
Single-Cell RNA Sequencing for Plant Research: Insights and Possible Benefits


{kind=link}
{kind=link}
Abstract
:1. Introduction
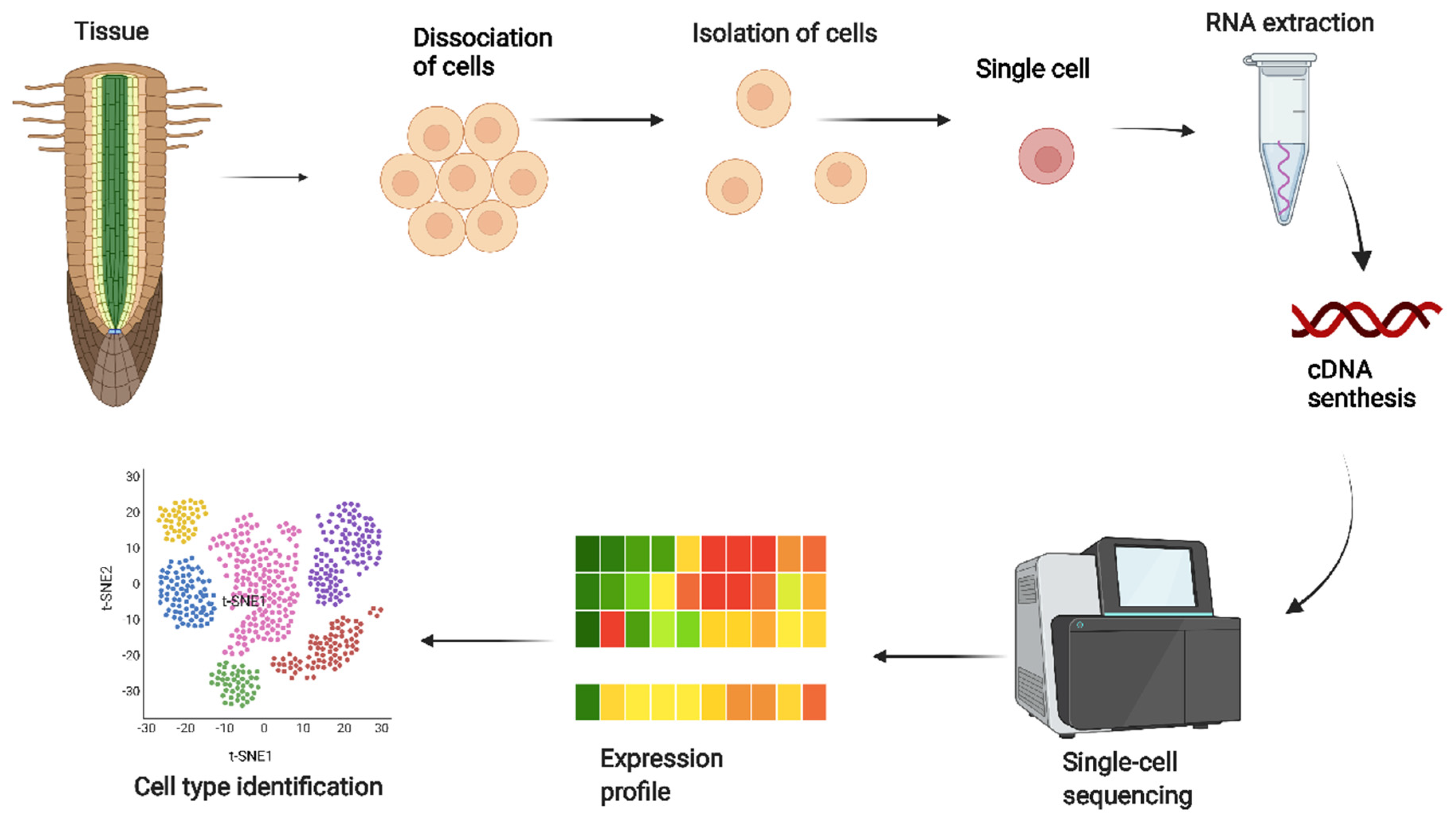
2. The Power of Single-Cell Sequencing Methodologies
3. Application of scRNA-seq in Plant Research
4. Why Consider Performing scRNA-seq in Plant Research?
5. scRNA-seq for Responses to Biotic and Abiotic Stresses
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fu, N.; Wang, Q.; Shen, H.-L. De Novo Assembly, Gene Annotation and Marker Development Using Illumina Paired-End Transcriptome Sequences in Celery (Apium graveolens L.). PLoS ONE 2013, 8, e57686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorizzo, M.; Senalik, D.A.; Grzebelus, D.; Bowman, M.; Cavagnaro, P.F.; Matvienko, M.; Ashrafi, H.; Van Deynze, A.; Simon, P.W. De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genom. 2011, 12, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutjahr, C.; Sawers, R.J.; Marti, G.; Andrés-Hernández, L.; Yang, S.Y.; Casieri, L.; Angliker, H.; Oakeley, E.J.; Wolfender, J.L.; Abreu-Goodger, C.; et al. Transcriptome diversity among rice root types during asymbiosis and interaction with arbuscular mycorrhizal fungi. Proc. Natl. Acad. Sci. USA 2015, 112, 6754–6759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takehisa, H.; Sato, Y.; Igarashi, M.; Abiko, T.; Antonio, B.A.; Kamatsuki, K.; Minami, H.; Namiki, N.; Inukai, Y.; Nakazono, M.; et al. Genome-wide transcriptome dissection of the rice root system: Implications for developmental and physiological functions. Plant J. Cell Mol. Biol. 2012, 69, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Van den Berge, K.; Perraudeau, F.; Soneson, C.; Love, M.I.; Risso, D.; Vert, J.P.; Robinson, M.D.; Dudoit, S.; Clement, L. Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications. Genome Biol. 2018, 19, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennecke, P.; Anders, S.; Kim, J.K.; Kołodziejczyk, A.A.; Zhang, X.; Proserpio, V.; Baying, B.; Benes, V.; Teichmann, S.A.; Marioni, J.C.; et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods 2013, 10, 1093–1095. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C. Defining cell types and states with single-cell genomics. Genome Res. 2015, 25, 1491–1498. [Google Scholar] [CrossRef] [Green Version]
- McFaline-Figueroa, J.L.; Trapnell, C.; Cuperus, J.T. The promise of single-cell genomics in plants. Curr. Opin. Plant Biol. 2020, 54, 114–121. [Google Scholar] [CrossRef]
- Rich-Griffin, C.; Stechemesser, A.; Finch, J.; Lucas, E.; Ott, S.; Schäfer, P. Single-Cell Transcriptomics: A High-Resolution Avenue for Plant Functional Genomics. Trends Plant Sci. 2020, 25, 186–197. [Google Scholar] [CrossRef]
- Shaw, R.; Tian, X.; Xu, J. Single-Cell Transcriptome Analysis in Plants: Advances and Challenges. Mol. Plant 2021, 14, 115–126. [Google Scholar] [CrossRef]
- Seyfferth, C.; Renema, J.; Wendrich, J.R.; Eekhout, T.; Seurinck, R.; Vandamme, N.; Blob, B.; Saeys, Y.; Helariutta, Y.; Birnbaum, K.D.; et al. Advances and Opportunities in Single-Cell Transcriptomics for Plant Research. Annu. Rev. Plant Biol. 2021, 72, 847–866. [Google Scholar] [CrossRef]
- Marand, A.P.; Chen, Z.; Gallavotti, A.; Schmitz, R.J. A cis-regulatory atlas in maize at single-cell resolution. Cell 2021, 184, 3041–3055.e21. [Google Scholar] [CrossRef]
- Denyer, T.; Ma, X.; Klesen, S.; Scacchi, E.; Nieselt, K.; Timmermans, M.C.P. Spatiotemporal Developmental Trajectories in the Arabidopsis Root Revealed Using High-Throughput Single-Cell RNA Sequencing. Dev. Cell 2019, 48, 840–852.e5. [Google Scholar] [CrossRef] [Green Version]
- Gala, H.P.; Lanctot, A.; Jean-Baptiste, K.; Guiziou, S.; Chu, J.C.; Zemke, J.E.; George, W.; Queitsch, C.; Cuperus, J.T.; Nemhauser, J.L. A single-cell view of the transcriptome during lateral root initiation in Arabidopsis thaliana. Plant Cell 2021, 33, 2197–2220. [Google Scholar] [CrossRef]
- Jean-Baptiste, K.; McFaline-Figueroa, J.L.; Alexandre, C.M.; Dorrity, M.W.; Saunders, L.; Bubb, K.L.; Trapnell, C.; Fields, S.; Queitsch, C.; Cuperus, J.T. Dynamics of Gene Expression in Single Root Cells of Arabidopsis thaliana. Plant Cell 2019, 31, 993–1011. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Huan, Q.; Li, K.; Qian, W. Single-cell transcriptome atlas of the leaf and root of rice seedlings. J. Genet. Genom. 2021, 48, 881–898. [Google Scholar] [CrossRef]
- Kim, J.Y.; Symeonidi, E.; Pang, T.Y.; Denyer, T.; Weidauer, D.; Bezrutczyk, M.; Miras, M.; Zöllner, N.; Hartwig, T.; Wudick, M.M.; et al. Distinct identities of leaf phloem cells revealed by single cell transcriptomics. Plant Cell 2021, 33, 511–530. [Google Scholar] [CrossRef]
- Ortiz-Ramírez, C.; Dias, P.; Zhang, S.; Demesa-Arévalo, E.; Yan, Z.; Xu, X.; Rahni, R.; Gingeras, T.; Jackson, D.; Gallagher, K.; et al. Ground Tissue Circuitry Regulates Organ Complexity in Cereal Roots. bioRxiv 2021. [Google Scholar] [CrossRef]
- Liu, Z.; Guo, C.; Wu, R.; Wang, J.; Zhou, Y.; Yu, X.; Zhang, Y.; Zhao, Z.; Liu, H.; Sun, S.; et al. Identification of the Regulators of Epidermis Development under Drought- and Salt-Stressed Conditions by Single-Cell RNA-Seq. Int. J. Mol. Sci. 2022, 23, 2759. [Google Scholar] [CrossRef]
- Tripathi, R.K.; Wilkins, O. Single cell gene regulatory networks in plants: Opportunities for enhancing climate change stress resilience. Plant Cell Environ. 2021, 44, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Choi, Y.; Kim, H.; Kim, S.-G. Single-cell RNA-sequencing of Nicotiana attenuata corolla cells reveals the biosynthetic pathway of a floral scent. New Phytol. 2022, 234, 527–544. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Lee, J.; Song, Q.; Li, Q.; Schiefelbein, J.; Zhao, B.; Li, S. Identification of new marker genes from plant single-cell RNA-seq data using interpretable machine learning methods. New Phytol. 2022, 234, 1507–1520. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, J.; Zhou, Y.; Zhang, Y.; Qin, A.; Yu, X.; Zhao, Z.; Wu, R.; Guo, C.; Bawa, G.; et al. Identification of Novel Regulators Required for Early Development of Vein Pattern in the Cotyledons by Single-cell RNA-seq. Plant J. 2022, 110, 7–22. [Google Scholar] [CrossRef]
- Tian, C.; Du, Q.; Xu, M.; Du, F.; Jiao, Y.J.B. Single-nucleus RNA-seq resolves spatiotemporal developmental trajectories in the tomato shoot apex. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ryu, K.H.; Huang, L.; Kang, H.M.; Schiefelbein, J. Single-Cell RNA Sequencing Resolves Molecular Relationships Among Individual Plant Cells. Plant Physiol. 2019, 179, 1444–1456. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhou, Y.; Guo, J.; Li, J.; Tian, Z.; Zhu, Z.; Wang, J.; Wu, R.; Zhang, B.; Hu, Y.; et al. Global Dynamic Molecular Profiling of Stomatal Lineage Cell Development by Single-Cell RNA Sequencing. Mol. Plant 2020, 13, 1178–1193. [Google Scholar] [CrossRef]
- Shulse, C.N.; Cole, B.J.; Ciobanu, D.; Lin, J.; Yoshinaga, Y.; Gouran, M.; Turco, G.M.; Zhu, Y.; O’Malley, R.C.; Brady, S.M.; et al. High-Throughput Single-Cell Transcriptome Profiling of Plant Cell Types. Cell Rep. 2019, 27, 2241–2247.e4. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.-Q.; Xu, Z.-G.; Shang, G.-D.; Wang, J.-W. A Single-Cell RNA Sequencing Profiles the Developmental Landscape of Arabidopsis Root. Mol. Plant 2019, 12, 648–660. [Google Scholar] [CrossRef]
- Kim, D.H.; Marinov, G.K.; Pepke, S.; Singer, Z.S.; He, P.; Williams, B.; Schroth, G.P.; Elowitz, M.B.; Wold, B.J. Single-Cell Transcriptome Analysis Reveals Dynamic Changes in lncRNA Expression during Reprogramming. Cell Stem Cell 2015, 16, 88–101. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhang, Y.; Fang, X.; Tran, S.; Zhai, N.; Yang, Z.; Guo, F.; Chen, L.; Yu, J.; Ison, M.S.; et al. Transcriptional landscapes of de novo root regeneration from detached Arabidopsis leaves revealed by time-lapse and single-cell RNA sequencing analyses. Plant Commun. 2022, 100306. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Gao, S.; Cui, F.; Chen, W.; Fan, L.; Qi, Y. Single-cell RNA sequencing reveals the landscape of maize root tips and assists in identification of cell type-specific nitrate-response genes. Crop J. 2022; in press. [Google Scholar] [CrossRef]
- Liu, Q.; Liang, Z.; Feng, D.; Jiang, S.; Wang, Y.; Du, Z.; Li, R.; Hu, G.; Zhang, P.; Ma, Y.; et al. Transcriptional landscape of rice roots at the single-cell resolution. Mol. Plant 2021, 14, 384–394. [Google Scholar] [CrossRef]
- Xie, J.; Li, M.; Zeng, J.; Li, X.; Zhang, D. Single-cell RNA sequencing profiles of stem-differentiating xylem in poplar. Plant Biotechnol. J. 2022, 20, 417–419. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Birnbaum, K.D.; Ehrhardt, D.W. Towards Building a Plant Cell Atlas. Trends Plant Sci. 2019, 24, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Ivakov, A.; Persson, S.J.E. Plant cell walls. eLS 2012. [Google Scholar] [CrossRef]
- Efroni, I.; Ip, P.L.; Nawy, T.; Mello, A.; Birnbaum, K.D. Quantification of cell identity from single-cell gene expression profiles. Genome Biol. 2015, 16, 9. [Google Scholar] [CrossRef] [Green Version]
- Efroni, I.; Mello, A.; Nawy, T.; Ip, P.L.; Rahni, R.; DelRose, N.; Powers, A.; Satija, R.; Birnbaum, K.D. Root Regeneration Triggers an Embryo-like Sequence Guided by Hormonal Interactions. Cell 2016, 165, 1721–1733. [Google Scholar] [CrossRef] [Green Version]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef] [Green Version]
- Wendrich, J.R.; Yang, B.; Vandamme, N.; Verstaen, K.; Smet, W.; Van de Velde, C.; Minne, M.; Wybouw, B.; Mor, E.; Arents, H.E.; et al. Vascular transcription factors guide plant epidermal responses to limiting phosphate conditions. Science 2020, 370, eaay4970. [Google Scholar] [CrossRef]
- Lopez-Anido, C.B.; Vatén, A.; Smoot, N.K.; Sharma, N.; Guo, V.; Gong, Y.; Anleu Gil, M.X.; Weimer, A.K.; Bergmann, D.C. Single-cell resolution of lineage trajectories in the Arabidopsis stomatal lineage and developing leaf. Dev. Cell 2021, 56, 1043–1055.e4. [Google Scholar] [CrossRef]
- Apelt, F.; Mavrothalassiti, E.; Gupta, S.; Machin, F.; Olas, J.J.; Annunziata, M.G.; Schindelasch, D.; Kragler, F. Shoot and root single cell sequencing reveals tissue- and daytime-specific transcriptome profiles. Plant Physiol. 2022, 188, 861–878. [Google Scholar] [CrossRef]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Turco, G.M.; Rodriguez-Medina, J.; Siebert, S.; Han, D.; Valderrama-Gómez, M.; Vahldick, H.; Shulse, C.N.; Cole, B.J.; Juliano, C.E.; Dickel, D.E.; et al. Molecular Mechanisms Driving Switch Behavior in Xylem Cell Differentiation. Cell Rep. 2019, 28, 342–351.e4. [Google Scholar] [CrossRef] [Green Version]
- Bergen, V.; Lange, M.; Peidli, S.; Wolf, F.A.; Theis, F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020, 38, 1408–1414. [Google Scholar] [CrossRef]
- Nelms, B.; Walbot, V. Defining the developmental program leading to meiosis in maize. Science 2019, 364, 52–56. [Google Scholar] [CrossRef]
- Satterlee, J.W.; Strable, J.; Scanlon, M.J. Plant stem-cell organization and differentiation at single-cell resolution. Proc. Natl. Acad. Sci. USA 2020, 117, 33689–33699. [Google Scholar] [CrossRef] [PubMed]
- Bezrutczyk, M.; Zöllner, N.R.; Kruse, C.P.S.; Hartwig, T.; Lautwein, T.; Köhrer, K.; Frommer, W.B.; Kim, J.Y. Evidence for phloem loading via the abaxial bundle sheath cells in maize leaves. Plant Cell 2021, 33, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Crow, M.; Rice, B.R.; Li, F.; Harris, B.; Liu, L.; Demesa-Arevalo, E.; Lu, Z.; Wang, L.; Fox, N.; et al. Single-cell RNA sequencing of developing maize ears facilitates functional analysis and trait candidate gene discovery. Dev. Cell 2021, 56, 557–568.e6. [Google Scholar] [CrossRef] [PubMed]
- Shahan, R.; Nolan, T.M.; Benfey, P.N. Single-cell analysis of cell identity in the Arabidopsis root apical meristem: Insights and opportunities. J. Exp. Bot. 2021, 72, 6679–6686. [Google Scholar] [CrossRef] [PubMed]
- Dorrity, M.W.; Alexandre, C.M.; Hamm, M.O.; Vigil, A.L.; Fields, S.; Queitsch, C.; Cuperus, J.T. The regulatory landscape of Arabidopsis thaliana roots at single-cell resolution. Nat. Commun. 2021, 12, 3334. [Google Scholar] [CrossRef] [PubMed]
- Farmer, A.; Thibivilliers, S.; Ryu, K.H.; Schiefelbein, J.; Libault, M. Single-nucleus RNA and ATAC sequencing reveals the impact of chromatin accessibility on gene expression in Arabidopsis roots at the single-cell level. Mol. Plant 2021, 14, 372–383. [Google Scholar] [CrossRef]
- Coate, J.E.; Farmer, A.D.; Schiefelbein, J.W.; Doyle, J.J. Expression Partitioning of Duplicate Genes at Single Cell Resolution in Arabidopsis Roots. Front. Genet. 2020, 11, 596150. [Google Scholar] [CrossRef]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; de Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O.; et al. CEL-Seq2: Sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016, 17, 77. [Google Scholar] [CrossRef] [Green Version]
- Picelli, S.; Faridani, O.R.; Björklund, A.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Hagemann-Jensen, M.; Ziegenhain, C.; Chen, P.; Ramsköld, D.; Hendriks, G.J.; Larsson, A.J.M.; Faridani, O.R.; Sandberg, R. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 2020, 38, 708–714. [Google Scholar] [CrossRef]
- Roszak, P.; Heo, J.-O.; Blob, B.; de Luis Balaguer, M.; Lau, W.; Hamey, F.; Cirrone, J.; Wang, X.; Ursache, R.; Tavares, H.; et al. Analysis of phloem trajectory links tissue maturation to cell specialization. bioRxiv 2021. [Google Scholar] [CrossRef]
- Serrano-Ron, L.; Perez-Garcia, P.; Sanchez-Corrionero, A.; Gude, I.; Cabrera, J.; Ip, P.L.; Birnbaum, K.D.; Moreno-Risueno, M.A. Reconstruction of lateral root formation through single-cell RNA sequencing reveals order of tissue initiation. Mol. Plant 2021, 14, 1362–1378. [Google Scholar] [CrossRef]
- Graeff, M.; Rana, S.; Wendrich, J.R.; Dorier, J.; Eekhout, T.; Aliaga Fandino, A.C.; Guex, N.; Bassel, G.W.; De Rybel, B.; Hardtke, C.S. A single-cell morpho-transcriptomic map of brassinosteroid action in the Arabidopsis root. Mol. Plant 2021, 14, 1985–1999. [Google Scholar] [CrossRef]
- Kubo, M.; Nishiyama, T.; Tamada, Y.; Sano, R.; Ishikawa, M.; Murata, T.; Imai, A.; Lang, D.; Demura, T.; Reski, R.; et al. Single-cell transcriptome analysis of Physcomitrella leaf cells during reprogramming using microcapillary manipulation. Nucleic Acids Res. 2019, 47, 4539–4553. [Google Scholar] [CrossRef] [Green Version]
- Omary, M.; Gil-Yarom, N.; Yahav, C.; Steiner, E.; Hendelman, A.; Efroni, I. A conserved superlocus regulates above- and belowground root initiation. Science 2022, 375, eabf4368. [Google Scholar] [CrossRef]
- Gulati, G.S.; Sikandar, S.S.; Wesche, D.J.; Manjunath, A.; Bharadwaj, A.; Berger, M.J.; Ilagan, F.; Kuo, A.H.; Hsieh, R.W.; Cai, S.; et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 2020, 367, 405–411. [Google Scholar] [CrossRef]
- Kester, L.; van Oudenaarden, A. Single-Cell Transcriptomics Meets Lineage Tracing. Cell Stem Cell 2018, 23, 166–179. [Google Scholar] [CrossRef] [Green Version]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.Q.; Chen, Y.; Liu, Y.; Lin, W.H.; Wang, J.W. Single-cell transcriptome atlas and chromatin accessibility landscape reveal differentiation trajectories in the rice root. Nat. Commun. 2021, 12, 2053. [Google Scholar] [CrossRef]
- Verhage, L. Single but not alone: The transcriptomes of 14,000 single cells from developing cotyledon veins. Plant J. 2022, 110, 5–6. [Google Scholar] [CrossRef]
- Birnbaum, K.; Shasha, D.E.; Wang, J.Y.; Jung, J.W.; Lambert, G.M.; Galbraith, D.W.; Benfey, P.N. A gene expression map of the Arabidopsis root. Science 2003, 302, 1956–1960. [Google Scholar] [CrossRef] [Green Version]
- Brady, S.M.; Orlando, D.A.; Lee, J.Y.; Wang, J.Y.; Koch, J.; Dinneny, J.R.; Mace, D.; Ohler, U.; Benfey, P.N. A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 2007, 318, 801–806. [Google Scholar] [CrossRef]
- Li, S.; Yamada, M.; Han, X.; Ohler, U.; Benfey, P.N. High-Resolution Expression Map of the Arabidopsis Root Reveals Alternative Splicing and lincRNA Regulation. Dev. Cell 2016, 39, 508–522. [Google Scholar] [CrossRef] [Green Version]
- Nawy, T.; Lee, J.Y.; Colinas, J.; Wang, J.Y.; Thongrod, S.C.; Malamy, J.E.; Birnbaum, K.; Benfey, P.N. Transcriptional profile of the Arabidopsis root quiescent center. Plant Cell 2005, 17, 1908–1925. [Google Scholar] [CrossRef] [Green Version]
- Giacomello, S. A new era for plant science: Spatial single-cell transcriptomics. Curr. Opin. Plant Biol. 2021, 60, 102041. [Google Scholar] [CrossRef]
- Zhang, G.; Guo, G.; Hu, X.; Zhang, Y.; Li, Q.; Li, R.; Zhuang, R.; Lu, Z.; He, Z.; Fang, X.; et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Wu, R.; Wee, C.; Xie, F.; Wei, X.; Chan, P.; Tham, C.; Duan, L.; Dinneny, J. A Spatio-Temporal Understanding of Growth Regulation during the Salt Stress Response in Arabidopsis. Plant Cell 2013, 25, 2132–2154. [Google Scholar] [CrossRef] [Green Version]
- Dinneny, J.; Long, T.; Wang, J.; Jung, J.; Mace, D.; Pointer, S.; Barron, C.; Brady, S.; Schiefelbein, J.; Benfey, P. Cell Identity Mediates the Response of Arabidopsis Roots to Abiotic Stress. Science 2008, 320, 942–945. [Google Scholar] [CrossRef] [Green Version]
- Mustroph, A.; Zanetti, M.E.; Jang, C.J.H.; Holtan, H.E.; Repetti, P.P.; Galbraith, D.W.; Girke, T.; Bailey-Serres, J. Profiling translatomes of discrete cell populations resolves altered cellular priorities during hypoxia in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 18843–18848. [Google Scholar] [CrossRef] [Green Version]
- Iyer-Pascuzzi, A.S.; Jackson, T.; Cui, H.; Petricka, J.J.; Busch, W.; Tsukagoshi, H.; Benfey, P.N. Cell identity regulators link development and stress responses in the Arabidopsis root. Dev. Cell 2011, 21, 770–782. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Ando, A.; Jiang, N.; Ikeda, Y.; Chen, Z.J. Single-cell RNA-seq analysis reveals ploidy-dependent and cell-specific transcriptome changes in Arabidopsis female gametophytes. Genome Biol. 2020, 21, 178. [Google Scholar] [CrossRef]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imdahl, F.; Vafadarnejad, E.; Homberger, C.; Saliba, A.E.; Vogel, J. Single-cell RNA-sequencing reports growth-condition-specific global transcriptomes of individual bacteria. Nat. Microbiol. 2020, 5, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M. A functional perspective on phenotypic heterogeneity in microorganisms. Nat. Rev. Microbiol. 2015, 13, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Gasch, A.P.; Yu, F.B.; Hose, J.; Escalante, L.E.; Place, M.; Bacher, R.; Kanbar, J.; Ciobanu, D.; Sandor, L.; Grigoriev, I.V.; et al. Single-cell RNA sequencing reveals intrinsic and extrinsic regulatory heterogeneity in yeast responding to stress. PLoS Biol. 2017, 15, e2004050. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, Y.; Lü, T.; Yang, X.; Liu, J.; Dong, Y.; Wang, Y. An Efficient and Universal Protoplast Isolation Protocol Suitable for Transient Gene Expression Analysis and Single-Cell RNA Sequencing. Int. J. Mol. Sci. 2022, 23, 3419. [Google Scholar] [CrossRef]
- Baccin, C.; Al-Sabah, J.; Velten, L.; Helbling, P.M.; Grünschläger, F.; Hernández-Malmierca, P.; Nombela-Arrieta, C.; Steinmetz, L.M.; Trumpp, A.; Haas, S. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol. 2020, 22, 38–48. [Google Scholar] [CrossRef]
- Lohoff, T.; Ghazanfar, S.; Missarova, A.; Koulena, N.; Pierson, N.; Griffiths, J.A.; Bardot, E.S.; Eng, C.H.L.; Tyser, R.C.V.; Argelaguet, R.; et al. Integration of spatial and single-cell transcriptomic data elucidates mouse organogenesis. Nat. Biotechnol. 2022, 40, 74–85. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bawa, G.; Liu, Z.; Yu, X.; Qin, A.; Sun, X. Single-Cell RNA Sequencing for Plant Research: Insights and Possible Benefits. Int. J. Mol. Sci. 2022, 23, 4497. https://doi.org/10.3390/ijms23094497
Bawa G, Liu Z, Yu X, Qin A, Sun X. Single-Cell RNA Sequencing for Plant Research: Insights and Possible Benefits. International Journal of Molecular Sciences. 2022; 23(9):4497. https://doi.org/10.3390/ijms23094497
Chicago/Turabian StyleBawa, George, Zhixin Liu, Xiaole Yu, Aizhi Qin, and Xuwu Sun. 2022. "Single-Cell RNA Sequencing for Plant Research: Insights and Possible Benefits" International Journal of Molecular Sciences 23, no. 9: 4497. https://doi.org/10.3390/ijms23094497
APA StyleBawa, G., Liu, Z., Yu, X., Qin, A., & Sun, X. (2022). Single-Cell RNA Sequencing for Plant Research: Insights and Possible Benefits. International Journal of Molecular Sciences, 23(9), 4497. https://doi.org/10.3390/ijms23094497