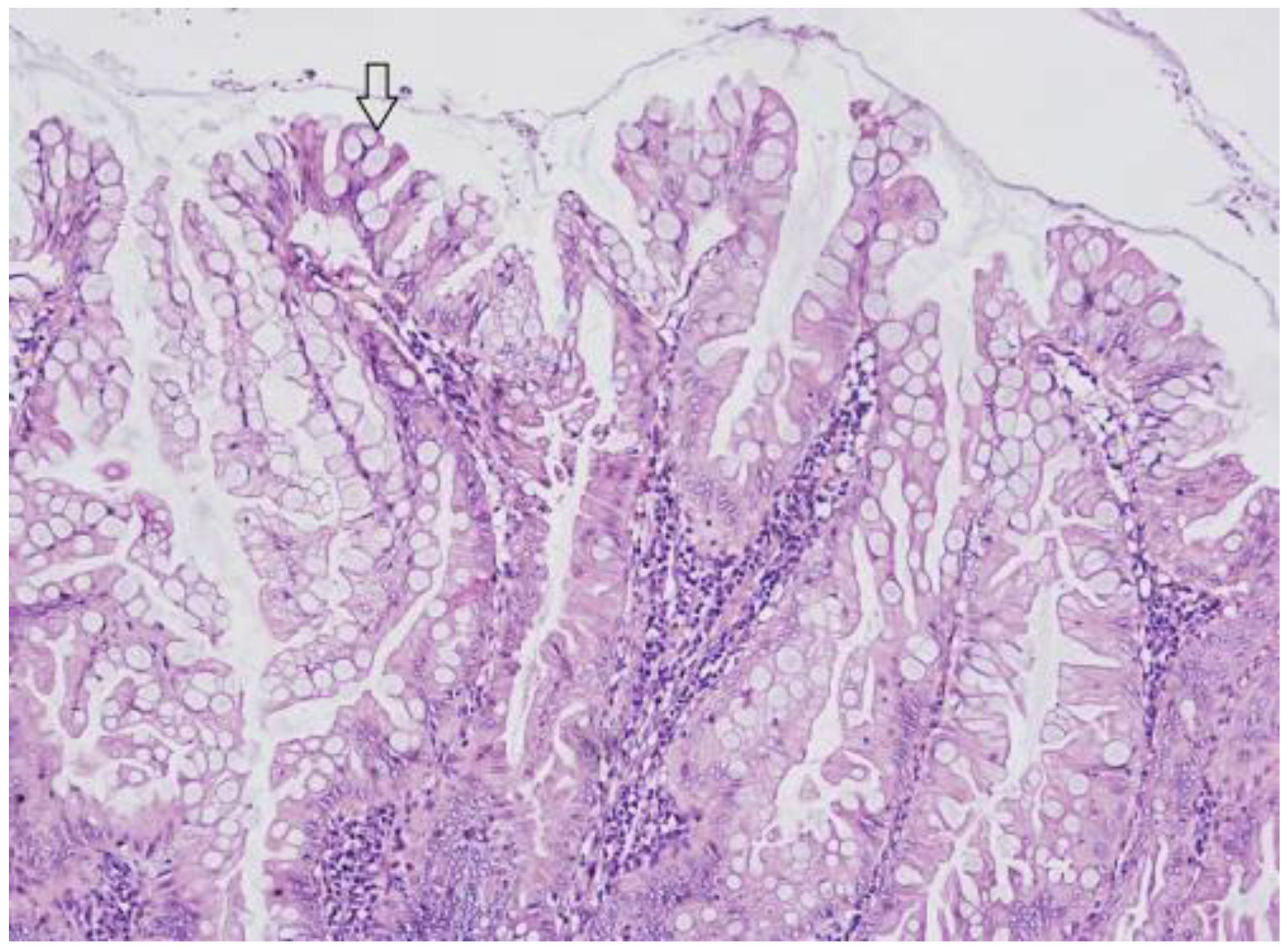
3.1. Hyperplastic Polyps (HP)
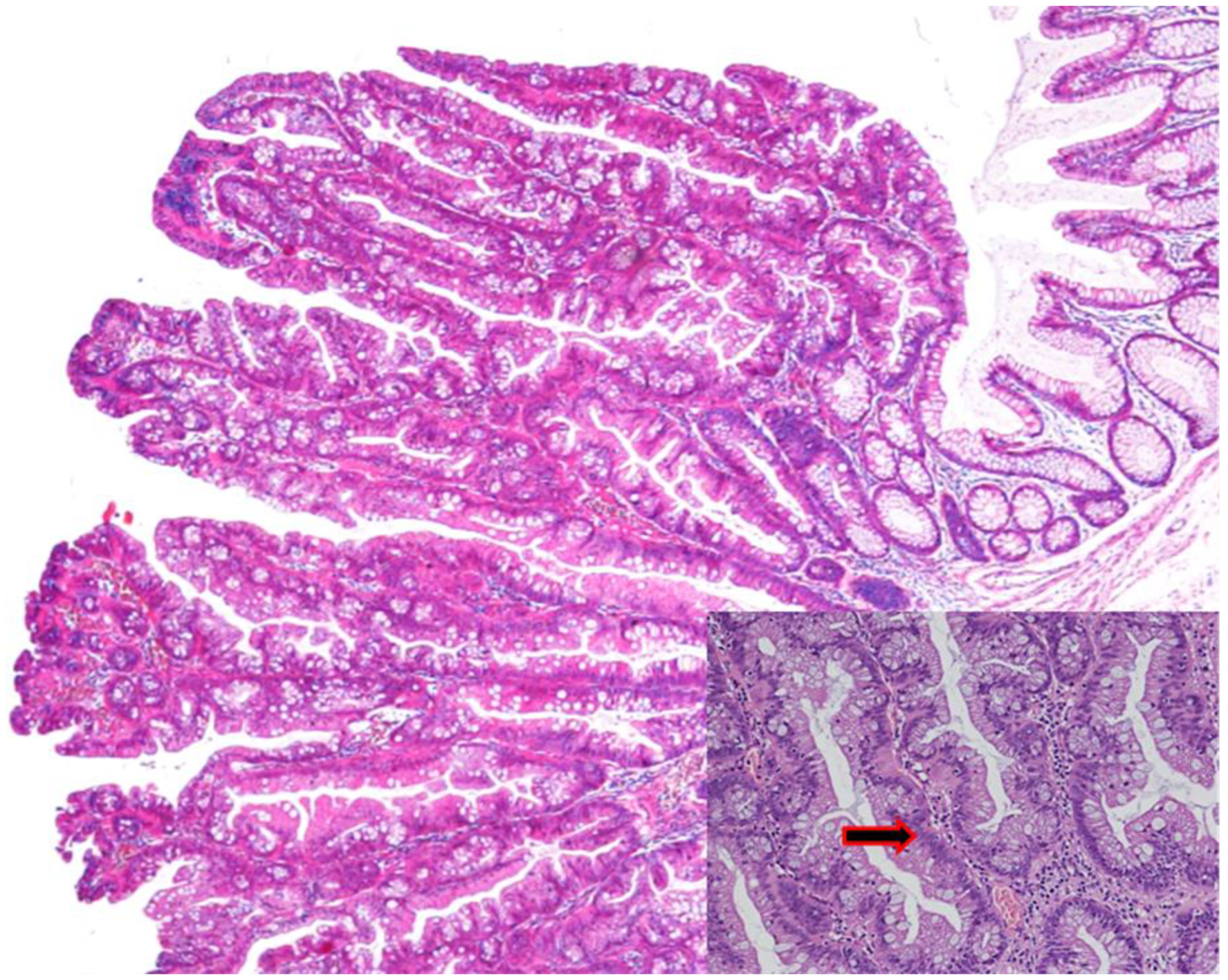
3.2. Sessile Serrated Lesions (SSLs)
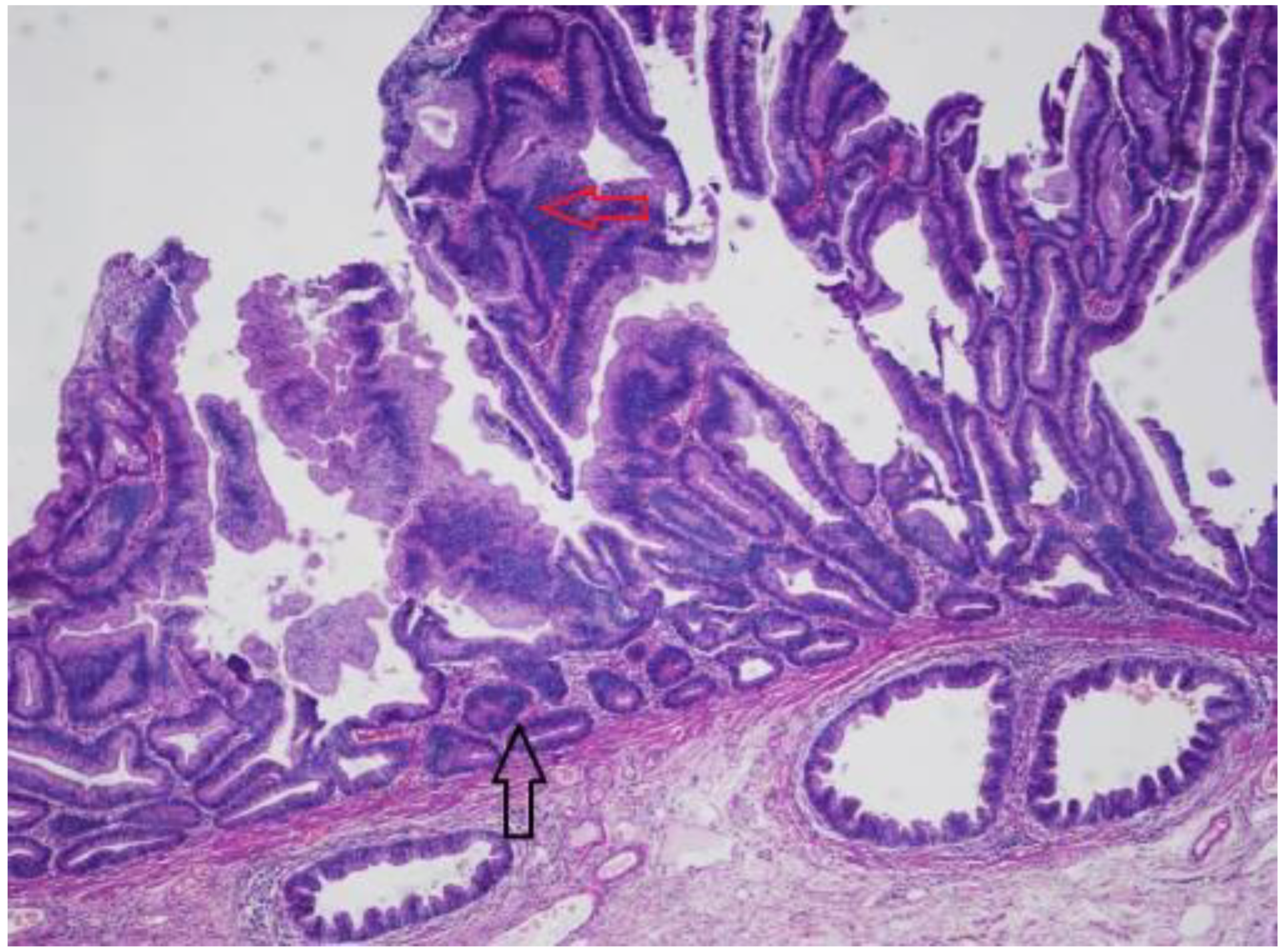
Sessile Serrated Lesions with Dysplasia (SSL-Ds)
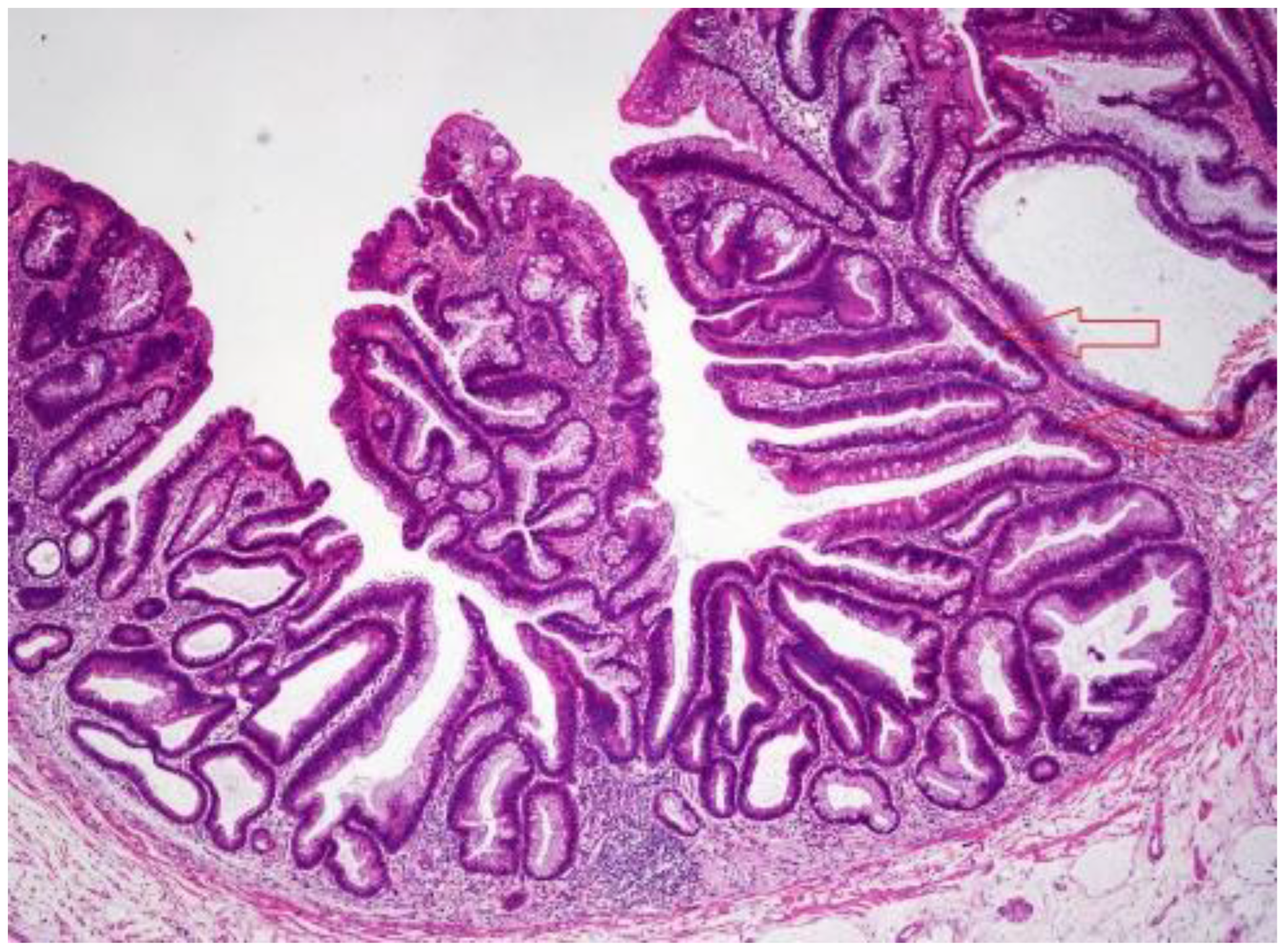
3.3. Traditional Serrated Adenoma (TSA)
3.4. Serrated Polyposis Syndrome (SPS)
4. Molecular Pathways Leading from Normal Mucosa to CRC
4.1. Hereditary CRCs
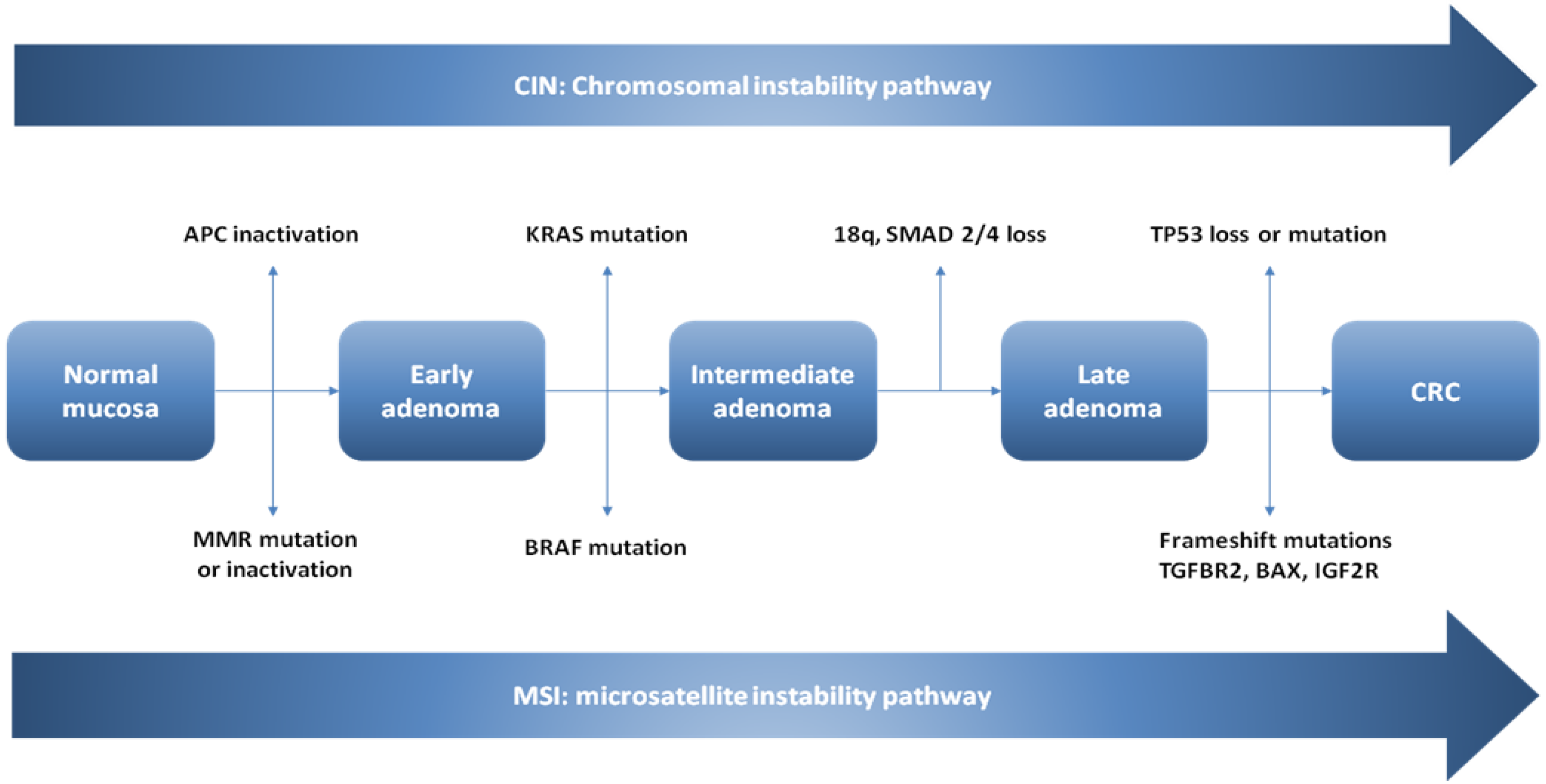
4.2. The Conventional Model of Colorectal Carcinogenesis in Sporadic CRCs
4.2.1. CIN Pathway
4.2.2. Microsatellite Instability
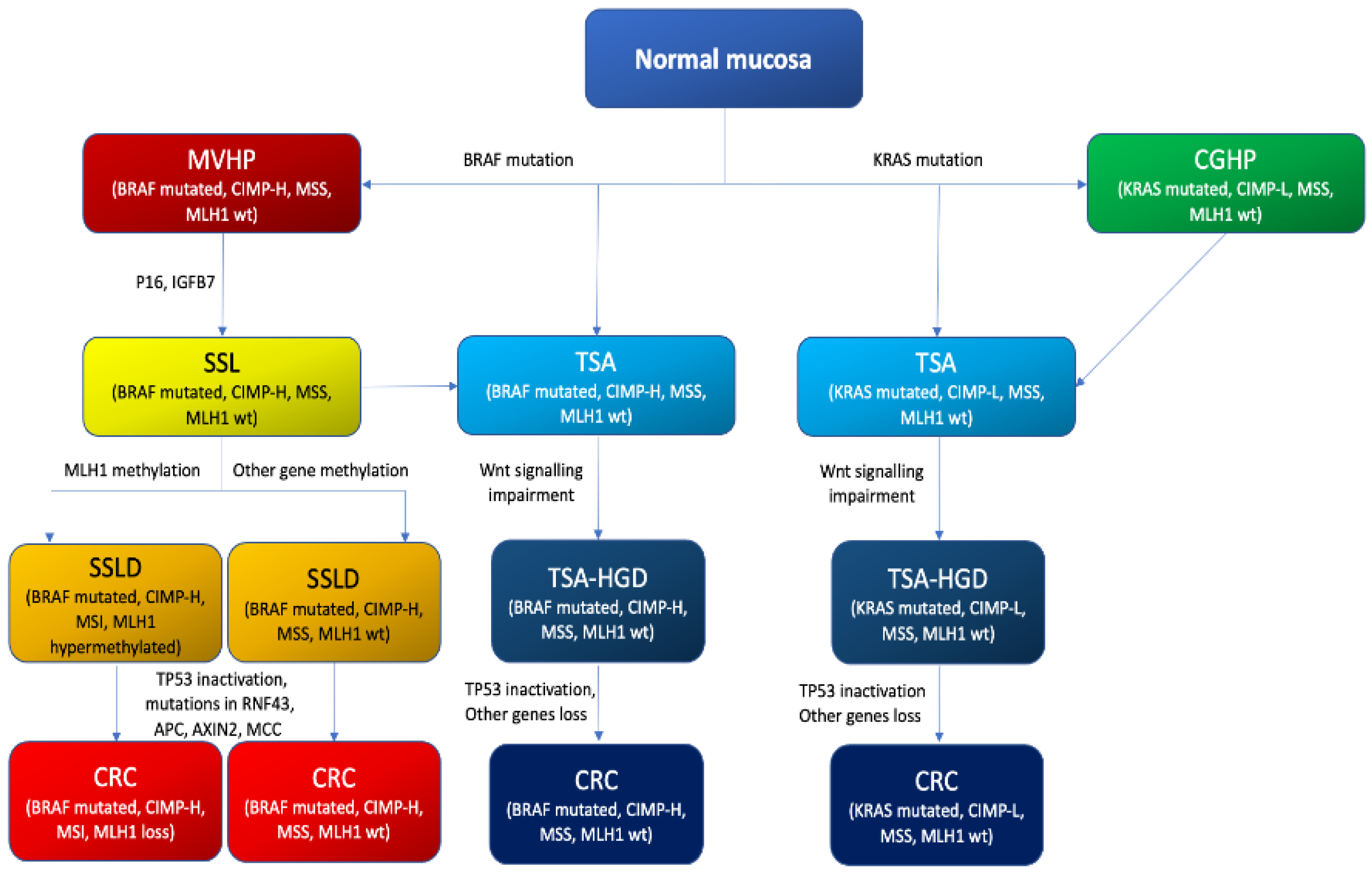
4.3. The Serrated Neoplasia Pathway
CpG Island Methylator Phenotype (CIMP)
5. Molecular Features of Serrated Colorectal Lesions
5.1. Hyperplastic Polyps
5.2. Sessile Serrated Lesions
5.3. Sessile Serrated Lesions with Dysplasia (SSLDs)
5.4. Traditional Serrated Adenomas (TSAs)
5.5. Serrated Polyposis Syndrome (SPS)
6. CRC Molecular Subtypes and the Role of Colorectal Serrated Lesions as Precursor
7. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, CA. Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strum, W.B. Colorectal adenomas. N. Engl. J. Med. 2016, 374, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Bettington, M.; Walker, N.; Rahman, T.; Vandeleur, A.; Whitehall, V.; Leggett, B.; Croese, J. High prevalence of sessile serrated adenomas in contemporary outpatient colonoscopy practice. Intern. Med. J. 2017, 47, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Spring, K.J.; Zhao, Z.Z.; Karamatic, R.; Walsh, M.D.; Whitehall, V.L.; Pike, T.; Simms, L.A.; Young, J.; James, M.; Montgomery, G.W.; et al. High prevalence of sessile serrated adenomas with BRAF mutations: A prospective study of patients undergoing colonoscopy. Gastroenterology 2006, 131, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Corley, D.A.; Jensen, C.D.; Marks, A.R.; Zhao, W.K.; Lee, J.K.; Doubeni, C.A.; Zauber, A.G.; de Boer, J.; Fireman, B.H.; Schottinger, J.E.; et al. Adenoma detection rate and risk of colorectal cancer and death. N. Engl. J. Med. 2014, 370, 1298–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schramm, C.; Janhsen, K.; Hofer, J.H.; Toermer, H.; Stelzer, A.; Stenschke, F.; Stollenwerk, M.; Scheller, I.; Lang, S.; Goeser, T.; et al. Detection of clinically relevant serrated polyps during screening colonoscopy: Results from seven cooperating centers within the German colorectal screening program. Endoscopy 2018, 50, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, D.A.; Williams, J.L.; Holub, J.L.; Morris, C.D.; Logan, J.R.; Eisen, G.M.; Carney, P. Race, ethnicity, and sex affect risk for polyps >9 mm in average-risk individuals. Gastroenterology 2014, 147, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Levin, T.R.; Lavin, P.; Lidgard, G.P.; Ahlquist, D.A.; Berger, B.M. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014, 370, 1287–1297. [Google Scholar] [CrossRef] [Green Version]
- Longacre, T.A.; Fenoglio-Preiser, C.M. Mixed hyperplastic adenomatous polyps/serrated adenomas. A distinct form of colorectal neoplasia. Am. J. Surg. Pathol. 1990, 14, 524–537. [Google Scholar] [CrossRef]
- World Health Organization, International Agency for Research on Cancer. WHO Classification of Tumours of the Digestive System, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2010. [Google Scholar]
- WHO Classification of Tumours Editorial Board. WHO Classification of Tumors: DIGESTIVE System Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2019. [Google Scholar]
- Odze, R.D. “Sessile Serrated Lesion”: The Art and Science of Naming a Disorder. Arch. Pathol. Lab. Med. 2021, 145, 1190–1191. [Google Scholar] [CrossRef]
- Pai, R.K.; Bettington, M.; Srivastava, A.; Rosty, C. An update on the morphology and molecular pathology of serrated colorectal polyps and associated carcinomas. Mod. Pathol. 2019, 32, 1390–1415. [Google Scholar] [CrossRef]
- Torlakovic, E.E.; Gomez, J.D.; Driman, D.K.; Parfitt, J.R.; Wang, C.; Benerjee, T.; Snover, D.C. Sessile serratedadenoma (SSA) vs. traditional serrated adenoma (TSA). Am. J. Surg. Pathol. 2008, 32, 21–29. [Google Scholar] [CrossRef] [PubMed]
- East, J.E.; Saunders, B.P.; Jass, J.R. Sporadic and syndromic hyperplastic polyps and serrated adenomas of the colon: Classification, molecular genetics, natural history, and clinical management. Gastroenterol. Clin. N. Am. 2008, 37, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Crockett, S.D.; Nagtegaal, I.D. Terminology, Molecular Features, Epidemiology, and Management of Serrated Colorectal Neoplasia. Gastroenterology 2019, 157, 949–966. [Google Scholar] [CrossRef] [Green Version]
- Sano, W.; Sano, Y.; Iwatate, M.; Hasuike, N.; Hattori, S.; Kosaka, H.; Ikumoto, T.; Kotaka, M.; Fujimori, T. Prospective evaluation of the proportion of sessile serrated adenoma/polyps in endoscopically diagnosed colorectal polyps with hyperplastic features. Endosc. Int. Open 2015, 3, 354–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwatate, M.; Sano, Y.; Tanaka, S.; Kudo, S.E.; Saito, S.; Matsuda, T.; Wada, Y.; Fujii, T.; Ikematsu, H.; Uraoka, T.; et al. Validation study for development of the Japan NBI Expert Team classification of colorectal lesions. Dig. Endosc. 2018, 30, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Hazewinkel, Y.; de Wijkerslooth, T.R.; Stoop, E.M.; Bossuyt, P.M.; Biermann, K.; van de Vijver, M.J.; Fockens, P.; van Leerdam, M.E.; Kuipers, E.J.; Dekker, E. Prevalence of serrated polyps and association with synchronous advanced neoplasia in screening colonoscopy. Endoscopy 2014, 46, 219–224. [Google Scholar] [CrossRef]
- Carr, N.J.; Mahajan, H.; Tan, K.L.; Hawkins, N.J.; Ward, R.L. Serrated and non-serrated polyps of the colorectum: Their prevalence in an unselected case series and correlation of BRAF mutation analysis with the diagnosis of sessile serrated adenoma. J. Clin. Pathol. 2009, 62, 516–518. [Google Scholar] [CrossRef]
- Abdeljawad, K.; Vemulapalli, K.C.; Kahi, C.J.; Cummings, O.W.; Snover, D.C.; Rex, D.K. Sessile serrated polyp prevalence determined by a colonoscopist with a high lesion detection rate and an experienced pathologist. Gastrointest. Endosc. 2015, 81, 517–524. [Google Scholar] [CrossRef]
- Wataru, S.; Daizen, H.; Akira, T.; Mineo, I.; Santa, H.; Mikio, F.; Yasushi, S. Serrated polyps of the colon and rectum: Remove or not? World J. Gastroenterol. 2020, 26, 2276–2285. [Google Scholar]
- Tadepalli, U.S.; Feihel, D.; Miller, K.M.; Itzkowitz, S.H.; Freedman, J.S.; Kornacki, S.; Cohen, L.B.; Bamji, N.D.; Bodian, C.A.; Aisenberg, J. A morphologic analysis of sessile serrated polyps observed during routine colonoscopy. Gastrointest. Endosc. 2011, 74, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Yamamoto, E.; Yamano, H.O.; Suzuki, H.; Kamimae, S.; Nojima, M.; Sawada, T.; Ashida, M.; Yoshikawa, K.; Takagi, R.; et al. A novel pit pattern identifies the precursor of colorectal cancer derived from sessile serrated adenoma. Am. J. Gastroenterol. 2012, 107, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.F.; Tang, S.J.; Lash, R.H.; Wu, R.; Yang, Q. Anatomic distribution of sessile serrated adenoma/polyp with and without cytologic dysplasia. Arch. Pathol. Lab. Med. 2015, 139, 388–393. [Google Scholar] [CrossRef]
- Liu, C.; Walker, N.I.; Leggett, B.A.; Whitehall, V.L.; Bettington, M.L.; Rosty, C. Sessile serrated adenomas with dysplasia: Morphological patterns and correlations with MLH1 immunohistochemistry. Mod. Pathol. 2017, 30, 1728–1738. [Google Scholar] [CrossRef]
- Cenaj, O.; Gibson, J.; Odze, R.D. Clinicopathologic and outcome study of sessile serrated adenomas/polyps with serrated versus intestinal dysplasia. Mod. Pathol. 2018, 31, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Bettington, M.L.; Walker, N.I.; Rosty, C.; Brown, I.S.; Clouston, A.D.; McKeone, D.M.; Pearson, S.A.; Klein, K.; Leggett, B.; Whitehall, V.L. A clinicopathological and molecular analysis of 200 traditional serrated adenomas. Mod. Pathol. 2015, 28, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Sano, W.; Fujimori, T.; Ichikawa, K.; Sunakawa, H.; Utsumi, T.; Iwatate, M.; Hasuike, N.; Hattori, S.; Kosaka, H.; Sano, Y. Clinical and endoscopic evaluations of sessile serrated adenoma/polyps with cytological dysplasia. J. Gastroenterol. Hepatol. 2018, 33, 1454–1460. [Google Scholar] [CrossRef]
- Murakami, T.; Sakamoto, N.; Ritsuno, H.; Shibuya, T.; Osada, T.; Mitomi, H.; Yao, T.; Watanabe, S. Distinct endoscopic characteristics of sessile serrated adenoma/polyp with and without dysplasia/carcinoma. Gastrointest. Endosc. 2017, 85, 590–600. [Google Scholar] [CrossRef]
- Tate, D.J.; Jayanna, M.; Awadie, H.; Desomer, L.; Lee, R.; Heitman, S.J.; Sidhu, M.; Goodrick, K.; Burgess, N.G.; Mahajan, H.; et al. A standardized imaging protocol for the endoscopic prediction of dysplasia within sessile serrated polyps. Gastrointest. Endosc. 2018, 87, 222–231. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, A.J.; Serra, S.; Chetty, R. Traditional serrated adenoma: An overview of pathology and emphasis on molecular pathogenesis. BMJ Open Gastroenterol. 2019, 6, 120–122. [Google Scholar] [CrossRef]
- Snover, D.C.; Jass, J.R.; Fenoglio-Preiser, C.; Batts, K.P. Serrated polyps of the large intestine: A morphologic and molecular review of an evolving concept. Am. J. Clin. Pathol. 2005, 124, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Saito, Y.; Fu, K.I.; Matsuda, T.; Uraoka, T.; Kobayashi, N.; Ito, H.; Machida, H.; Iwasaki, J.; Emura, F.; et al. Efficacy of magnifying chromoendoscopy for the differential diagnosis of colorectal lesions. Dig. Endosc. 2005, 17, 105–116. [Google Scholar] [CrossRef]
- Edelstein, D.L.; Axilbund, J.E.; Hylind, L.M.; Romans, K.; Griffin, C.A.; Cruz-Correa, M.; Giardiello, F.M. Serrated polyposis: Rapid and relentless development of colorectal neoplasia. Gut 2013, 62, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Carballal, S.; Rodríguez-Alcalde, D.; Moreira, L.; Hernández, L.; Rodríguez, L.; Rodríguez-Moranta, F.; Gonzalo, V.; Bujanda, L.; Bessa, X.; Poves, C.; et al. Colorectal cancer risk factors in patients with serrated polyposis syndrome: A large multicentre study. Gut 2016, 65, 1829–1837. [Google Scholar] [CrossRef]
- IJspeert, J.E.; Rana, S.A.; Atkinson, N.S.; van Herwaarden, Y.J.; Bastiaansen, B.A.; van Leerdam, M.E.; Sanduleanu, S.; Bisseling, T.M.; Spaander, M.C.; Clark, S.K.; et al. Clinical risk factors of colorectal cancer in patients with serrated polyposis syndrome: A multicentre cohort analysis. Gut 2017, 66, 278–284. [Google Scholar] [CrossRef] [Green Version]
- MacPhail, M.E.; Thygesen, S.B.; Patel, N.; Broadley, H.M.; Rex, D.K. Endoscopic control of polyp burden and expansion of surveillance intervals in serrated polyposis syndrome. Gastrointest. Endosc. 2019, 90, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Burt, R. Inheritance of colorectal cancer. Drug Discov. Today Dis. Mech. 2007, 4, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.T.; Duong, H.Q. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy. Oncol. Lett. 2018, 16, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Half, E.; Bercovich, D.; Rozen, P. Familial adenomatous polyposis. Orphanet J. Rare Dis. 2009, 12, 4–22. [Google Scholar] [CrossRef] [Green Version]
- Lynch, H.T.; De la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef]
- Sinicrope, F.A. Lynch Syndrome-Associated Colorectal Cancer. N. Engl. J. Med. 2018, 379, 764–773. [Google Scholar] [CrossRef]
- Kastrinos, F.; Sapna, S. Inherited colorectal cancer syndromes. Cancer J. 2011, 17, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Grady, W.M. Epigenetic events in the colorectum and in colon cancer. Biochem. Soc. Trans. 2005, 33, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef]
- Gupta, R.; Sinha, S.; Paul, R.N. The impact of microsatellite stability status in colorectal cancer. Curr. Probl. Cancer 2018, 42, 548–559. [Google Scholar] [CrossRef]
- Pino, M.S.; Chung, D.C. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [Green Version]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [Green Version]
- Tsang, A.H.; Cheng, K.H.; Wong, A.S.; Ng, S.S.; Ma, B.B.; Chan, C.M.; Tsui, N.B.; Chan, L.W.; Yung, B.Y.; Wong, S.C. Current and future molecular diagnostics in colorectal cancer and colorectal adenoma. World J. Gastroenterol. 2014, 20, 3847–3857. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Thiagalingam, S.; Lengauer, C.; Leach, F.S.; Schutte, M.; Hahn, S.A.; Overhauser, J.; Willson, J.K.; Markowitz, S.; Hamilton, S.R.; Kern, S.E.; et al. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat. Genet. 1996, 13, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Delker, D.A.; McGettigan, B.M.; Kanth, P.; Pop, S.; Neklason, D.W.; Bronner, M.P.; Burt, R.W.; Hagedorn, C.H. RNA Sequencing of Sessile Serrated Colon Polyps Identifies Differentially Expressed Genes and Immunohistochemical Markers. PLoS ONE 2014, 9, e88367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell. Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Pal, T.; Permuth-Wey, J.; Sellers, T.A. A review of the clinical relevance of mismatch-repair deficiency in ovarian cancer. Cancer 2008, 113, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Meyerhardt, J.A.; Loda, M.; Giovannucci, E.L.; Fuchs, C.S. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 2009, 58, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, H.; Kuroda, H.; Imai, Y.; Hiraishi, H. Molecular pathogenesis of sporadic colorectal cancers. Chin. J. Cancer 2016, 35, 76–79. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.; Ferber, M.; Mao, R.; Samowitz, W.; Ganguly, A. Working Group of the American College of Medical Genetics and Genomics (ACMG) Laboratory Quality Assurance Committee. ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis). Genet. Med. 2014, 16, 101–116. [Google Scholar]
- Jass, J.R. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007, 50, 113–130. [Google Scholar] [CrossRef]
- Perucho, M. Cancer of the microsatellite mutator phenotype. Biol. Chem. 1996, 377, 675–684. [Google Scholar] [PubMed]
- Mori, Y.; Yin, J.; Rashid, A.; Leggett, B.A.; Young, J.; Simms, L.; Kuehl, P.M.; Langenberg, P.; Meltzer, S.J.; Stine, O.C. Instabilotyping: Comprehensive identification of frameshift mutations caused by coding region microsatellite instability. Cancer Res. 2001, 61, 6046–6049. [Google Scholar] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Rex, D.K.; Ahnen, D.J.; Baron, J.A.; Batts, K.P.; Burke, C.A.; Burt, R.W.; Goldblum, J.R.; Guillem, J.G.; Kahi, C.J.; Kalady, M.F.; et al. Serrated Lesions of the Colorectum: Review and Recommendations from an Expert Panel. Am. J. Gastroenterol. 2012, 107, 1315–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patai, A.V.; Molnár, B.; Tulassay, Z.; Sipos, F. Serrated pathway: Alternative route to colorectal cancer. World J. Gastroenterol. 2013, 19, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Leggett, B.; Whitehall, V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology 2010, 138, 2088–2100. [Google Scholar] [CrossRef]
- Nazemalhosseini Mojarad, E.; Kuppen, P.J.; Aghdaei, H.A.; Zali, M.R. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol. Hepatol. Bed Bench 2013, 6, 120–128. [Google Scholar]
- Lao, V.V.; Grady, W.M. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Nati. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [Green Version]
- Hughes, L.A.; Khalid-de Bakker, C.A.; Smits, K.M.; van den Brandt, P.A.; Jonkers, D.; Ahuja, N.; Herman, J.G.; Weijenberg, M.P.; van Engeland, M. The CpG island methylator phenotype in colorectal cancer: Progress and problems. Biochim. Acta 2012, 1825, 77–85. [Google Scholar] [CrossRef]
- Yokoi, K.; Harada, H.; Yokota, K.; Ishii, S.; Tanaka, T.; Nishizawa, N.; Shimazu, M.; Kojo, K.; Miura, H.; Yamanashi, T.; et al. Epigenetic Status of CDO1 Gene May Reflect Chemosensitivity in Colon Cancer with Postoperative Adjuvant Chemotherapy. Ann. Surg. Oncol. 2019, 26, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Kokelaar, R.F.; Jones, H.; Beynon, J.; Evans, M.E.; Harris, D.A. Meta-analysis of the prognostic value of CpGisland methylator phenotype in rectal cancer. Int. J. Colorectal. Dis. 2018, 33, 995–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, N.; Norrie, M.; Cheong, K.; Mokany, E.; Ku, S.L.; Meagher, A.; O’Connor, T.; Ward, R. CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology 2002, 122, 1376–1387. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Catalano, P.J.; Benson, A.B., 3rd; O’Dwyer, P.; Hamilton, S.R.; Issa, J.P. Association between DNA methylation and shortened survival in patients with advanced colorectal cancer treated with 5-fluorouracil based chemotherapy. Clin. Cancer Res. 2007, 13, 6093–6098. [Google Scholar] [CrossRef] [Green Version]
- Ogino, S.; Cantor, M.; Kawasaki, T.; Brahmandam, M.; Kirkner, G.J.; Weisenberger, D.J.; Campan, M.; Laird, P.W.; Loda, M.; Fuchs, C.S. CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut 2006, 55, 1000–1006. [Google Scholar] [CrossRef]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Kraft, P.; Loda, M.; Fuchs, C.S. Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J. Mol. Diagn. 2007, 9, 305–314. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, M.J.; Yang, S.; Mack, C.; Xu, H.; Huang, C.S.; Mulcahy, E.; Amorosino, M.; Farraye, F.A. Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am. J. Surg. Pathol. 2006, 30, 1491–1501. [Google Scholar] [CrossRef]
- Gibson, J.A.; Hahn, H.P.; Shahsafaei, A.; Odze, R.D. MUC expression in hyperplastic and serrated colonic polyps: Lack of specificity of MUC6. Am. J. Surg. Pathol. 2011, 35, 742–749. [Google Scholar] [CrossRef]
- Murakami, T.; Mitomi, H.; Saito, T.; Takahashi, M.; Sakamoto, N.; Fukui, N.; Yao, T.; Watanabe, S. Distinct WNT/β-catenin signaling activation in the serrated neoplasia pathway and the adenoma-carcinoma sequence of the colorectum. Mod. Pathol. 2015, 28, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Rhee, Y.Y.; Kim, K.J.; Kang, G.H. CpG island methylator phenotype-high colorectal cancers and their prognostic implications and relationships with the serrated neoplasia pathway. Gut Liver 2017, 11, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Kang, B.; Petkovich, D.A.; Bhandari, Y.R.; In, J.; Stein-O’Brien, G.; Kong, X.; Xie, W.; Zachos, N.; Maegawa, S.; et al. Aging-like spontaneous epigenetic silencing facilitates Wnt activation, stemness, and BRAF(V600E)-induced tumorigenesis. Cancer Cell 2019, 35, 315–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Bettington, M.L.; Walker, N.I.; Dwine, J.; Hartel, G.F.; Leggett, B.A.; Whitehall, V.L.J. CpG island methylation in sessile serrated adenomas increases with age, indicating lower risk of malignancy in young patients. Gastroenterology 2018, 155, 1362–1365. [Google Scholar] [CrossRef]
- Mesteri, I.; Bayer, G.; Meyer, J.; Capper, D.; Schoppmann, S.F.; von Deimling, A.; Birner, P. Improved molecular classification of serrated lesions of the colon by immunohistochemical detection of BRAF V600E. Mod. Pathol. 2014, 27, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Caruso, M.; Moore, J.; Goodall, G.J.; Thomas, M.; Phillis, S.; Tyskin, A.; Cheetham, G.; Lerda, N.; Takahashi, H.; Ruszkiewicz, A. Over-expression of cathepsin E and trefoil factor 1 in sessile serrated adenomas of the colorectum identified by gene expression analysis. Virchows Arch. 2009, 454, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, K.J.; Rhee, Y.Y.; Bae, J.M.; Cho, N.Y.; Lee, H.S.; Kang, G.H. Gastric-type expression signature in serrated pathway-associated colorectal tumors. Hum. Pathol. 2015, 46, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Fennell, L.J.; Jamieson, S.; McKeone, D.; Corish, T.; Rohdmann, M.; Furner, T.; Bettington, M.; Liu, C.; Kawamata, F.; Bond, C.; et al. MLH1-93 G/a polymorphism is associated with MLH1 promoter methylation and protein loss in dysplastic sessile serrated adenomas with BRAFV600E mutation. BMC Cancer 2018, 18, 35. [Google Scholar] [CrossRef] [Green Version]
- Rickelt, S.; Condon, C.; Mana, M.; Whittaker, C.; Pfirschke, C.; Roper, J.; Patil, D.T.; Brown, I.; Mattia, A.R.; Zukerberg, L.; et al. Agrin in the muscularis mucosa serves as a biomarker distinguishing hyperplastic polyps from sessile serrated lesions. Clin. Cancer Res. 2020, 26, 277–287. [Google Scholar] [CrossRef]
- Cui, M.; Awadallah, A.; Liu, W.; Zhou, L.; Xin, W. Loss of Hes1 Differentiates Sessile Serrated Adenoma/Polyp from Hyperplastic Polyp. Am. J. Surg. Pathol. 2016, 40, 113–119. [Google Scholar] [CrossRef]
- Beggs, A.D.; Jones, A.; Shepherd, N.; Arnaout, A.; Finlayson, C.; Abulafi, A.M.; Morton, D.G.; Matthews, G.M.; Hodgson, S.V.; Tomlinson, I.P. Loss of Expression and Promoter Methylation of SLIT2 Are Associated with Sessile Serrated Adenoma Formation. PLoS Genet. 2013, 9, 87–89. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Akazawa, Y.; Yatagai, N.; Hiromoto, T.; Sasahara, N.; Saito, T.; Sakamoto, N.; Nagahara, A.; Yao, T. Molecular characterization of sessile serrated adenoma/polyps with dysplasia/carcinoma based on immunohistochemistry, next-generation sequencing, and microsatellite instability testing: A case series study. Diagn. Pathol. 2018, 13, 88. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kang, G.H. Molecular and prognostic heterogeneity of microsatellite-unstable colorectal cancer. World J. Gastroenterol. 2014, 20, 4230–4243. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Park, H.E.; Yoo, S.Y.; Jeong, S.; Cho, N.Y.; Kang, G.H.; Kim, J.H. CpG island methylation in sessile serrated adenoma/polyp of the colorectum: Implications for differential diagnosis of molecularly high-risk lesions among non-dysplastic sessile serrated adenomas/polyps. J. Pathol. Transl. Med. 2019, 53, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Bettington, M.; Brown, I.; Rosty, C.; Walker, N.; Liu, C.; Croese, J.; Rahman, T.; Pearson, S.A.; McKeone, D.; Leggett, B.; et al. Sessile serrated adenomas in young patients may have limited risk of malignant progression. J. Clin. Gastroenterol. 2019, 53, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Yamashita, S.; Yoshida, H.; Taniguchi, H.; Ushijima, T.; Yamada, T.; Saito, Y.; Ochiai, A.; Sekine, S.; Hiraoka, N. WNT Pathway Gene Mutations Are Associated with the Presence of Dysplasia in Colorectal Sessile Serrated Adenoma/Polyps. Am. J. Surg. Pathol. 2017, 41, 1188–1197. [Google Scholar] [CrossRef]
- Bond, C.E.; McKeone, D.M.; Kalimutho, M.; Bettington, M.L.; Pearson, S.A.; Dumenil, T.D.; Wockner, L.F.; Burge, M.; Leggett, B.A.; Whitehall, V.L. RNF43 and ZNRF3 are commonly altered in serrated pathway colorectal tumorigenesis. Oncotarget 2016, 7, 70589–70600. [Google Scholar] [CrossRef]
- Koinuma, K.; Yamashita, Y.; Liu, W.; Hatanaka, H.; Kurashina, K.; Wada, T.; Takada, S.; Kaneda, R.; Choi, Y.L.; Fujiwara, S.I.; et al. Epigenetic silencing of AXIN2 in colorectal carcinoma with microsatellite instability. Oncogene 2006, 25, 139–146. [Google Scholar] [CrossRef] [Green Version]
- East, J.E.; Atkin, W.S.; Bateman, A.C.; Clark, S.K.; Dolwani, S.; Ket, S.N.; Leedham, S.J.; Phull, P.S.; Rutter, M.D.; Shepherd, N.A.; et al. British Society of Gastroenterology position statement on serrated polyps in the colon and rectum. Gut 2017, 66, 1181–1196. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.H.; Liau, J.Y.; Lin, Y.L.; Lin, L.I.; Cheng, Y.C.; Cheng, M.L.; Jeng, Y.M. Traditional serrated adenoma has two pathways of neoplastic progression that are distinct from the sessile serrated pathway of colorectal carcinogenesis. Mod. Pathol. 2014, 27, 1375–1385. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Lee, E.J.; Suh, J.P.; Chun, S.M.; Jang, S.J.; Kim, D.S.; Lee, D.H.; Lee, S.H.; Youk, E.G. Traditional serrated adenoma of the colorectum: Clinicopathologic implications and endoscopic findings of the precursor lesions. Am. J. Clin. Pathol. 2013, 140, 898–911. [Google Scholar] [CrossRef] [Green Version]
- Sekine, S.; Yamashita, S.; Tanabe, T.; Hashimoto, T.; Yoshida, H.; Taniguchi, H.; Kojima, M.; Shinmura, K.; Saito, Y.; Hiraoka, N.; et al. Frequent PTPRK-RSPO3 fusions and RNF43 mutations in colorectal traditional serrated adenoma. J. Pathol. 2016, 239, 133–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, S.; Ogawa, R.; Hashimoto, T.; Motohiro, K.; Yoshida, H.; Taniguchi, H.; Saito, Y.; Yasuhiro, O.; Ochiai, A.; Hiraoka, N. Comprehensive characterization of RSPO fusions in colorectal traditional serrated adenomas. Histopathology 2017, 71, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Liau, J.Y.; Yuan, C.T.; Lin, Y.L.; Tseng, L.H.; Cheng, M.L.; Jeng, Y.M. RNF43 is an early and specific mutated gene in the serrated pathway, with increased frequency in traditional serrated adenoma and its associated malignancy. Am. J. Surg. Pathol. 2016, 40, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Sohier, P.; Sanson, R.; Leduc, M.; Audebourg, A.; Broussard, C.; Salnot, V.; Just, P.A.; Pasmant, E.; Mayeux, P.; Guillonneau, F.; et al. Proteome analysis of formalin-fixed paraffin-embedded colorectal adenomas reveals the heterogeneous nature of traditional serrated adenomas compared to other colorectal adenomas. J. Pathol. 2020, 250, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Gala, M.K.; Mizukami, Y.; Le, L.P.; Moriichi, K.; Austin, T.; Yamamoto, M.; Lauwers, G.Y.; Bardeesy, N.; Chung, D.C. Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology 2014, 146, 520–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokko, A.; Laiho, P.; Lehtonen, R.; Korja, S.; Carvajal-Carmona, L.; Jarvinen, H.; Mecklin, J.P.; Eng, C.; Schleutker, J.; Tomlinson, I.P.; et al. EPHB2 germline variants in patients with colorectal cancer or hyperplastic polyposis. BMC Cancer 2006, 6, 145. [Google Scholar] [CrossRef] [Green Version]
- Phipps, A.I.; Limburg, P.J.; Baron, J.A.; Burnett-Hartman, A.N.; Weisenberger, D.J.; Laird, P.W.; Sinicrope, F.A.; Rosty, C.; Buchanan, D.D.; Potter, J.D.; et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology 2015, 148, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Phipps, A.I.; Alwers, E.; Harrison, T.; Banbury, B.; Brenner, H.; Campbell, P.T.; Chang-Claude, J.; Buchanan, D.; Chan, A.T.; Farris, A.B.; et al. Association between molecular subtypes of colorectal tumors and patient survival, based on pooled analysis of 7 international studies. Gastroenterology 2020, 158, 2158–2168. [Google Scholar] [CrossRef]
- Kim, J.H.; Bae, J.M.; Cho, N.Y.; Kang, G.H. Distinct features between MLH1-methylated and unmethylated colorectal carcinomas with the CpG island methylator phenotype: Implication in the serrated neoplasia pathway. Oncotarget 2016, 7, 14095–14111. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kang, G.H. Evolving pathologic concepts of serrated lesions of the colorectum. J. Pathol. Transl. Med. 2020, 54, 276–289. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serrated Colorectal Lesions Classification (2010 WHO 4th Edition) | |
|---|---|
| Histological type | Histological sub-type |
| Hyperplastic polyp (HP) |
|
| Sessile serrated adenoma/polyp (SSA/P) |
|
| Traditional serrated adenoma (TSA) | |
| Serrated Colorectal Lesions Classification (2019 WHO 5th Edition) | |
|---|---|
| Histological type | Histological subtype |
| Hyperplastic polyp (HP) |
|
| Sessile serrated lesion (SSL) |
|
| Traditional serrated adenoma (TSA) | |
| Serrated adenoma, unclassified | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mezzapesa, M.; Losurdo, G.; Celiberto, F.; Rizzi, S.; d’Amati, A.; Piscitelli, D.; Ierardi, E.; Di Leo, A. Serrated Colorectal Lesions: An Up-to-Date Review from Histological Pattern to Molecular Pathogenesis. Int. J. Mol. Sci. 2022, 23, 4461. https://doi.org/10.3390/ijms23084461
Mezzapesa M, Losurdo G, Celiberto F, Rizzi S, d’Amati A, Piscitelli D, Ierardi E, Di Leo A. Serrated Colorectal Lesions: An Up-to-Date Review from Histological Pattern to Molecular Pathogenesis. International Journal of Molecular Sciences. 2022; 23(8):4461. https://doi.org/10.3390/ijms23084461
Chicago/Turabian StyleMezzapesa, Martino, Giuseppe Losurdo, Francesca Celiberto, Salvatore Rizzi, Antonio d’Amati, Domenico Piscitelli, Enzo Ierardi, and Alfredo Di Leo. 2022. "Serrated Colorectal Lesions: An Up-to-Date Review from Histological Pattern to Molecular Pathogenesis" International Journal of Molecular Sciences 23, no. 8: 4461. https://doi.org/10.3390/ijms23084461
APA StyleMezzapesa, M., Losurdo, G., Celiberto, F., Rizzi, S., d’Amati, A., Piscitelli, D., Ierardi, E., & Di Leo, A. (2022). Serrated Colorectal Lesions: An Up-to-Date Review from Histological Pattern to Molecular Pathogenesis. International Journal of Molecular Sciences, 23(8), 4461. https://doi.org/10.3390/ijms23084461