
Hemin-Induced Endothelial Dysfunction and Endothelial to Mesenchymal Transition in the Pathogenesis of Pulmonary Hypertension Due to Chronic Hemolysis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
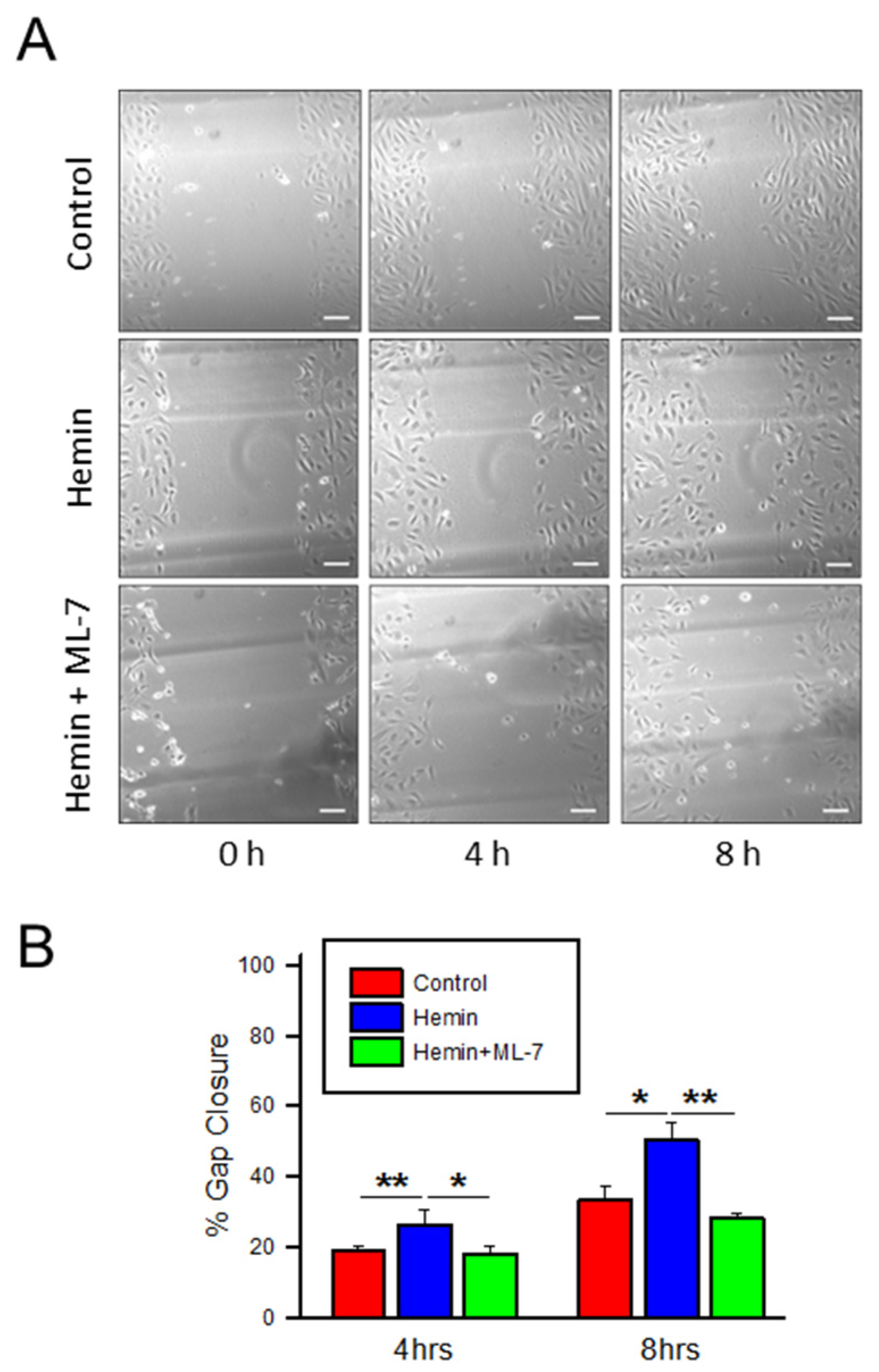{kind=link}
Abstract
:1. Introduction
2. Results
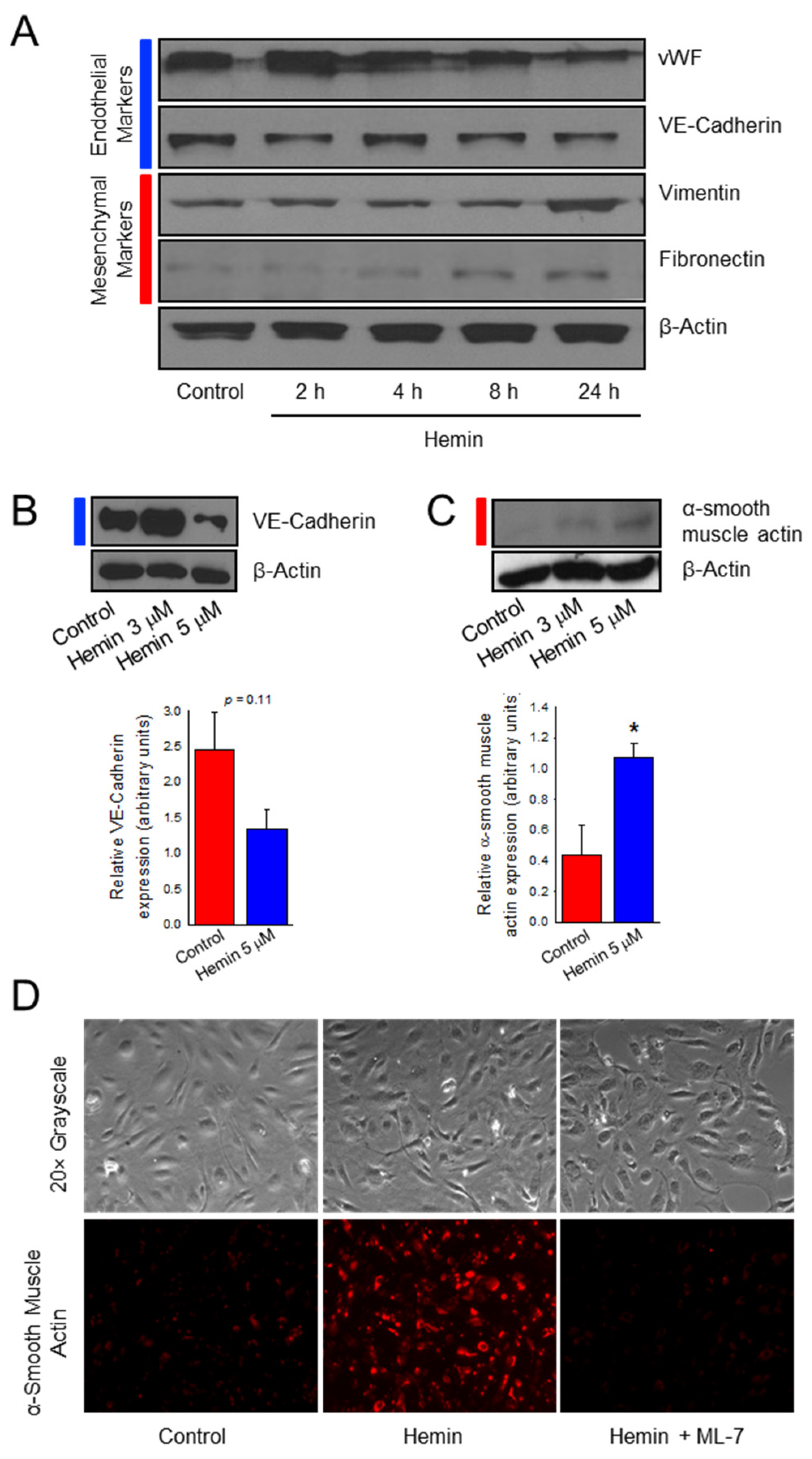
2.1. Hemin Downregulates Endothelial Marker Expression and Upregulates Mesenchymal Marker Expression in Pulmonary Artery Endothelial Cells
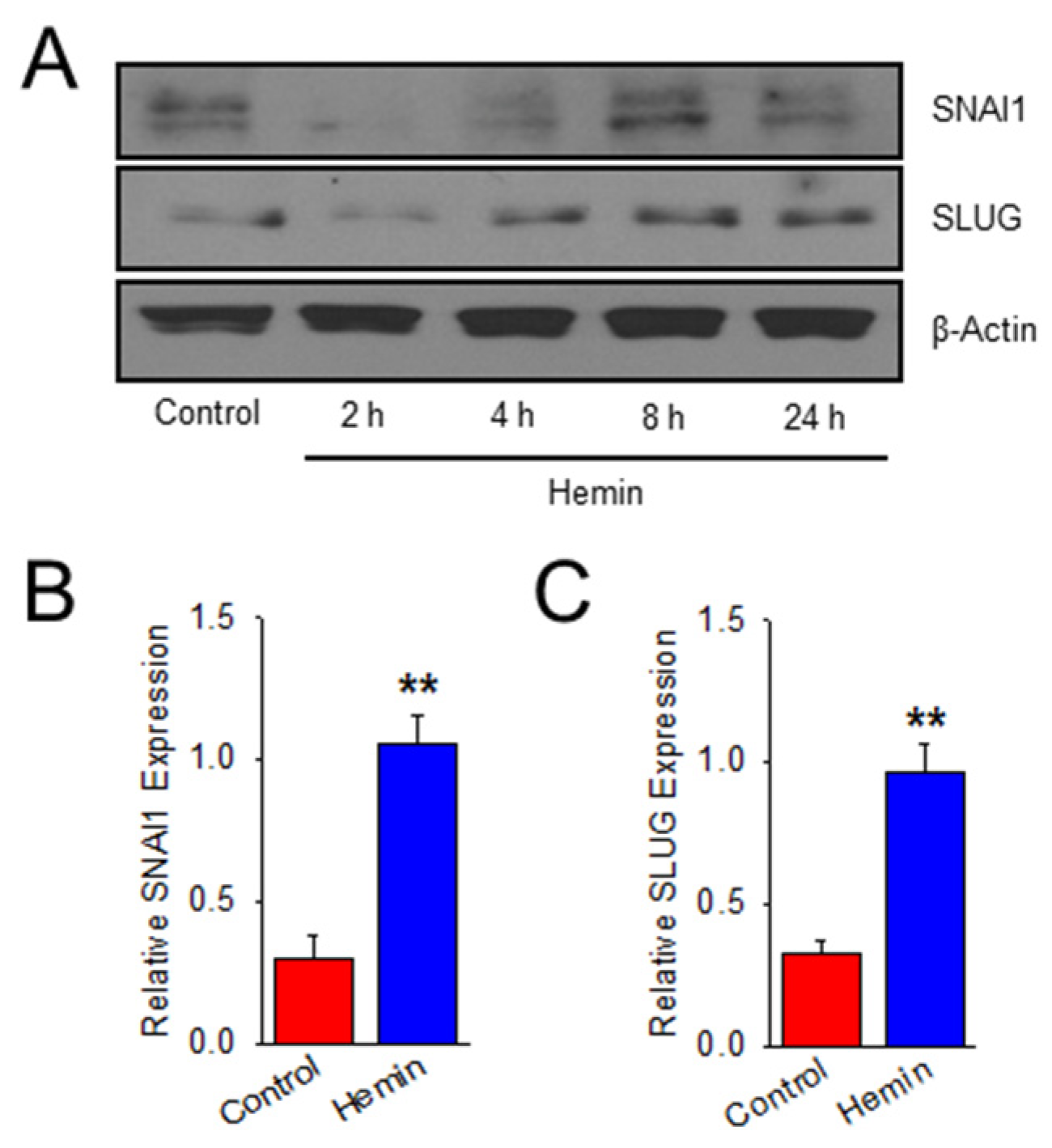
2.2. Hemin Upregulates EndoMT Transcription Factors
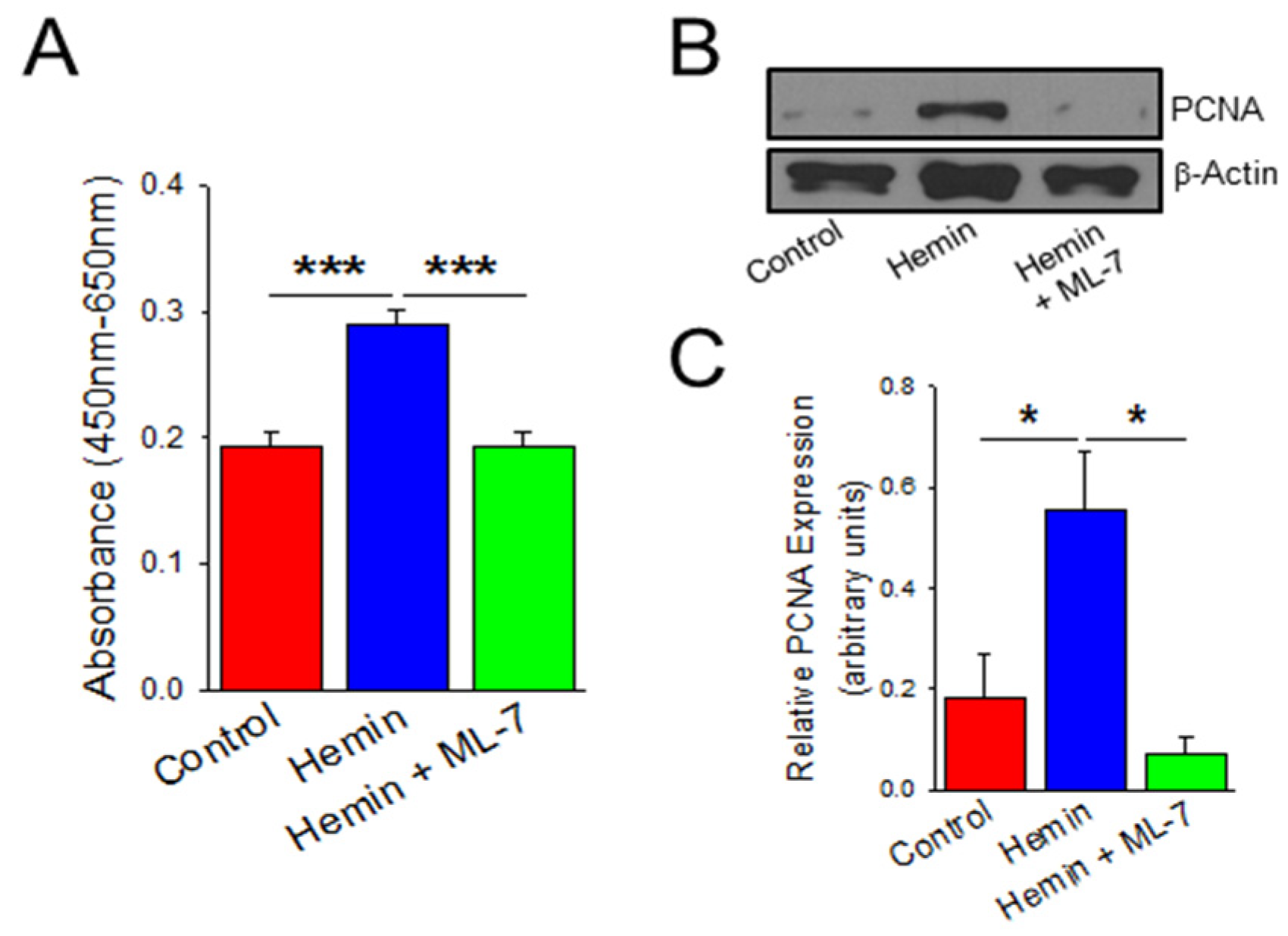
2.3. Hemin Induces Endothelial Cell Viability and Proliferation Which Is Prevented by a Myosin Light Chain Kinase Inhibitor
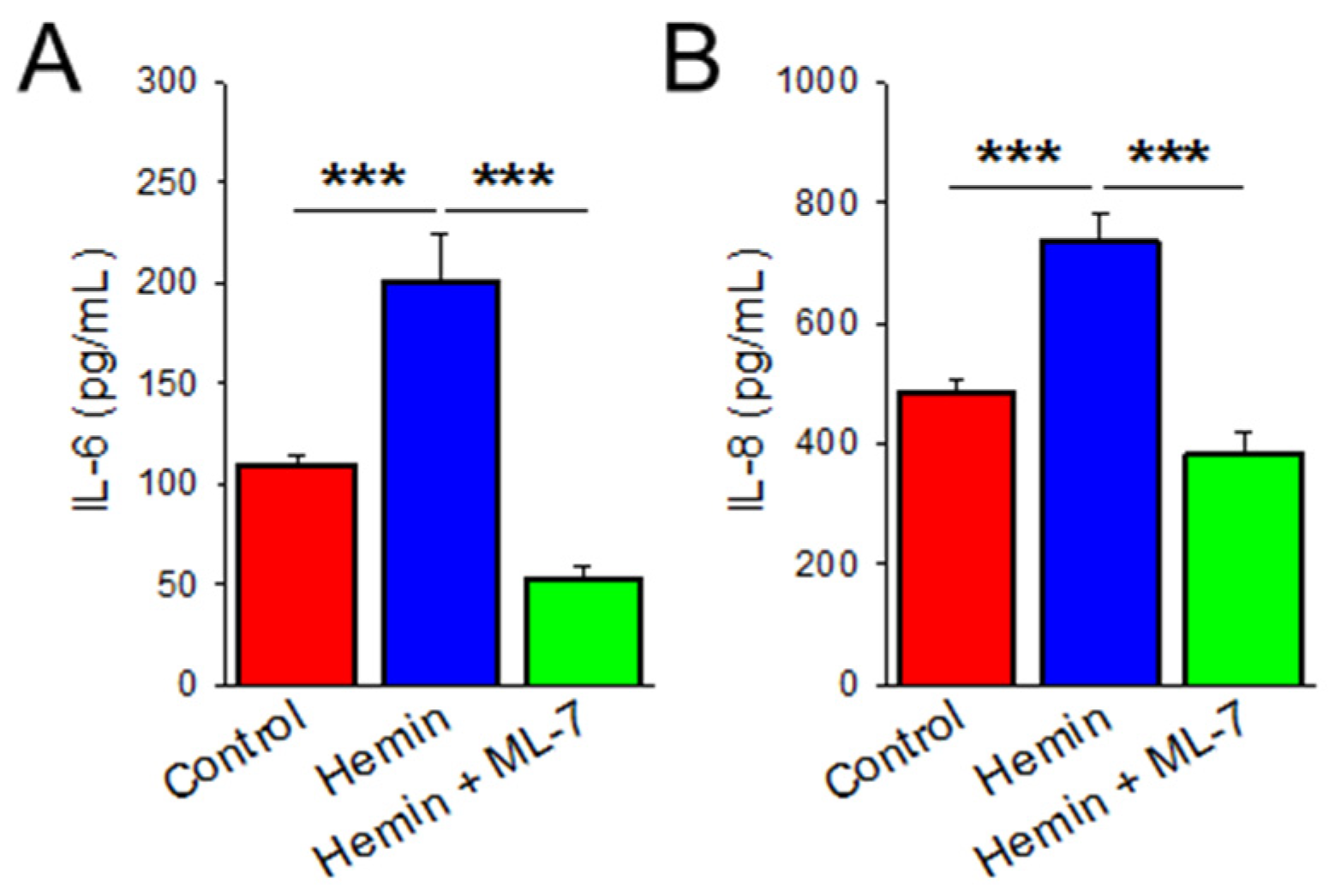
2.4. Hemin Induces Cytokine Release Which Is Prevented by MLCK Inhibition
2.5. Hemin Induces Endothelial Cell Migration and Myosin Light Chain Kinase Inhibitor Negates the Hemin-Induced Hyper Migration
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Proliferation and Viability Assay
4.4. Enzyme Linked Immunosorbent Assay (ELISA)
4.5. Electrical Cell Impedance Sensing (ECIS) Wound Assay
4.6. Scratch Assay
4.7. Immunofluorescence Cell Staining
4.8. Western Blotting
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mehari, A.; Gladwin, M.T.; Tian, X.; Machado, R.F.; Kato, G.J. Mortality in Adults with Sickle Cell Disease and Pulmonary Hypertension. JAMA 2012, 307, 1254–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordeuk, V.R.; Castro, O.L.; Machado, R.F. Pathophysiology and treatment of pulmonary hypertension in sickle cell disease. Blood 2016, 127, 820–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gbotosho, O.T.; Kapetanaki, M.G.; Ghosh, S.; Villanueva, F.S.; Ofori-Acquah, S.F.; Kato, G.J. Heme Induces IL-6 and Cardiac Hypertrophy Genes Transcripts in Sickle Cell Mice. Front. Immunol. 2020, 11, 1910. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef]
- Ingoglia, G.; Sag, C.M.; Rex, N.; De Franceschi, L.; Vinchi, F.; Cimino, J.; Petrillo, S.; Wagner, S.; Kreitmeier, K.; Silengo, L.; et al. Hemopexin counteracts systolic dysfunction induced by heme-driven oxidative stress. Free Radic. Biol. Med. 2017, 108, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Murakami, J.; Shimizu, Y. Hepatic manifestations in hematological disorders. Int. J. Hepatol. 2013, 2013, 484903. [Google Scholar] [CrossRef] [Green Version]
- Petrillo, S.; Chiabrando, D.; Genova, T.; Fiorito, V.; Ingoglia, G.; Vinchi, F.; Mussano, F.; Carossa, S.; Silengo, L.; Altruda, F.; et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ. 2018, 25, 573–588. [Google Scholar] [CrossRef]
- Rother, R.P.; Bell, L.; Hillmen, P.; Gladwin, M.T. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: A novel mechanism of human disease. JAMA 2005, 293, 1653–1662. [Google Scholar] [CrossRef]
- Sawicki, K.T.; Chang, H.C.; Ardehali, H. Role of heme in cardiovascular physiology and disease. J. Am. Heart Assoc. 2015, 4, e001138. [Google Scholar] [CrossRef] [Green Version]
- Tracz, M.J.; Alam, J.; Nath, K.A. Physiology and pathophysiology of heme: Implications for kidney disease. J. Am. Soc. Nephrol. 2007, 18, 414–420. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O.; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Liu, S.C.; Zhai, S.; Palek, J. Detection of hemin release during hemoglobin S denaturation. Blood 1988, 71, 1755–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, G.J.; McGowan, V.; Machado, R.F.; Little, J.A.; Taylor, J.t.; Morris, C.R.; Nichols, J.S.; Wang, X.; Poljakovic, M.; Morris, S.M., Jr.; et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood 2006, 107, 2279–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, L.L.; Champion, H.C.; Campbell-Lee, S.A.; Bivalacqua, T.J.; Manci, E.A.; Diwan, B.A.; Schimel, D.M.; Cochard, A.E.; Wang, X.; Schechter, A.N.; et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood 2007, 109, 3088–3098. [Google Scholar] [CrossRef]
- Klings, E.S.; Machado, R.F.; Barst, R.J.; Morris, C.R.; Mubarak, K.K.; Gordeuk, V.R.; Kato, G.J.; Ataga, K.I.; Gibbs, J.S.; Castro, O.; et al. An official American Thoracic Society clinical practice guideline: Diagnosis, risk stratification, and management of pulmonary hypertension of sickle cell disease. Am. J. Respir. Crit. Care Med. 2014, 189, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Budhiraja, R.; Tuder, R.M.; Hassoun, P.M. Endothelial dysfunction in pulmonary hypertension. Circulation 2004, 109, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Harvey, L.D.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.-J.; Perez, V.d.J. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045893217752912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Montani, D.; Perros, F.; Dorfmüller, P.; Adnot, S.; Eddahibi, S. Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vascul. Pharmacol. 2008, 49, 113–118. [Google Scholar] [CrossRef]
- Dai, Z.; Zhu, M.M.; Peng, Y.; Machireddy, N.; Evans, C.E.; Machado, R.; Zhang, X.; Zhao, Y.Y. Therapeutic Targeting of Vascular Remodeling and Right Heart Failure in Pulmonary Arterial Hypertension with a HIF-2α Inhibitor. Am. J. Respir. Crit. Care Med. 2018, 198, 1423–1434. [Google Scholar] [CrossRef]
- Derada Troletti, C.; Fontijn, R.D.; Gowing, E.; Charabati, M.; van Het Hof, B.; Didouh, I.; van der Pol, S.M.A.; Geerts, D.; Prat, A.; van Horssen, J.; et al. Inflammation-induced endothelial to mesenchymal transition promotes brain endothelial cell dysfunction and occurs during multiple sclerosis pathophysiology. Cell Death Dis. 2019, 10, 45. [Google Scholar] [CrossRef] [Green Version]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Alvandi, Z.; Bischoff, J. Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2357–2369. [Google Scholar] [CrossRef] [PubMed]
- Man, S.; Sanchez Duffhues, G.; Ten Dijke, P.; Baker, D. The therapeutic potential of targeting the endothelial-to-mesenchymal transition. Angiogenesis 2019, 22, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Péchoux, C.; Bogaard, H.J.; Dorfmüller, P.; Remy, S.; Lecerf, F.; Planté, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L256–L275. [Google Scholar] [CrossRef] [PubMed]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef]
- Isobe, S.; Kataoka, M.; Endo, J.; Moriyama, H.; Okazaki, S.; Tsuchihashi, K.; Katsumata, Y.; Yamamoto, T.; Shirakawa, K.; Yoshida, N.; et al. Endothelial-Mesenchymal Transition Drives Expression of CD44 Variant and xCT in Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2019, 61, 367–379. [Google Scholar] [CrossRef]
- Arciniegas, E.; Frid, M.G.; Douglas, I.S.; Stenmark, K.R. Perspectives on endothelial-to-mesenchymal transition: Potential contribution to vascular remodeling in chronic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1–L8. [Google Scholar] [CrossRef] [Green Version]
- Ciszewski, W.M.; Wawro, M.E.; Sacewicz-Hofman, I.; Sobierajska, K. Cytoskeleton Reorganization in EndMT-The Role in Cancer and Fibrotic Diseases. Int. J. Mol. Sci. 2021, 22, 11607. [Google Scholar] [CrossRef]
- Platel, V.; Faure, S.; Corre, I.; Clere, N. Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J. Oncol. 2019, 2019, 8361945. [Google Scholar] [CrossRef] [PubMed]
- Kurakula, K.; Smolders, V.; Tura-Ceide, O.; Jukema, J.W.; Quax, P.H.A.; Goumans, M.J. Endothelial Dysfunction in Pulmonary Hypertension: Cause or Consequence? Biomedicines 2021, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wang, X.; Wan, Y.; Zhou, Q.; Zhu, H.; Wang, Y. Myosin light chain kinase inhibitor ML7 improves vascular endothelial dysfunction via tight junction regulation in a rabbit model of atherosclerosis. Mol. Med. Rep. 2015, 12, 4109–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.G.N.; Verin, A.D.; Schaphorst, K.; Siddiqui, R.; Patterson, C.E.; Csortos, C.; Natarajan, V. Regulation of endothelial cell myosin light chain kinase by Rho, cortactin, and p60 src. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1999, 276, L989–L998. [Google Scholar] [CrossRef]
- Shen, Q.; Rigor, R.R.; Pivetti, C.D.; Wu, M.H.; Yuan, S.Y. Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc. Res. 2010, 87, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Kato, G.J.; Martyr, S.; Blackwelder, W.C.; Nichols, J.S.; Coles, W.A.; Hunter, L.A.; Brennan, M.L.; Hazen, S.L.; Gladwin, M.T. Levels of soluble endothelium-derived adhesion molecules in patients with sickle cell disease are associated with pulmonary hypertension, organ dysfunction, and mortality. Br. J. Haematol. 2005, 130, 943–953. [Google Scholar] [CrossRef] [Green Version]
- Rafikova, O.; Williams, E.R.; McBride, M.L.; Zemskova, M.; Srivastava, A.; Nair, V.; Desai, A.A.; Langlais, P.R.; Zemskov, E.; Simon, M.; et al. Hemolysis-induced Lung Vascular Leakage Contributes to the Development of Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2018, 59, 334–345. [Google Scholar] [CrossRef]
- Ghosh, S.; Adisa, O.A.; Chappa, P.; Tan, F.; Jackson, K.A.; Archer, D.R.; Ofori-Acquah, S.F. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J. Clin. Investig. 2013, 123, 4809–4820. [Google Scholar] [CrossRef] [Green Version]
- Singla, S.; Sysol, J.R.; Dille, B.; Jones, N.; Chen, J.; Machado, R.F. Hemin Causes Lung Microvascular Endothelial Barrier Dysfunction by Necroptotic Cell Death. Am. J. Respir. Cell Mol. Biol. 2017, 57, 307–314. [Google Scholar] [CrossRef]
- Vichinsky, E.P.; Styles, L.A.; Colangelo, L.H.; Wright, E.C.; Castro, O.; Nickerson, B. Acute chest syndrome in sickle cell disease: Clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood 1997, 89, 1787–1792. [Google Scholar] [CrossRef] [Green Version]
- Janciauskiene, S.; Vijayan, V.; Immenschuh, S. TLR4 Signaling by Heme and the Role of Heme-Binding Blood Proteins. Front. Immunol. 2020, 11, 1964. [Google Scholar] [CrossRef] [PubMed]
- Piazza, M.; Damore, G.; Costa, B.; Gioannini, T.L.; Weiss, J.P.; Peri, F. Hemin and a metabolic derivative coprohemin modulate the TLR4 pathway differently through different molecular targets. Innate Immun. 2011, 17, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Tada, Y.; Nishimura, R.; Kawasaki, T.; Sekine, A.; Urushibara, T.; Kato, F.; Kinoshita, T.; Ikari, J.; West, J.; et al. Endothelial-to-mesenchymal transition in lipopolysaccharide-induced acute lung injury drives a progenitor cell-like phenotype. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L1185–L1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Canalli, A.A.; Franco-Penteado, C.F.; Traina, F.; Saad, S.T.; Costa, F.F.; Conran, N. Role for cAMP-protein kinase A signalling in augmented neutrophil adhesion and chemotaxis in sickle cell disease. Eur. J. Haematol. 2007, 79, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Lanaro, C.; Franco-Penteado, C.F.; Albuqueque, D.M.; Saad, S.T.; Conran, N.; Costa, F.F. Altered levels of cytokines and inflammatory mediators in plasma and leukocytes of sickle cell anemia patients and effects of hydroxyurea therapy. J. Leukoc. Biol. 2009, 85, 235–242. [Google Scholar] [CrossRef]
- Belcher, J.D.; Bryant, C.J.; Nguyen, J.; Bowlin, P.R.; Kielbik, M.C.; Bischof, J.C.; Hebbel, R.P.; Vercellotti, G.M. Transgenic sickle mice have vascular inflammation. Blood 2003, 101, 3953–3959. [Google Scholar] [CrossRef] [Green Version]
- Sarray, S.; Saleh, L.R.; Lisa Saldanha, F.; Al-Habboubi, H.H.; Mahdi, N.; Almawi, W.Y. Serum IL-6, IL-10, and TNFalpha levels in pediatric sickle cell disease patients during vasoocclusive crisis and steady state condition. Cytokine 2015, 72, 43–47. [Google Scholar] [CrossRef]
- Garcia, J.G.; Davis, H.W.; Patterson, C.E. Regulation of endothelial cell gap formation and barrier dysfunction: Role of myosin light chain phosphorylation. J. Cell Physiol. 1995, 163, 510–522. [Google Scholar] [CrossRef]
- Belvitch, P.; Adyshev, D.; Elangovan, V.R.; Brown, M.E.; Naureckas, C.; Rizzo, A.N.; Siegler, J.H.; Garcia, J.G.; Dudek, S.M. Proline-rich region of non-muscle myosin light chain kinase modulates kinase activity and endothelial cytoskeletal dynamics. Microvasc. Res. 2014, 95, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Dudek, S.M.; Chiang, E.T.; Camp, S.M.; Guo, Y.; Zhao, J.; Brown, M.E.; Singleton, P.A.; Wang, L.; Desai, A.; Arce, F.T.; et al. Abl tyrosine kinase phosphorylates nonmuscle Myosin light chain kinase to regulate endothelial barrier function. Mol. Biol. Cell 2010, 21, 4042–4056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Liu, Y.; You, J.; Zhang, H.; Zhang, X.; Ye, L. Myosin light-chain kinase contributes to the proliferation and migration of breast cancer cells through cross-talk with activated ERK1/2. Cancer Lett. 2008, 270, 312–327. [Google Scholar] [CrossRef] [PubMed]
- Gerthoffer, W.T. Mechanisms of vascular smooth muscle cell migration. Circ. Res. 2007, 100, 607–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totsukawa, G.; Wu, Y.; Sasaki, Y.; Hartshorne, D.J.; Yamakita, Y.; Yamashiro, S.; Matsumura, F. Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J. Cell Biol. 2004, 164, 427–439. [Google Scholar] [CrossRef] [Green Version]
- Kishi, H.; Mikawa, T.; Seto, M.; Sasaki, Y.; Kanayasu-Toyoda, T.; Yamaguchi, T.; Imamura, M.; Ito, M.; Karaki, H.; Bao, J.; et al. Stable Transfectants of Smooth Muscle Cell Line Lacking the Expression of Myosin Light Chain Kinase and Their Characterization with Respect to the Actomyosin System. J. Biol. Chem. 2000, 275, 1414–1420. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Yang, Y.; Yang, T.; Jiang, Q.; Yang, L.; Tan, H. Inhibition of MLCK attenuates pulmonary artery hypertension. Int. J. Clin. Exp. Pathol. 2016, 9, 7961–7968. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzales, J.; Holbert, K.; Czysz, K.; George, J.; Fernandes, C.; Fraidenburg, D.R. Hemin-Induced Endothelial Dysfunction and Endothelial to Mesenchymal Transition in the Pathogenesis of Pulmonary Hypertension Due to Chronic Hemolysis. Int. J. Mol. Sci. 2022, 23, 4763. https://doi.org/10.3390/ijms23094763
Gonzales J, Holbert K, Czysz K, George J, Fernandes C, Fraidenburg DR. Hemin-Induced Endothelial Dysfunction and Endothelial to Mesenchymal Transition in the Pathogenesis of Pulmonary Hypertension Due to Chronic Hemolysis. International Journal of Molecular Sciences. 2022; 23(9):4763. https://doi.org/10.3390/ijms23094763
Chicago/Turabian StyleGonzales, Janae, Kelsey Holbert, Kamryn Czysz, Joseph George, Caroline Fernandes, and Dustin R. Fraidenburg. 2022. "Hemin-Induced Endothelial Dysfunction and Endothelial to Mesenchymal Transition in the Pathogenesis of Pulmonary Hypertension Due to Chronic Hemolysis" International Journal of Molecular Sciences 23, no. 9: 4763. https://doi.org/10.3390/ijms23094763
APA StyleGonzales, J., Holbert, K., Czysz, K., George, J., Fernandes, C., & Fraidenburg, D. R. (2022). Hemin-Induced Endothelial Dysfunction and Endothelial to Mesenchymal Transition in the Pathogenesis of Pulmonary Hypertension Due to Chronic Hemolysis. International Journal of Molecular Sciences, 23(9), 4763. https://doi.org/10.3390/ijms23094763