
Molecular Alterations of the Endocannabinoid System in Psychiatric Disorders

 ,
,  ,
,  , ,
, , 

Abstract
:1. Introduction
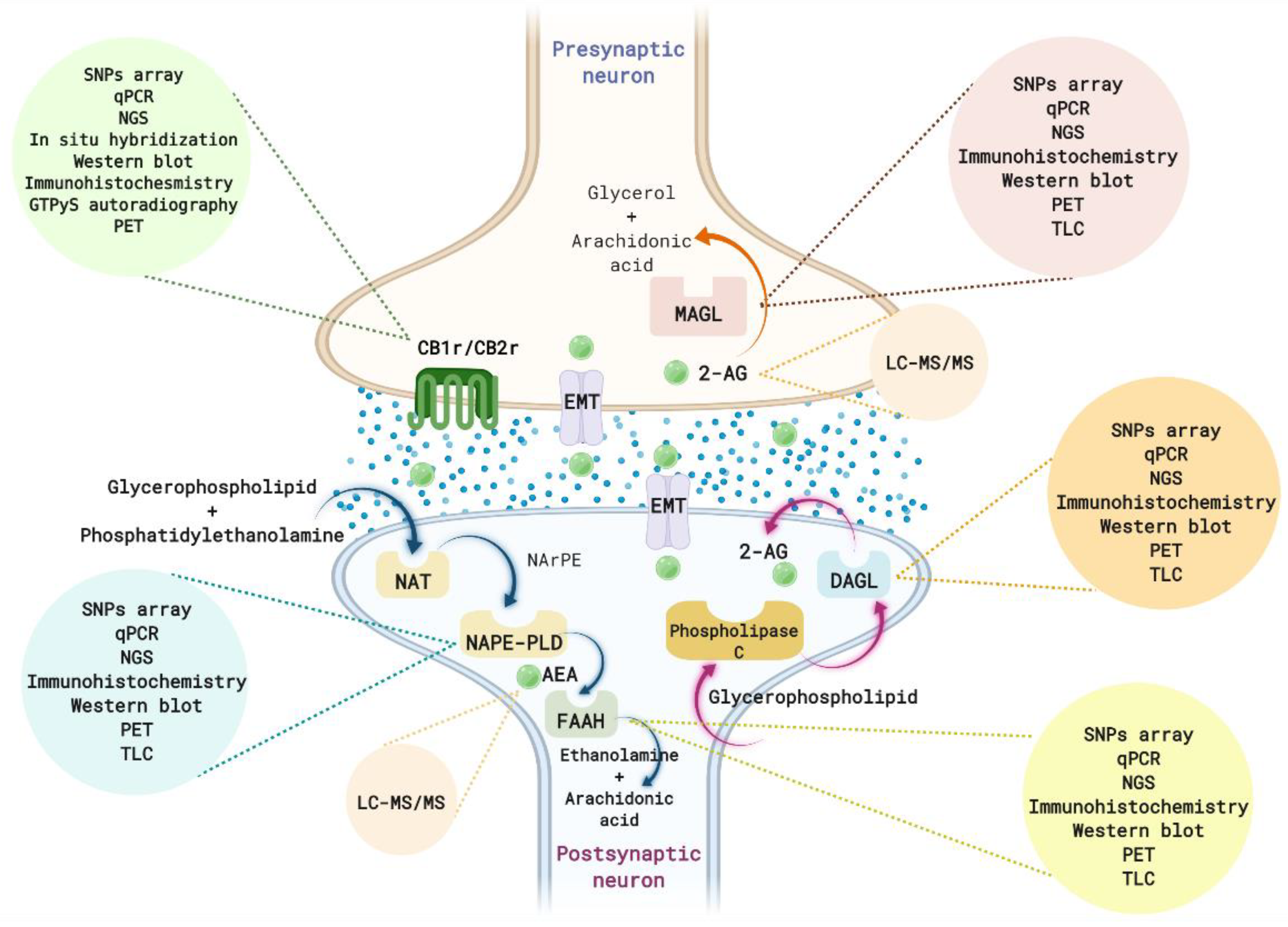
2. A Brief Overview of the ECS
3. Methods to Identify Alterations in the ECS
3.1. Alterations in Endocannabinoid Ligands
3.2. Alterations in the Enzymes of Synthesis and Metabolization
3.2.1. Genomic Alterations
3.2.2. Epigenetic Alterations
3.2.3. Gene Expression Alterations
3.2.4. Protein Level Alterations
3.2.5. Alterations in Protein Activity
3.2.6. Functional Alterations by Neuroimaging Techniques
3.3. Alterations in Cannabinoid Receptors
3.3.1. Genomic Alterations
3.3.2. Epigenetic Alterations
3.3.3. Gene Expression Alterations
3.3.4. Protein Level Alterations
3.3.5. Alterations in Protein Activity
3.3.6. Functional Alterations by Neuroimaging Techniques
4. Anxiety-Related Disorders
4.1. Generalized Anxiety Disorder
4.1.1. Clinical Studies
4.1.2. Animal Studies
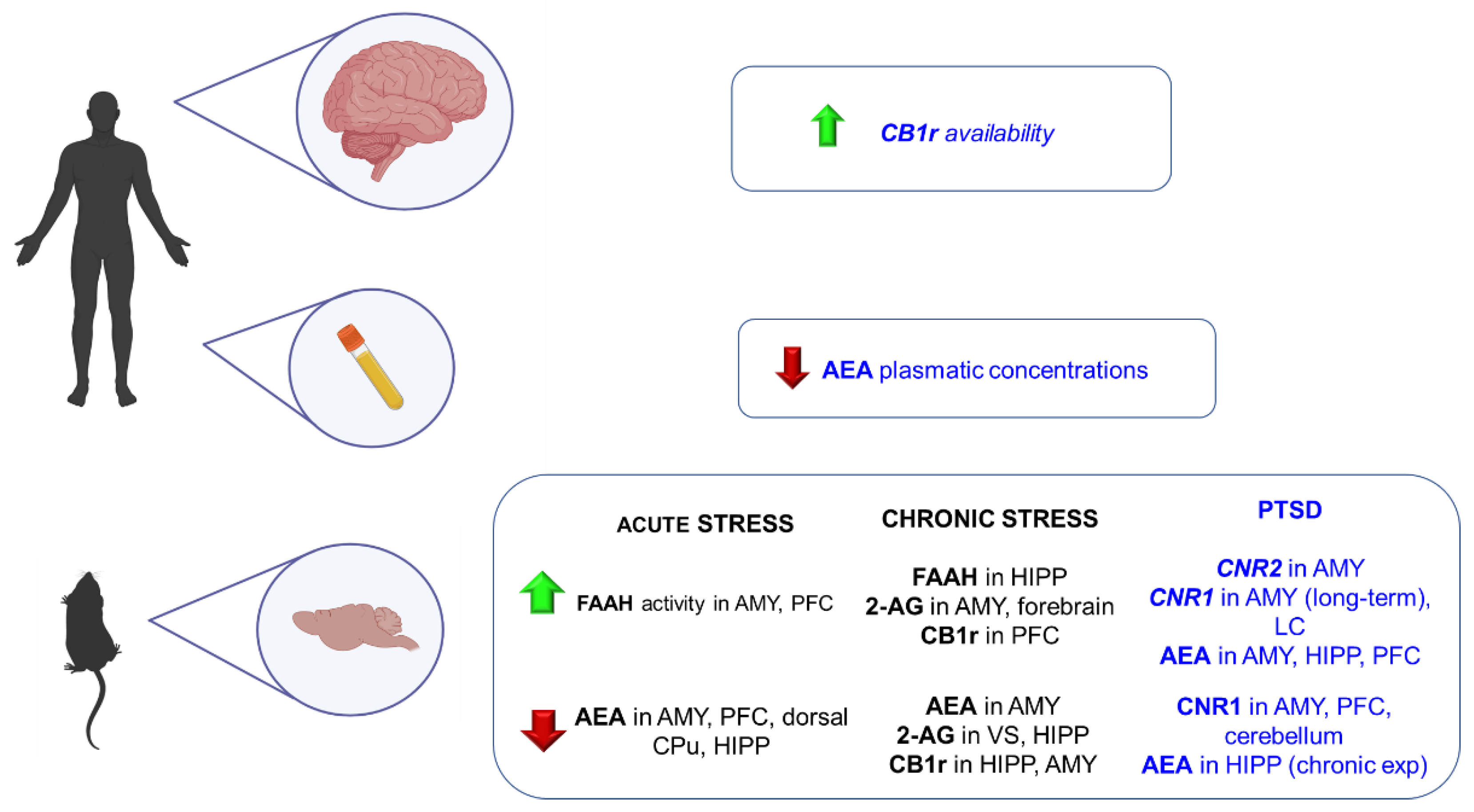
4.2. Post-Traumatic Stress Disorder (PTSD)
4.2.1. Clinical Studies
4.2.2. Animal Studies
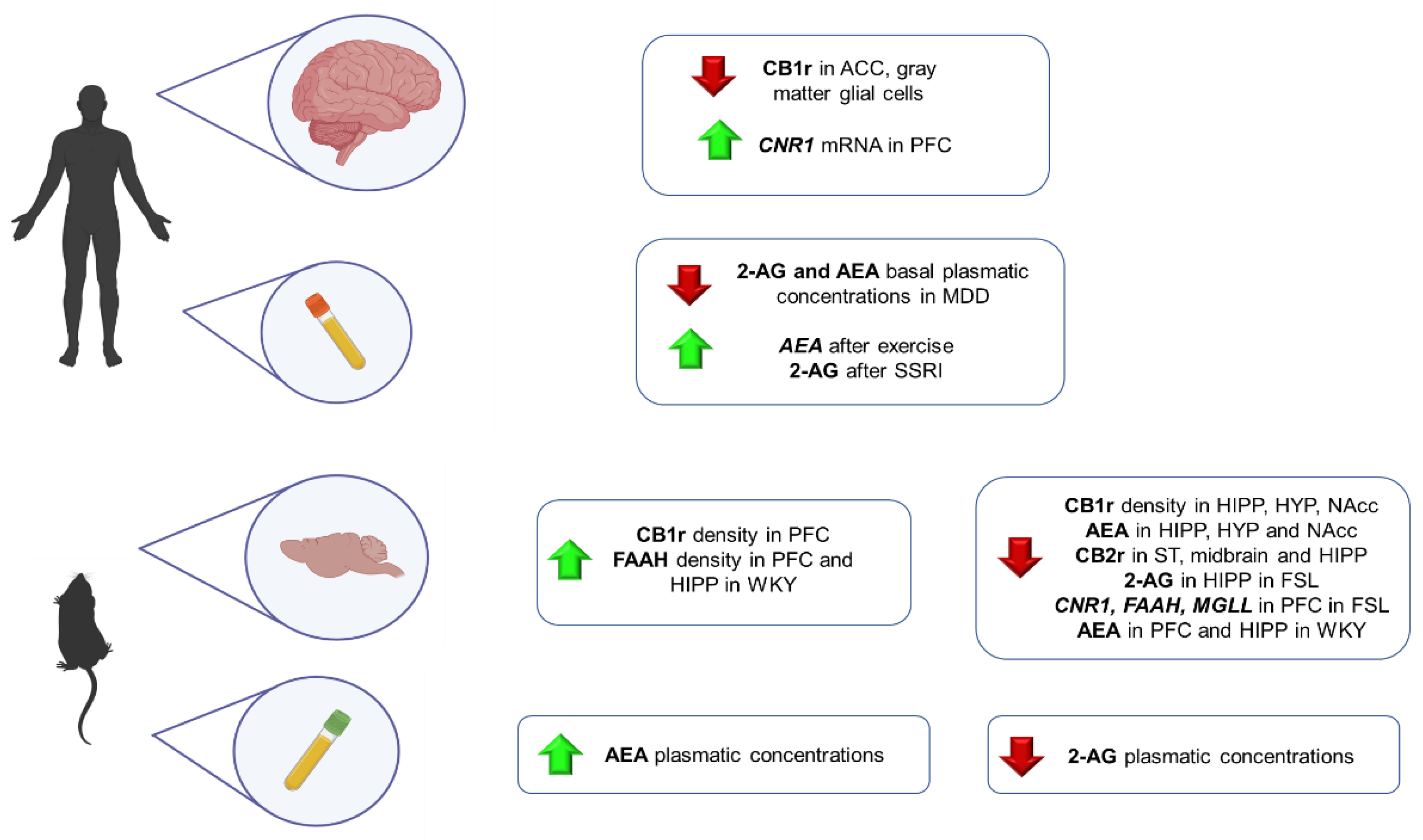
5. Depression
5.1. Clinical Studies
5.2. Animal Studies
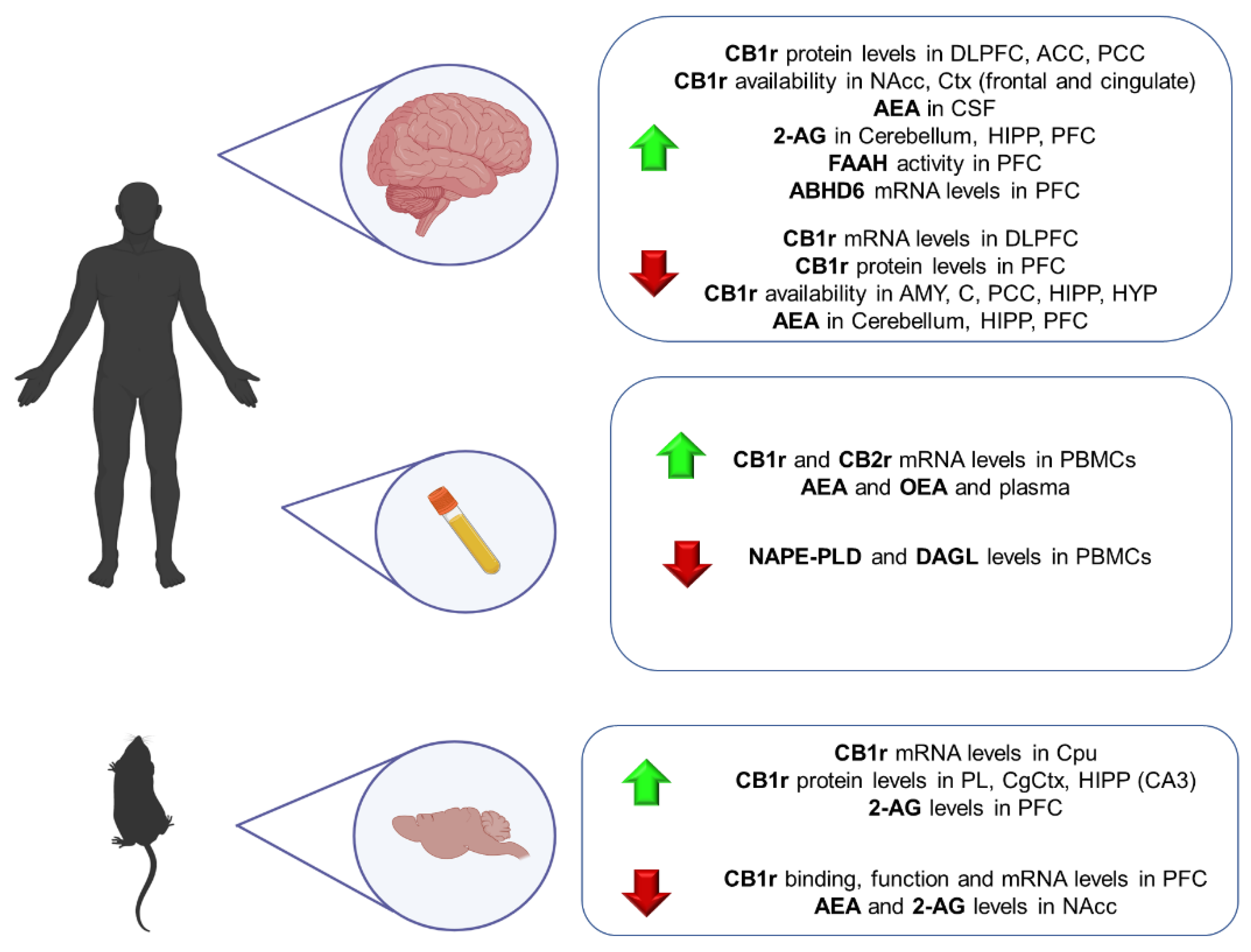
6. Schizophrenia
6.1. Clinical Studies
6.2. Animal Studies
7. Autism Spectrum Disorder (ASD)
7.1. Clinical Studies
7.2. Animal Studies
8. Attention Deficit Hyperactivity Disorder (ADHD)
8.1. Clinical Studies
8.2. Animal Studies
9. Eating Disorders (ED)
9.1. Clinical Studies
9.2. Animal Studies
10. Substance Use Disorders
10.1. Nicotine Use Disorders
10.1.1. Clinical Studies
Neuroimaging Studies
Genetic Studies
10.1.2. Animal Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nicotine Use Disorder | |||
|---|---|---|---|
| Authors | Type of Sample | Type of Evaluation | Outcomes |
| [417] | Humans | Neuroimaging (fMRI) | ↓ reward anticipation activity in the NAcc after THC administration in NUDs |
| [418] | Humans | Neuroimaging (PET) | ↓ CB1r in all brain areas in NUDs |
| [422] | Animals | CB1r KO vs WT mice | Nicotine rewarding effects in WT mice but not in CB1r KO mice No significant differences in the severity of nicotine withdrawal between WT and CB1r KO mice |
| [423] | Animals | CB1r KO vs FAAH KO vs WT mice | CB1r KO mice blocked nicotine reward FAAH KO mice had an enhanced expression of nicotine reward Nicotine withdrawal was unaffected in CB1r KO mice, FAAH KO mice displayed increased nicotine withdrawal |
| [424] | Animals | MAGL KO vs WT mice | MAGL KO mice failed to develop a nicotine CPP compared to WT mice |
| [425] | Animals | CB2r KO vs WT mice | CB2r KO mice did not show nicotine-induced PCC and hardly self-administered nicotine compared to WT mice Somatic signs of nicotine withdrawal ↑ in WT but were absent in CB2r KO mice |
| [426] | Animals | CB2r KO vs WT mice | Nicotine-induced CPP was absent in CB2r KO WT, and CB2r KO nicotine-dependent mice showed a similar response during nicotine withdrawal |
| [427] | Animals | DAT-CNR2 KO vs WT mice | Compared to WT, DAT-CNR2 KO mice showed the absence of nicotine-induced CPP. |
10.2. Alcohol Use Disorders (AUD)
10.2.1. Clinical Studies
Neuroimaging Studies
Genetic Studies
Post-Mortem Studies
10.2.2. Animal studies
| Alcohol Use Disorders | |||
|---|---|---|---|
| References | Type of Sample | Type of Evaluation | Outcomes |
| [428] | Humans | Neuroimaging (PET) | AUD showed ↑ CB1r binding in a circuit that included the AMY, HIPP, PT, insula, anterior and posterior cingulate cortices, and OFC. |
| [433] | Humans | Neuroimaging (PET) | AD subjects showed ↓ CB1r binding during early abstinence (3–7 days), which remained reduced during protracted abstinence (2–4 weeks). |
| [429] | Humans | Neuroimaging (PET) | Acute alcohol consumption resulted in a ↑ CB1r availability Chronic alcohol drinking resulted in a ↓ CB1r availability that remained unaltered after abstinence (1 month). |
| [437] | Humans | Post-mortem | Cloninger type 1 alcohol dependent subjects showed ↑ DHEA levels in the AMY and a negative correlation between AEA concentrations and mGlu1/5 receptor density in the HIPP compared to Cloninger type 2 alcohol-dependent subjects and controls. |
| [438] | Humans | Post-mortem | CB1r protein expression in the PFC of the suicidal alcohol-dependent group Alcohol-dependent subjects, regardless of the cause of death, ↓ MAGL activity, ↓ ERK, and ↓ CREB levels. |
| [154] | Humans | Post-mortem | Alcohol-dependent subjects presented hyper-functional CB1r in the caudate nucleus Non-suicidal alcohol-dependent subjects showed hypofunctional CB1r in the cerebellum. |
| [439] | Animals | CB1R KO vs WT mice | CB1r KO mice exhibited voluntary alcohol consumption and completely lacked alcohol-induced DA release in the NAcc compared to WT mice. |
| [440] | Animals | CB1R KO vs WT mice | CB1r KO mice displayed ↓ OH-induced CPP compared to WT mice. This ↓ OH-induced CPP exhibited by CB1r KO mice was correlated with an increase in striatum D2/D3 receptors. |
| [441] | Animals | CB1r KO vs WT mice | CB1r KO mice ↓OH consumption and preference, compared to WT mice CB1r KO mice were more sensitive to the acute alcohol effects than WT mice. The severity of alcohol withdrawal was also increased in CB1r KO mice |
| [139,436] | Animals | C57/BJ6 male mice | Mice with high-alcohol preference had a lower gene expression of CNR2 at the ventral midbrain |
| [442] | Animals | CB2r KO vs WT mice | CB2r KO mice presented ↑ a response to alcohol effects, OH-induced CPP, voluntary OH intake and preference, acquisition of alcohol self-administration, and motivation to drink alcohol compared to WT mice. |
| [443] | Animals | FAAH gene KO vs WT mice | FAAH KO mice showed a ↑ preference for alcohol and consumed more alcohol than WT mice There were no significant differences between FAAH KO and WT mice in the severity of alcohol induced acute withdrawal, CPP, or sensitivity to the hypnotic effect of alcohol. FAAH KO mice showed a shorter duration and a faster recovery from intoxicating effects induced by alcohol. |
| [444] | Animals | FAAH gene KO vs WT mice | Female FAAH KO mice had an ↑ alcohol intake and preference, were less sensitive to the effects of acute alcohol, and no CB1r levels and function down-regulation after voluntary alcohol consumption, compared to male FAAH KO, and male and female WT mice. |
| [445] | Animals | Male Wistar rats exposed to continuous OH access vs intermittent OH access | Alcohol withdrawal was associated with significant ↓ mRNA expression FAAH, MAGL, CB1r, CB2r, and GPR55r in the AMY. ↓ MAGL, CB1r, CB2r, and GPR55r were more pronounced following intermittent alcohol exposure. |
| [446] | Animals | Male Wistar rats exposed to intermittent OH access | Alcohol-exposed rats expressed ↑ mRNA levels of NAPE-PLD and DGL in the mPFC and the AMY, respectively, and ↓mRNA levels of CB1r, CB2r, and PPARα in the striatum. |
10.3. Cannabis Use Disorders
10.3.1. Clinical Studies
Neuroimaging Studies
Genetic Studies
Post-Mortem Studies
10.3.2. Animal Studies
| Cannabis Use Disorders | |||
|---|---|---|---|
| References | Type of Sample | Type of Evaluation | Outcomes |
| [447] | Humans | Neuroimaging (PET) | CB1r downregulation in years of THC smokers After 4 weeks of abstinence, CB1r density returned to normal levels. |
| [293] | Humans | Neuroimaging (PET) | THC users showed an ↓ in CB1r availability, significant in the temporal lobe, the anterior and PCC, and in the NAcc. |
| [449] | Humans | Neuroimaging (HRRT) | THC-dependent subjects showed ↓ CB1r availability Differences in CB1r availability were no longer evident after 2 days of abstinence, and no significant group differences in CB1r availability after 28 days of abstinence. |
| [455] | Human | Post-mortem | In chronic cannabis users, CB1r binding was ↓ in the HIPP, caudate nucleus, PT, and NAcc. |
| [456] | Human | Post-mortem | In THC-dependent subjects, regions with higher MAGL expression are more vulnerable to cortical thinning. |
| [457] | Animal | Rats exposed to THC | CB1r mRNA levels were increased in the cerebellum and HIPP and reduced in the striatum until day 14. CB1r expression in all three brain areas returned to control levels by day 21 of THC treatment once behavioral tolerance had been developed. |
| [458] | Animals | THC-tolerant rats | THC-tolerant rats exhibited an ↓in CB1r and [35S]GTPγS binding in most brain areas, except the limbic forebrain. AEA ↑ in the limbic forebrain, and AEA and 2-AG ↓in the striatum. |
| [459] | Animals | CB1r KO vs WT mice | Long-term depression of VTA GABA neurons was absent in CB1r KO but preserved in WT mice. THC produced a long-term depression in the WT but not in CB1r KO mice. |
10.4. Cocaine and Other Stimulant Use Disorders
10.4.1. Clinical Studies
Genetic Studies
Post-Mortem Studies
10.4.2. Animal Studies
| Cocaine Use Disorders | |||
|---|---|---|---|
| References | Type of Sample | Type of Evaluation | Outcomes |
| [462] | Human | Post-mortem | ↓ CB1r and GRK2/3/5 in the PFC in CoUDs |
| [463] | Animals | CB1r KO vs WT mice | 25% of the CB1r KO mice compared to the 75% of their WT littermates acquired a reliable operant responding to self-administration of cocaine, and the number of sessions required to attain this behavior was ↑ in CB1r KO mice. |
| [464] | Animals | Glu-CB1r vs GABA-CB1r KO vs WT | CB1r expression in forebrain GABAergic neurons-controlled sensitivity to cocaine, while CB1r expression in cortical glutamatergic neurons controlled the associative learning processes. |
| [465] | Animals | D1-CNR1 KO vs A2a-CNR1 KO vs WT mice | D1-CNR1 KO mice did not display hyperlocomotion in response to acute cocaine dosing. D1-CNR1 and A2a-CNR1 KO mice exhibited blunted locomotor activity across repeated cocaine doses A2a-CNR1 KO mice did not express a preference for cocaine paired environments in a two-choice place preference task. |
| [466] | Animals | Male Long Evans rats | Systemic cocaine increased premature responding, a measure of impulsivity. |
| [467] | Animals | Transgenic mice overexpressing the CB2r vs WT littermates | Overexpression of the CB2r significantly ↓ motor response to acute administration of cocaine cocaine-induced motor sensitization, CPP, and cocaine self-administration. |
| [427] | Animals | DAT-CNR2 KO vs WT mice | DAT-CNR2 KO mice enhanced psychostimulant-induced hyperactivity but an absence of psychostimulant-induced sensitization compared to WT mice. |
| [468] | Animals | Male Wistar rats | Following cocaine self-administration, a ↑ CB1r expression in the VTA and a ↓ CB1r expression in the PFC, dorsal striatum, and AMY. Cocaine abstinence, ↑CB1r expression in the SN and the AMY, and a ↓ CB2r expression in the PFC, NAcc, and medial globus pallidus. |
10.5. Opiate Use Disorders
10.5.1. Clinical Studies
Plasma Studies
Genetic Studies
10.5.2. Animal Studies
| Opiate Use Disorders | |||
|---|---|---|---|
| Authors | Type of Sample | Type of Evaluation | Outcomes |
| [469] | Humans | Peripheral Plasma | In morphine abusers, CB2r were upregulated in the PBMCs. |
| [471] | Animals | CB1r KO vs WT mice | CB1r KO mice, the reinforcing properties of morphine and the severity of the morphine withdrawal syndrome were strongly ↓. |
| [472] | Animals | CB1r KO vs WT mice | The sensitization to the locomotor response induced by chronic morphine treatment was abolished in CB1r KO mice. Morphine induced a CPP in WT mice but failed to produce any response in CB1r KO mice |
| [469] | Animals | Sprague-Dawley rats under morphine exposure vs control rats | Rats under morphine exposure exhibited CB2r upregulation in the spleen and PBMCs |
| [473] | Animals | CBr2 KO vs KO mice | In WT mice, LY2828360 blocked morphine-induced reward in a CPP paradigm, whereas morphine-induced reward was absent in CB2r KO mice. LY2828360 partially attenuated naloxone-precipitated opioid withdrawal in morphine-dependent WT mice, whereas this withdrawal was markedly exacerbated in CB2r KO mice |
| [474] | Animals | Maternally deprived adolescent rats | Maternally deprived adolescent rats exhibited ↑ AEA in the NAcc, the Cpu nucleus, and the mesencephalon Maternally deprived adult rats, showed ↑ AEA and 2-AG in the NAcc, and ↑ 2-AG in the CPu nucleus, |
| Gene | SNP | Disorder | Authors |
|---|---|---|---|
| CNR1 | rs110402 rs7209436C rs242924G rs7766029 rs1049353 rs2180619 rs806366 rs806367 rs806368 rs806369 rs806370 rs806371 rs806379 rs806380 rs2023239 rs6454674 rs1049353 rs12720071 rs1535255 | Anxiety Anxiety Anxiety Anxiety, Schizophrenia PTSD, Depression, Schizophrenia, ADHD, ED PTSD Depression, Schizophrenia Depression Depression, ADHD, ED, OUD, AUD, CaUDs, CoUDs ED Depression Depression NUD, AUD CaUDs Depression, Schizophrenia, ADHD, NUD, OUD, AUD, CaUDs Depression, AUD, CoUDs Schizophrenia, ADHD, AUD Schizophrenia ADHD, AUD | [172,174] [174] [174] [177,273,275] [193,195,196,230,231,233,269,271,271,276,375,376,396,397,430,434] [134] [234,271,396] [234] [234,269,270,375,396,430,434,451,460,470] [396] [234] [157,230] [269,376,419,435] [450,451] [133,232,274,276,375,419,432,433,435,452,453] [234,430,434,460] [193,195,196,230,231,233,269,272,277,375,376,396,397,430,434] [273,275] [277,279,419,435] |
| CNR2 | rs2501432 | Depression, Schizophrenia | [139,233,299,300] |
| rs12744386 | Schizophrenia | [135,299] | |
| rs35761398 | Schizophrenia | [135] | |
| Q63R | AUD | [299,436] | |
| rs2229579 | CaUds | [454] | |
| FAAH | rs324420 | PTSD, Depression, Schizophrenia, CaUDs, CoUDs | [95,172,173,174,175,181,196,310,434,453,461] |
| rs2295633 | ADHD | [377] | |
| 385 A/A genotype | ED | [407] |
11. Concluding Remarks
12. Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Cannabis | Cannabis sativa L. |
| 1-AG | 1-arachidonoylglycerol |
| 2-AG | 2-arachidonoylglycerol |
| 2-MAGs | 2-monoacylglycerols |
| 5HT2Ar | Serotoninergic 2A receptor |
| ABA | Activity-based anorexia |
| ABHD12 | α/β-hydrolase domain 12 |
| ABHD6 | α/β-hydrolase domain 6 |
| ACC | Anterior cingulate cortex |
| AD | Anxiety disorders |
| ADHD | Attention deficit hyperactivity disorder |
| AEA | Anandamide |
| AMY | Amygdala |
| AN | Anorexia Nervosa |
| ASD | Autism spectrum disorder |
| AUD | Alcohol use disorders |
| BED | Binge eating disorder |
| BMI | Body mass index |
| BN | Bulimia Nervosa |
| CaUD | Cannabis Use Disorder |
| CB1r | Cannabinoid receptor 1 |
| CB2r | Cannabinoid receptor 2 |
| CBD | Cannabidiol |
| CNR1 | Gene encoding cannabinoid receptor 1 |
| CNR2 | Gene encoding cannabinoid receptor 2 |
| CNR2A | Gene encoding cannabinoid receptor 2 isoform A |
| CNR2B | Gene encoding cannabinoid receptor 2 isoform B |
| CNS | Central Nervous System |
| CPP | Conditioned Place Preference |
| CoUD | Cocaine use disorder |
| CpG | cytosine-guanine dinucleotide |
| CPP | conditioned place preference |
| CPu | Caudate putamen |
| CSF | Cerebrospinal fluid |
| d8-2AG | Deuterium-labeled 2AG |
| d8-AA | Deuterium-labeled AA |
| D1 | Dopamine receptor 1 |
| DA | Dopamine |
| DAGL | Diacylglycerol lipase |
| DAT | Dopamine transporter |
| DD | Depressive disorders |
| DLPFC | Dorsolateral frontal cortex |
| DNA | Desoxyribonucleic acid |
| DSM-V | Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition |
| EA | Eating disorders |
| eCBs | Endocannabinoids |
| ECS | Endocannabinoid system |
| EMT | Endocannabinoid Membrane Transporter |
| EOPF | Octylphosphonofluoridate |
| FAAH | fatty acid amide hydrolase |
| FD | Functional dyspepsia |
| GABA | γ-aminobutyric acid |
| GAD | Generalized anxiety disorder |
| GPCRs | Gq protein-coupled receptors |
| GPR55r | G protein coupled receptor 55 |
| GTPγS | GTPgammaS o Guanosine 5′-O-(γ-thio) triphosphate |
| GWAS | Genome-wide association studies |
| HIPP | Hippocampus |
| HPLC-MS/MS | High-performance liquid chromatography-tandem mass spectrometry |
| HYP | Hypothalamus |
| LC | Locus coeruleus |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
| MAGs | Monoacylglycerols |
| MAGL | Monoacylglycerol lipase |
| MGLL | Gene encoding monoacylglycerol lipase |
| mRNA | Ribonucleic acid messenger |
| MSDB | Medial septum-diagonal band of Broca area |
| NAcc | Nucleus accumbens |
| NAEs | N-acyl-ethanolamines |
| NAPE-PLD | N-acylphosphatidylethanolamine specific phospholipase D |
| NarPE | N-arachidonoyl phosphatidylethanolamine |
| NGS | Next-generation or massively parallel sequencing |
| NUD | Nicotine use disorder |
| OB | Obesity |
| OEA | Oleoyl-ethanolamide |
| OFC | Orbitofrontal cortex |
| ON | Olfactory neuroepithelium |
| OUD | Opioid Use Disorder |
| PPARα | Proliferator-activated receptor-α |
| PBMCs | Peripheral blood mononuclear cells |
| PCC | Posterior cingulate cortex |
| PCR | Polymerase chain reaction |
| PEA | Palmitoyl-ethanolamide |
| PET | Positron emission tomography |
| PFC | Prefrontal cortex |
| PLB | Placebo |
| PPI | Prepulse inhibition |
| PT | Putamen |
| PTMs | Post-translational modifications |
| PTSD | Post-traumatic stress disorder |
| Q | Glutamine |
| q-PCR | Quantitative Real-Time Polymerase chain reaction |
| R | Arginine |
| SCUD | Synthetic cannabinoid use disorder |
| SHR | Spontaneously hypertensive rats |
| SN | Substantia nigra |
| SNPs | Single nucleotide polymorphisms |
| STG | Superior temporal gyrus |
| STRs | Short sequence repeats |
| SUD | Substance use disorders |
| THC | Δ9-tetrahydrocannabinol |
| TLC | Thin-layer chromatography |
| UFLC-MS/MS | Ultra-fast liquid chromatography coupled with tandem mass spectrometry |
| UPLC-MS/MS | Ultra-performance liquid chromatography coupled to mass spectrometry |
| UPLC-TOF/MS | Ultra-high-performance liquid chromatography-quadrupole time-of-flight mass spectrometry |
| VS | Ventral striatum |
| WES | Whole-exome sequencing |
| WGS | Whole-genome sequencing |
| WKY | Wistar Kyoto rats |
References
- ElSohly, M.A.; Radwan, M.M.; Gul, W.; Chandra, S.; Galal, A. Phytochemistry of Cannabis sativa L. Prog. Chem. Org. Nat. Prod. 2017, 103, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Savage, S.R.; Romero-Sandoval, A.; Schatman, M.; Wallace, M.; Fanciullo, G.; McCarberg, B.; Ware, M. Cannabis in Pain Treatment: Clinical and Research Considerations. J. Pain 2016, 17, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Abdi, S. Editorial: Cannabis and cannabinoids for pain: A long way to go (or not): Time will tell. Curr. Opin. Anaesthesiol. 2020, 33, 823–824. [Google Scholar] [CrossRef]
- Andrade, C. Cannabis and Neuropsychiatry, 1: Benefits and Risks. J. Clin. Psychiatry 2016, 77, e551–e554. [Google Scholar] [CrossRef] [Green Version]
- Cameron, C.; Watson, D.; Robinson, J. Use of a synthetic cannabinoid in a correctional population for posttraumatic stress disorder-related insomnia and nightmares, chronic pain, harm reduction, and other indications: A retrospective evaluation. J. Clin. Psychopharmacol. 2014, 34, 559–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, D.G.; Fetcho, R.N.; Jing, D.; Li, A.; Glatt, C.E.; Drysdale, A.T.; Cohen, A.O.; Dellarco, D.V.; Yang, R.R.; Dale, A.M.; et al. Individual differences in frontolimbic circuitry and anxiety emerge with adolescent changes in endocannabinoid signaling across species. Proc. Natl. Acad. Sci. USA 2016, 113, 4500–4505. [Google Scholar] [CrossRef] [Green Version]
- Bruijnzeel, A.W.; Knight, P.; Panunzio, S.; Xue, S.; Bruner, M.M.; Wall, S.C.; Pompilus, M.; Febo, M.; Setlow, B. Effects in rats of adolescent exposure to cannabis smoke or THC on emotional behavior and cognitive function in adulthood. Psychopharmacology 2019, 236, 2773–2784. [Google Scholar] [CrossRef] [PubMed]
- APA: Textbook of Substance Abuse Treatment; DSM-5; The American Psychiatric Association: Arlington, VA, USA, 2014.
- Dragt, S.; Nieman, D.H.; Schultze-Lutter, F.; van der Meer, F.; Becker, H.; de Haan, L.; Dingemans, P.M.; Birchwood, M.; Patterson, P.; Salokangas, R.K.R.; et al. Cannabis use and age at onset of symptoms in subjects at clinical high risk for psychosis. Acta Psychiatr. Scand. 2012, 125, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Fried, P.; Watkinson, B.; Gray, R. Neurocognitive consequences of marihuana—a comparison with pre-drug performance. Neurotoxicol. Teratol. 2005, 27, 231–239. [Google Scholar] [CrossRef]
- Manzanares, J.; Cabañero, D.; Puente, N.; García-Gutiérrez, M.S.; Grandes, P.; Maldonado, R. Role of the endocannabinoid system in drug addiction. Biochem. Pharmacol. 2018, 157, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Marsicano, G.; Lutz, B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur. J. Neurosci. 1999, 11, 4213–4225. [Google Scholar] [CrossRef]
- Terzian, A.L.; Drago, F.; Wotjak, C.T.; Micale, V. The Dopamine and Cannabinoid Interaction in the Modulation of Emotions and Cognition: Assessing the Role of Cannabinoid CB1 Receptor in Neurons Expressing Dopamine D1 Receptors. Front. Behav. Neurosci. 2011, 5, 49. [Google Scholar] [CrossRef] [Green Version]
- García-Gutiérrez, M.S.; García-Bueno, B.; Zoppi, S.; Leza, J.C.; Manzanares, J. Chronic blockade of cannabinoid CB2 receptors induces anxiolytic-like actions associated with alterations in GABA(A) receptors. Br. J. Pharmacol. 2012, 165, 951–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Gutiérrez, M.S.; Manzanares, J. Overexpression of CB2 cannabinoid receptors decreased vulnerability to anxiety and impaired anxiolytic action of alprazolam in mice. J. Psychopharmacol. 2011, 25, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Gutiérrez, M.; Pérez-Ortiz, J.; Gutiérrez-Adán, A.; Manzanares, J. Depression-resistant endophenotype in mice overexpressing cannabinoid CB2 receptors. J. Cereb. Blood Flow Metab. 2010, 160, 1773–1784. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Doods, H.; Treede, R.-D.; Ceci, A. Depression-like behaviour in rats with mononeuropathy is reduced by the CB2-selective agonist GW405833. Pain 2009, 143, 206–212. [Google Scholar] [CrossRef]
- Bedse, G.; Bluett, R.J.; Patrick, T.A.; Romness, N.K.; Gaulden, A.D.; Kingsley, P.J.; Plath, N.; Marnett, L.J.; Patel, S. Therapeutic endocannabinoid augmentation for mood and anxiety disorders: Comparative profiling of FAAH, MAGL and dual inhibitors. Transl. Psychiatry 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Mulder, J.; Aguado, T.; Keimpema, E.; Barabás, K.; Rosado, C.J.B.; Nguyen, L.; Monory, K.; Marsicano, G.; Di Marzo, V.; Hurd, Y.L.; et al. Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proc. Natl. Acad. Sci. USA 2008, 105, 8760–8765. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez de Fonseca, F.; Ramos, J.A.; Bonnin, A.; Fernández-Ruiz, J.J. Presence of cannabinoid binding sites in the brain from early postnatal ages. Neuroreport 1993, 4, 135–138. [Google Scholar] [CrossRef]
- Berrendero, F.; Sepe, N.; Di Marzo, V. Analysis of cannabinoid receptor binding and mRNA expression and endogenous cannabinoid contents in the developing rat brain during late gestation and early postnatal period. Synapse 1999, 33, 181–191. [Google Scholar] [CrossRef]
- Glass, M.; Faull, R.; Dragunow, M. Cannabinoid receptors in the human brain: A detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 1997, 77, 299–318. [Google Scholar] [CrossRef]
- Wang, X.; Dow-Edwards, D.; Keller, E.; Hurd, Y. Preferential limbic expression of the cannabinoid receptor mRNA in the human fetal brain. Neuroscience 2003, 118, 681–694. [Google Scholar] [CrossRef]
- Bluett, R.J.; Gamble-George, J.C.; Hermanson, D.J.; Hartley, N.D.; Marnett, L.J.; Patel, S. Central anandamide deficiency predicts stress-induced anxiety: Behavioral reversal through endocannabinoid augmentation. Transl. Psychiatry 2014, 4, e408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.R.; Stanley, C.M.; Foss, T.; Boles, R.G.; McKernan, K. Rare genetic variants in the endocannabinoid system genes CNR1 and DAGLA are associated with neurological phenotypes in humans. PLoS ONE 2017, 12, e0187926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller-Vahl, K.R.; Emrich, H.M. Cannabis and schizophrenia: Towards a cannabinoid hypothesis of schizophrenia. Expert Rev. Neurother. 2008, 8, 1037–1048. [Google Scholar] [CrossRef]
- Mechoulam, R.; Parker, L.A. The Endocannabinoid System and the Brain. Annu. Rev. Psychol. 2013, 64, 21–47. [Google Scholar] [CrossRef] [Green Version]
- Watkins, B.A.; Kim, J. The endocannabinoid system: Directing eating behavior and macronutrient metabolism. Front. Psychol. 2014, 5, 1506. [Google Scholar] [CrossRef]
- Carbone, E.; Manduca, A.; Cacchione, C.; Vicari, S.; Trezza, V. Healing autism spectrum disorder with cannabinoids: A neuroinflammatory story. Neurosci. Biobehav. Rev. 2021, 121, 128–143. [Google Scholar] [CrossRef]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef] [Green Version]
- Katona, I.; Freund, T.F. Multiple Functions of Endocannabinoid Signaling in the Brain. Annu. Rev. Neurosci. 2012, 35, 529–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackie, K. Distribution of Cannabinoid Receptors in the Central and Peripheral Nervous System. Handb. Exp. Pharmacol. 2005, 168, 299–325. [Google Scholar] [CrossRef]
- Atkinson, D.L.; Abbott, J.K. Cannabinoids and the Brain: The Effects of Endogenous and Exogenous Cannabinoids on Brain Systems and Function. In The Complex Connection between Cannabis and Schizophrenia; Academic Press: Cambridge, MA, USA, 2018; pp. 37–74. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Alger, B.E.; Kim, J. Supply and demand for endocannabinoids. Trends Neurosci. 2011, 34, 304–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [Green Version]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-Mediated Control of Synaptic Transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef]
- Okamoto, Y.; Morishita, J.; Tsuboi, K.; Tonai, T.; Ueda, N. Molecular Characterization of a Phospholipase D Generating Anandamide and Its Congeners. J. Biol. Chem. 2004, 279, 5298–5305. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, V.; Stella, N.; Zimmer, A. Endocannabinoid signalling and the deteriorating brain. Nat. Rev. Neurosci. 2014, 16, 30–42. [Google Scholar] [CrossRef] [Green Version]
- Ueda, N. Endocannabinoid hydrolases. Prostaglandins Other Lipid Mediat. 2002, 68–69, 521–534. [Google Scholar] [CrossRef]
- Egertová, M.; Cravatt, B.; Elphick, M. Comparative analysis of fatty acid amide hydrolase and cb1 cannabinoid receptor expression in the mouse brain: Evidence of a widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience 2003, 119, 481–496. [Google Scholar] [CrossRef] [Green Version]
- Fezza, F.; Bari, M.; Florio, R.; Talamonti, E.; Feole, M.; Maccarrone, M. Endocannabinoids, Related Compounds and Their Metabolic Routes. Molecules 2014, 19, 17078–17106. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Vasilyev, D.V.; Goncalves, M.B.; Howell, F.V.; Hobbs, C.; Reisenberg, M.; Shen, R.; Zhang, M.-Y.; Strassle, B.W.; Lu, P.; et al. Loss of Retrograde Endocannabinoid Signaling and Reduced Adult Neurogenesis in Diacylglycerol Lipase Knock-out Mice. J. Neurosci. 2010, 30, 2017–2024. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, A.; Yamazaki, M.; Hashimotodani, Y.; Uchigashima, M.; Kawata, S.; Abe, M.; Kita, Y.; Hashimoto, K.; Shimizu, T.; Watanabe, M.; et al. The Endocannabinoid 2-Arachidonoylglycerol Produced by Diacylglycerol Lipase α Mediates Retrograde Suppression of Synaptic Transmission. Neuron 2010, 65, 320–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinh, T.P.; Carpenter, D.; Leslie, F.M.; Freund, T.F.; Katona, I.; Sensi, S.L.; Kathuria, S.; Piomelli, D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. USA 2002, 99, 10819–10824. [Google Scholar] [CrossRef] [Green Version]
- Tsou, K.; Brown, S.; Sañudo-Peña, M.; Mackie, K.; Walker, J. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 1998, 83, 393–411. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Gutiérrez-Rodríguez, A.; Puente, N.; Elezgarai, I.; Ruehle, S.; Lutz, B.; Reguero, L.; Gerrikagoitia, I.; Marsicano, G.; Grandes, P. Anatomical characterization of the cannabinoid CB1receptor in cell-type-specific mutant mouse rescue models. J. Comp. Neurol. 2017, 525, 302–318. [Google Scholar] [CrossRef]
- Piazza, P.V.; Cota, D.; Marsicano, G. The CB1 Receptor as the Cornerstone of Exostasis. Neuron 2017, 93, 1252–1274. [Google Scholar] [CrossRef]
- Busquets-Garcia, A.; Bains, J.; Marsicano, G. CB1 Receptor Signaling in the Brain: Extracting Specificity from Ubiquity. Neuropsychopharmacology 2018, 43, 4–20. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Galiegue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carriere, D.; Carayon, P.; Bouaboula, M.; Shire, D.; Le Fur, G.; Casellas, P. Expression of Central and Peripheral Cannabinoid Receptors in Human Immune Tissues and Leukocyte Subpopulations. Eur. J. Biochem. 1995, 232, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Cabral, G.A.; Griffin-Thomas, L. Emerging role of the cannabinoid receptor CB2 in immune regulation: Therapeutic prospects for neuroinflammation. Expert Rev. Mol. Med. 2009, 11, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benito, C.; Núñez, E.; Tolón, R.M.; Carrier, E.J.; Rábano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J. Neurosci. 2003, 23, 11136–11341. [Google Scholar] [CrossRef] [Green Version]
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006, 6, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzmán, M.; Sanchez, C.; Galve-Roperh, I. Control of the cell survival/death decision by cannabinoids. Klin. Wochenschr. 2000, 78, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and Functional Characterization of Brainstem Cannabinoid CB2 Receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.-P.; Onaivi, E.S.; Ishiguro, H.; Liu, Q.-R.; Tagliaferro, P.A.; Brusco, A.; Uhl, G.R. Cannabinoid CB2 receptors: Immunohistochemical localization in rat brain. Brain Res. 2006, 1071, 10–23. [Google Scholar] [CrossRef]
- Onaivi, E.S. Neuropsychobiological Evidence for the Functional Presence and Expression of Cannabinoid CB2 Receptors in the Brain. Neuropsychobiology 2006, 54, 231–246. [Google Scholar] [CrossRef]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.P.; Patel, S.; Perchuk, A.; Meozzi, P.A.; Myers, L.; Mora, Z.; Tagliaferro, P.; Gardner, E.; et al. Discovery of the Presence and Functional Expression of Cannabinoid CB2 Receptors in Brain. Ann. N. Y. Acad. Sci. 2006, 1074, 514–536. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Gao, M.; Liu, Q.-R.; Bi, G.-H.; Li, X.; Yang, H.-J.; Gardner, E.L.; Wu, J.; Xi, Z.-X. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E5007–E5015. [Google Scholar] [CrossRef] [Green Version]
- Cabral, G.A.; Raborn, E.S.; Griffin, L.; Dennis, J.; Marciano-Cabral, F. CB2 receptors in the brain: Role in central immune function. Br. J. Pharmacol. 2008, 153, 240–251. [Google Scholar] [CrossRef] [Green Version]
- García-Gutiérrez, M.S.; Navarrete, F.; Navarro, G.; Reyes-Resina, I.; Franco, R.; Lanciego, J.L.; Giner, S.; Manzanares, J. Alterations in Gene and Protein Expression of Cannabinoid CB2 and GPR55 Receptors in the Dorsolateral Prefrontal Cortex of Suicide Victims. Neurotherapeutics 2018, 15, 796–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.-R.; Canseco-Alba, A.; Zhang, H.-Y.; Tagliaferro, P.; Chung, M.; Dennis, E.; Sanabria, B.; Schanz, N.; Escosteguy-Neto, J.C.; Ishiguro, H.; et al. Cannabinoid type 2 receptors in dopamine neurons inhibits psychomotor behaviors, alters anxiety, depression and alcohol preference. Sci. Rep. 2017, 7, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, F.; García-Gutiérrez, M.S.; Aracil-Fernández, A.; Lanciego, J.L.; Manzanares, J. Cannabinoid CB1 and CB2 Receptors, and Monoacylglycerol Lipase Gene Expression Alterations in the Basal Ganglia of Patients with Parkinson’s Disease. Neurotherapeutics 2018, 15, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. J. Cereb. Blood Flow Metab. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. GPR55: A new member of the cannabinoid receptor clan? J. Cereb. Blood Flow Metab. 2007, 152, 984–986. [Google Scholar] [CrossRef] [Green Version]
- Pistis, M.; Melis, M. From surface to nuclear receptors: The endocannabinoid family extends its assets. Curr. Med. Chem. 2010, 17, 1450–1467. [Google Scholar] [CrossRef]
- E O’Sullivan, S. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. J. Cereb. Blood Flow Metab. 2007, 152, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Pistis, M.; O’Sullivan, S.E. The Role of Nuclear Hormone Receptors in Cannabinoid Function. Stud. Surf. Sci. Catal. 2017, 80, 291–328. [Google Scholar] [CrossRef]
- Lago-Fernandez, A.; Zarzo-Arias, S.; Jagerovic, N.; Morales, P. Relevance of Peroxisome Proliferator Activated Receptors in Multitarget Paradigm Associated with the Endocannabinoid System. Int. J. Mol. Sci. 2021, 22, 1001. [Google Scholar] [CrossRef]
- Gaitán, A.V.; Wood, J.T.; Zhang, F.; Makriyannis, A.; Lammi-Keefe, C.J. Endocannabinoid Metabolome Characterization of Transitional and Mature Human Milk. Nutrients 2018, 10, 1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinitz, S.; Basolo, A.; Piaggi, P.; Piomelli, D.; Von Schwartzenberg, R.J.; Krakoff, J. Peripheral Endocannabinoids Associated With Energy Expenditure in Native Americans of Southwestern Heritage. J. Clin. Endocrinol. Metab. 2017, 103, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Bystrowska, B.; Frankowska, M.; Smaga, I.; Niedzielska-Andres, E.; Pomierny-Chamioło, L.; Filip, M. Cocaine-Induced Reinstatement of Cocaine Seeking Provokes Changes in the Endocannabinoid and N-Acylethanolamine Levels in Rat Brain Structures. Molecules 2019, 24, 1125. [Google Scholar] [CrossRef] [Green Version]
- Röhrig, W.; Achenbach, S.; Deutsch, B.; Pischetsrieder, M. Quantification of 24 circulating endocannabinoids, endocannabinoid-related compounds, and their phospholipid precursors in human plasma by UHPLC-MS/MS. J. Lipid Res. 2019, 60, 1475–1488. [Google Scholar] [CrossRef]
- Datta, P.; Melkus, M.; Rewers-Felkins, K.; Patel, D.; Bateman, T.; Baker, T.; Hale, T. Human Milk Endocannabinoid Levels as a Function of Obesity and Diurnal Rhythm. Nutrients 2021, 13, 2297. [Google Scholar] [CrossRef] [PubMed]
- Forte, D.; Fanelli, F.; Mezzullo, M.; Barone, M.; Corradi, G.; Auteri, G.; Bartoletti, D.; Martello, M.; Ottaviani, E.; Terragna, C.; et al. Disease-Specific Derangement of Circulating Endocannabinoids and N-Acylethanolamines in Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2020, 21, 3399. [Google Scholar] [CrossRef]
- Agrawal, K.; Hassoun, L.A.; Foolad, N.; Pedersen, T.L.; Sivamani, R.K.; Newman, J.W. Sweat lipid mediator profiling: A noninvasive approach for cutaneous research. J. Lipid Res. 2017, 58, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Fukumori, R.; Takeda, T.; Song, Y.; Morimoto, S.; Kikura-Hanajiri, R.; Yamaguchi, T.; Watanabe, K.; Aritake, K.; Tanaka, Y.; et al. Elevation of endocannabinoids in the brain by synthetic cannabinoid JWH-018: Mechanism and effect on learning and memory. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Voegel, C.D.; Baumgartner, M.R.; Kraemer, T.; Wüst, S.; Binz, T.M. Simultaneous quantification of steroid hormones and endocannabinoids (ECs) in human hair using an automated supported liquid extraction (SLE) and LC-MS/MS—Insights into EC baseline values and correlation to steroid concentrations. Talanta 2021, 222, 121499. [Google Scholar] [CrossRef]
- Castonguay-Paradis, S.; Lacroix, S.; Rochefort, G.; Parent, L.; Perron, J.; Martin, C.; Lamarche, B.; Raymond, F.; Flamand, N.; Di Marzo, V.; et al. Dietary fatty acid intake and gut microbiota determine circulating endocannabinoidome signaling beyond the effect of body fat. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Bashashati, M.; Leishman, E.; Bradshaw, H.; Sigaroodi, S.; Tatro, E.; Bright, T.; McCallum, R.; Sarosiek, I. Plasma endocannabinoids and cannabimimetic fatty acid derivatives are altered in gastroparesis: A sex- and subtype-dependent observation. Neurogastroenterol. Motil. 2021, 33, e13961. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Friuli, M.; Del Coco, L.; Longo, S.; Vergara, D.; Del Boccio, P.; Valentinuzzi, S.; Cicalini, I.; Fanizzi, F.P.; Gaetani, S.; et al. Chronic Oleoylethanolamide Treatment Decreases Hepatic Triacylglycerol Level in Rat Liver by a PPARgamma/SREBP-Mediated Suppression of Fatty Acid and Triacylglycerol Synthesis. Nutrients 2021, 13, 394. [Google Scholar] [CrossRef]
- Dempsey, S.K.; Gesseck, A.M.; Ahmad, A.; Daneva, Z.; Ritter, J.K.; Poklis, J.L. Formation of HETE-EAs and dihydroxy derivatives in mouse kidney tissue and analysis by high-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B 2019, 1126–1127, 121748. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A. Population genetics—making sense out of sequence. Nat. Genet. 1999, 21, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.G.; Fan, J.-B.; Siao, C.-J.; Berno, A.; Young, P.; Sapolsky, R.; Ghandour, G.; Perkins, N.; Winchester, E.; Spencer, J.; et al. Large-Scale Identification, Mapping, and Genotyping of Single-Nucleotide Polymorphisms in the Human Genome. Science 1998, 280, 1077–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.-W.; Lin, Y.-T.; Ding, S.-T.; Lo, L.-L.; Wang, P.-H.; Lin, E.-C.; Liu, F.-W.; Lu, Y.-W. Efficient SNP Discovery by Combining Microarray and Lab-on-a-Chip Data for Animal Breeding and Selection. Microarrays 2015, 4, 570–595. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.; Wilson, R.K.; Mardis, E.R. The Next-Generation Sequencing Revolution and Its Impact on Genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef] [Green Version]
- EMEA (European Medicines Agenc). Definitions for Genomic Biomarkers, Pharmacogenomics, Pharmacogenetics, Genomic Data and Sample Coding Categories; EMEA/CHMP/ICH: Amsterdam, The Netherlands, 2007.
- Kim, D.S.; Burt, A.A.; Ranchalis, J.E.; Wilmot, B.; Smith, J.D.; Patterson, K.E.; Coe, B.P.; Li, Y.K.; Bamshad, M.J.; Nikolas, M.; et al. Sequencing of sporadic Attention-Deficit Hyperactivity Disorder (ADHD) identifies novel and potentially pathogenic de novo variants and excludes overlap with genes associated with autism spectrum disorder. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2017, 174, 381–389. [Google Scholar] [CrossRef]
- Wangensteen, T.; Akselsen, H.; Holmen, J.; Undlien, D.; Retterstøl, L. A Common Haplotype in NAPEPLD Is Associated With Severe Obesity in a Norwegian Population-Based Cohort (the HUNT Study). Obesity 2011, 19, 612–617. [Google Scholar] [CrossRef]
- Dincheva, I.; Drysdale, A.T.; Hartley, C.A.; Johnson, D.C.; Jing, D.; King, E.C.; Ra, S.; Gray, J.M.; Yang, R.; DeGruccio, A.M.; et al. FAAH genetic variation enhances fronto-amygdala function in mouse and human. Nat. Commun. 2015, 6, 6395. [Google Scholar] [CrossRef] [Green Version]
- Mayo, L.M.; Asratian, A.; Lindé, J.; Holm, L.; Nätt, D.; Augier, G.; Stensson, N.; Vecchiarelli, H.A.; Balsevich, G.; Aukema, R.J.; et al. Protective effects of elevated anandamide on stress and fear-related behaviors: Translational evidence from humans and mice. Mol. Psychiatry 2020, 25, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.C.; Chiang, K.; Gerber, A.L.; Beutler, E.; Cravatt, B.F. A missense mutation in human fatty acid amide hydrolase associated with problem drug use. Proc. Natl. Acad. Sci. USA 2002, 99, 8394–8399. [Google Scholar] [CrossRef] [Green Version]
- Monteleone, P.; Bifulco, M.; Maina, G.; Tortorella, A.; Gazzerro, P.; Proto, M.C.; Di Filippo, C.; Monteleone, F.; Canestrelli, B.; Buonerba, G. Investigation of CNR1 and FAAH endocannabinoid gene polymorphisms in bipolar disorder and major depression. Pharmacol. Res. 2010, 61, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Cinar, O.G.; MacPherson, K.P.; Cinar, R.; Gamble-George, J.; Sugden, K.; Williams, B.; Godlewski, G.; Ramikie, T.S.; Gorka, A.X.; O Alapafuja, S.; et al. Convergent translational evidence of a role for anandamide in amygdala-mediated fear extinction, threat processing and stress-reactivity. Mol. Psychiatry 2013, 18, 813–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veyrieras, J.-B.; Kudaravalli, S.; Kim, S.Y.; Dermitzakis, E.T.; Gilad, Y.; Stephens, M.; Pritchard, J.K. High-Resolution Mapping of Expression-QTLs Yields Insight into Human Gene Regulation. PLoS Genet. 2008, 4, e1000214. [Google Scholar] [CrossRef] [Green Version]
- Huertas, E.; López-Moreno, J.A.; Fernández, V.; Echeverry-Alzate, V.; Bühler, K.-M. Associations between experimental substance use, FAAH-gene variations, impulsivity and sensation seeking. Psicothema 2019, 31, 239–245. [Google Scholar]
- Elkrief, L.; Spinney, S.; Vosberg, D.E.; Banaschewski, T.; Bokde, A.; Quinlan, E.B.; Conrod, P.; Nees, F.; Hohmann, S.; Whelan, R.; et al. Endocannabinoid Gene x Gene Interaction Association to Alcohol Use Disorder in Two Adolescent Cohorts. Front. Psychiatry 2021, 12, 645746. [Google Scholar] [CrossRef]
- Carey, C.E.; Agrawal, A.; Zhang, B.; Conley, E.D.; Degenhardt, L.; Heath, A.C.; Li, D.; Lynskey, M.T.; Martin, N.G.; Montgomery, G.W.; et al. Monoacylglycerol lipase (MGLL) polymorphism rs604300 interacts with childhood adversity to predict cannabis dependence symptoms and amygdala habituation: Evidence from an endocannabinoid system-level analysis. J. Abnorm. Psychol. 2015, 124, 860–877. [Google Scholar] [CrossRef]
- Meccariello, R.; Santoro, A.; D’Angelo, S.; Morrone, R.; Fasano, S.; Viggiano, A.; Pierantoni, R. The Epigenetics of the Endocannabinoid System. Int. J. Mol. Sci. 2020, 21, 1113. [Google Scholar] [CrossRef]
- Deans, C.; Maggert, K.A. What Do You Mean, “Epigenetic”? Genetics 2015, 199, 887–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henikoff, S.; Smith, M.M. Histone Variants and Epigenetics. Cold Spring Harb. Perspect. Biol. 2015, 7, a019364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Addario, C.; Di Francesco, A.; Arosio, B.; Gussago, C.; Dell’Osso, B.; Bari, M.; Galimberti, D.; Scarpini, E.; Altamura, A.C.; Mari, D.; et al. Epigenetic Regulation of Fatty Acid Amide Hydrolase in Alzheimer Disease. PLoS ONE 2012, 7, e39186. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Hutchison, K.E.; Bryan, A.D.; Filbey, F.M.; Calhoun, V.D.; Claus, E.D.; Lin, D.; Sui, J.; Du, Y.; Liu, J. Opposite Epigenetic Associations With Alcohol Use and Exercise Intervention. Front. Psychiatry 2018, 9, 594. [Google Scholar] [CrossRef]
- Pucci, M.; Micioni Di Bonaventura, M.V.; Zaplatic, E.; Bellia, F.; Maccarrone, M.; Cifani, C.; D’Addario, C. Transcriptional regulation of the endocannabinoid system in a rat model of binge-eating behavior reveals a selective modulation of the hypothalamic fatty acid amide hydrolase gene. Int. J. Eat. Disord. 2019, 52, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Peirson, S.N.; Butler, J.N. Quantitative polymerase chain reaction. Methods Mol. Biol. 2007, 362, 349–362. [Google Scholar]
- Kirkedal, C.; Elfving, B.; Müller, H.K.; Moreira, F.; Bindila, L.; Lutz, B.; Wegener, G.; Liebenberg, N. Hemisphere-dependent endocannabinoid system activity in prefrontal cortex and hippocampus of the Flinders Sensitive Line rodent model of depression. Neurochem. Int. 2019, 125, 7–15. [Google Scholar] [CrossRef]
- Amoako, A.; Marczylo, T.H.; Marczylo, E.L.; Elson, J.; Willets, J.M.; Taylor, A.H.; Konje, J.C. Anandamide modulates human sperm motility: Implications for men with asthenozoospermia and oligoasthenoteratozoospermia. Hum. Reprod. 2013, 28, 2058–2066. [Google Scholar] [CrossRef] [Green Version]
- Cueto, C.R.; Hernández, M.L.; Hillard, C.J.; Maciel, P.; Valdeolivas, S.; Ramos, J.A.; Ruiz, M.G.; Fernández-Ruiz, J. Altered striatal endocannabinoid signaling in a transgenic mouse model of spinocerebellar ataxia type-3. PLoS ONE 2017, 12, e0176521. [Google Scholar] [CrossRef]
- Munawar, N.; Oriowo, M.A.; Masocha, W. Antihyperalgesic Activities of Endocannabinoids in a Mouse Model of Antiretroviral-Induced Neuropathic Pain. Front. Pharmacol. 2017, 8, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.-C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.-T.; Ma, X.-J.; Luo, Y. RNAscope: A Novel in Situ RNA Analysis Platform for Formalin-Fixed, Paraffin-Embedded Tissues. J. Mol. Diagn. 2012, 14, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vickstrom, C.R.; Liu, X.; Liu, S.; Hu, M.-M.; Mu, L.; Hu, Y.; Yu, H.; Love, S.L.; Hillard, C.J.; Liu, Q.-S. Role of endocannabinoid signaling in a septohabenular pathway in the regulation of anxiety- and depressive-like behavior. Mol. Psychiatry 2021, 26, 3178–3191. [Google Scholar] [CrossRef] [PubMed]
- Gavini, K.; Parameshwaran, K. Western Blot; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Pillai-Kastoori, L.; Schutz-Geschwender, A.R.; Harford, J.A. A systematic approach to quantitative Western blot analysis. Anal. Biochem. 2020, 593, 113608. [Google Scholar] [CrossRef] [PubMed]
- Bioque, M.; Ctr Invest Biomed Red Salud From the FLAMM-PEPs study—Centro de Investigación Biomédica en Red de Salud Mental (CIBERSAM); Bueno, B.G.; MacDowell, K.S.; Meseguer, A.; A Saiz, P.; Parellada, M.; González-Pinto, A.M.; Rodríguez-Jiménez, R.; Lobo, A.; et al. Peripheral Endocannabinoid System Dysregulation in First-Episode Psychosis. Neuropsychopharmacology 2013, 38, 2568–2577. [Google Scholar] [CrossRef]
- Magaki, S.; Hojat, S.A.; Wei, B.; So, A.; Yong, W.H. An Introduction to the Performance of Immunohistochemistry. Methods Mol. Biol. 2018, 1897, 289–298. [Google Scholar] [CrossRef]
- Ayakannu, T.; Taylor, A.H.; Bari, M.; Mastrangelo, N.; Maccarrone, M.; Konje, J.C. Expression and Function of the Endocannabinoid Modulating Enzymes Fatty Acid Amide Hydrolase and N-Acylphosphatidylethanolamine-Specific Phospholipase D in Endometrial Carcinoma. Front. Oncol. 2019, 9, 1363. [Google Scholar] [CrossRef] [Green Version]
- Matalon, S.T.; Azar, S.; Meiri, D.; Hadar, R.; Nemirovski, A.; Abu Jabal, N.; Konikoff, F.M.; Drucker, L.; Tam, J.; Naftali, T. Endocannabinoid Levels in Ulcerative Colitis Patients Correlate With Clinical Parameters and Are Affected by Cannabis Consumption. Front. Endocrinol. 2021, 12, 685289. [Google Scholar] [CrossRef]
- Kucera, R.; Bouskila, J.; Elkrief, L.; Fink-Jensen, A.; Palmour, R.; Bouchard, J.-F.; Ptito, M. Expression and localization of CB1R, NAPE-PLD, and FAAH in the vervet monkey nucleus accumbens. Sci. Rep. 2018, 8, 8689. [Google Scholar] [CrossRef]
- Pirone, A.; Lazzarini, G.; Lenzi, C.; Giannessi, E.; Miragliotta, V. Immunolocalization of cannabinoid receptor 1 (CB1), monoglyceride lipase (MGL) and fatty-acid amide hydrolase 1 (FAAH) in the pig claustrum. J. Chem. Neuroanat. 2020, 109, 101843. [Google Scholar] [CrossRef]
- Nielsen, J.E.; Rolland, A.; Meyts, E.R.-D.; Janfelt, C.; Jørgensen, A.; Winge, S.B.; Kristensen, D.M.; Juul, A.; Chalmel, F.; Jegou, B.; et al. Characterisation and localisation of the endocannabinoid system components in the adult human testis. Sci. Rep. 2019, 9, 12866. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gao, S.; Li, W.; Liu, Z.; Shi, Z.; Qiu, C.; Jiang, J. Effect of monoacylglycerol lipase on the tumor growth in endometrial cancer. J. Obstet. Gynaecol. Res. 2019, 45, 2043–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisogno, T. Assay of DAGLalpha/beta Activity. Methods Mol. Biol. 2016, 1412, 149–156. [Google Scholar] [PubMed]
- Fezza, F.; Mastrangelo, N.; Maccarrone, M. Assay of NAPE-PLD Activity. Methods Pharmacol. Toxicol. 2016, 1412, 123–130. [Google Scholar] [CrossRef]
- Kayacelebi, A.A.; Schauerte, C.; Kling, K.; Herbers, J.; Beckmann, B.; Engeli, S.; Jordan, J.; Zoerner, A.A.; Tsikas, D. Cross-validated stable-isotope dilution GC–MS and LC–MS/MS assays for monoacylglycerol lipase (MAGL) activity by measuring arachidonic acid released from the endocannabinoid 2-arachidonoyl glycerol. J. Chromatogr. B 2017, 1047, 151–159. [Google Scholar] [CrossRef]
- van Der Wel, T.; Janssen, F.J.; Baggelaar, M.P.; Deng, H.; den Dulk, H.; Overkleeft, H.S.; van der Stelt, M. A natural substrate-based fluorescence assay for inhibitor screening on diacylglycerol lipase alpha. J. Lipid Res. 2015, 56, 927–935. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Hou, L.; Gan, J.; Cai, Q.; Ye, W.; Chen, J.; Tan, Z.; Zheng, C.; Li, G.; Xu, H.; et al. Synthesis and preliminary evaluation of a novel positron emission tomography (PET) ligand for imaging fatty acid amide hydrolase (FAAH). Bioorg. Med. Chem. Lett. 2020, 30, 127513. [Google Scholar] [CrossRef]
- Hou, L.; Rong, J.; Haider, A.; Ogasawara, D.; Varlow, C.; Schafroth, M.A.; Mu, L.; Gan, J.; Xu, H.; Fowler, C.J.; et al. Positron Emission Tomography Imaging of the Endocannabinoid System: Opportunities and Challenges in Radiotracer Development. J. Med. Chem. 2021, 64, 123–149. [Google Scholar] [CrossRef]
- Chukwueke, C.C.; Kowalczyk, W.J.; Gendy, M.; Taylor, R.; Tyndale, R.F.; Le Foll, B.; Heishman, S.J. The CB1R rs2023239 receptor gene variant significantly affects the reinforcing effects of nicotine, but not cue reactivity, in human smokers. Brain Behav. 2021, 11, e01982. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, Y.; Zhao, J.; Ma, Y.; Su, K.; Yuan, W.; Ma, J.Z.; Payne, T.J.; Li, M.D. Detection of Significant Association Between Variants in Cannabinoid Receptor 1 Gene (CNR1) and Personality in African–American Population. Front. Genet. 2018, 9, 199. [Google Scholar] [CrossRef]
- Arias Horcajadas, F.; Davila Piriz, J.R.; Parra Gonzalez, A.; Sanchez Romero, S.; Sanchez-Morla, E.; Ampuero Sanchez, I.; Ramos Atance, J.A. Cannabinoid receptor type 2 gene is associated with comorbidity of schizophrenia and cannabis dependence and fatty acid amide hydrolase gene is associated with cannabis dependence in the Spanish population. Adicciones 2021, 2021, 1587. [Google Scholar]
- Okahisa, Y.; Kodama, M.; Takaki, M.; Inada, T.; Uchimura, N.; Yamada, M.; Iwata, N.; Iyo, M.; Sora, I.; Ozaki, N.; et al. Association Study of Two Cannabinoid Receptor Genes, CNR1 and CNR2, with Methamphetamine Dependence. Curr. Neuropharmacol. 2011, 9, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.-P.; Patel, S.; Meozzi, P.A.; Myers, L.; Perchuk, A.; Mora, Z.; Tagliaferro, P.A.; Gardner, E.; et al. Functional expression of brain neuronal CB2 cannabinoid receptors are involved in the effects of drugs of abuse and in depression. Ann. N. Y. Acad. Sci. 2008, 1139, 434–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahamtan, A.; Rezaiy, S.; Samadizadeh, S.; Moradi, A.; Tabarraei, A.; Javid, N.; Oladnabi, M.; Naeimi, M.H. Cannabinoid CB2 Receptor Functional Variation (Q63R) Is Associated with Multiple Sclerosis in Iranian Subjects. J. Mol. Neurosci. 2020, 70, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.-P.; Patel, S.; Meozzi, P.A.; Myers, L.; Perchuk, A.; Mora, Z.; Tagliaferro, P.A.; Gardner, E.; et al. Brain Neuronal CB2 Cannabinoid Receptors in Drug Abuse and Depression: From Mice to Human Subjects. PLoS ONE 2008, 3, e1640. [Google Scholar] [CrossRef] [Green Version]
- Laprairie, R.; Kelly, M.; Denovan-Wright, E. The dynamic nature of type 1 cannabinoid receptor (CB1) gene transcription. J. Cereb. Blood Flow Metab. 2012, 167, 1583–1595. [Google Scholar] [CrossRef] [Green Version]
- Rotter, A.; Bayerlein, K.; Hansbauer, M.; Weiland, J.; Sperling, W.; Kornhuber, J.; Biermann, T. CB1 and CB2 Receptor Expression and Promoter Methylation in Patients with Cannabis Dependence. Eur. Addict. Res. 2013, 19, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Li, C.; Jaffe, A.E.; Shin, J.H.; Deep-Soboslay, A.; Yamin, R.E.; Weinberger, D.R.; Hyde, T.M.; Kleinman, J.E. Cannabinoid receptor CNR1 expression and DNA methylation in human prefrontal cortex, hippocampus and caudate in brain development and schizophrenia. Transl. Psychiatry 2020, 10, 158. [Google Scholar] [CrossRef]
- Subbanna, S.; Nagre, N.N.; Umapathy, N.S.; Pace, B.; Basavarajappa, B.S. Ethanol Exposure Induces Neonatal Neurodegeneration by Enhancing CB1R Exon1 Histone H4K8 Acetylation and Up-regulating CB1R Function causing Neurobehavioral Abnormalities in Adult Mice. Int. J. Neuropsychopharmacol. 2015, 18, pyu028. [Google Scholar] [CrossRef]
- Nagre, N.N.; Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Basavarajappa, B.S. CB1-receptor knockout neonatal mice are protected against ethanol-induced impairments of DNMT1, DNMT3A, and DNA methylation. J. Neurochem. 2014, 132, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Correa, F.; De Laurentiis, A.; Franchi, A.M. Ethanol downregulates N- acyl phosphatidylethanolamine-phospholipase D expression in BV2 microglial cells via epigenetic mechanisms. Eur. J. Pharmacol. 2016, 786, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S.; Ishiguro, H.; Gu, S.; Liu, Q.-R. CNS effects of CB2 cannabinoid receptors: Beyond neuro-immuno-cannabinoid activity. J. Psychopharmacol. 2011, 26, 92–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabban, E.L.; Serova, L.I.; Newman, E.; Aisenberg, N.; Akirav, I. Changes in Gene Expression in the Locus Coeruleus-Amygdala Circuitry in Inhibitory Avoidance PTSD Model. Cell. Mol. Neurobiol. 2017, 38, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.M.; Seo, S.; Park, D.; Kim, S.; Lamichhane, S.; Han, K.-M.; Kim, Y.-H.; Lee, S.; Hong, J.T.; Cha, H.J.; et al. Cannabinoid Receptor Type 1 Regulates Drug Reward Behavior via Glutamate Decarboxylase 67 Transcription. Int. J. Mol. Sci. 2021, 22, 10486. [Google Scholar] [CrossRef]
- Muguruza, C.; Morentin, B.; Meana, J.J.; Alexander, S.; Callado, L.F. Endocannabinoid system imbalance in the postmortem prefrontal cortex of subjects with schizophrenia. J. Psychopharmacol. 2019, 33, 1132–1140. [Google Scholar] [CrossRef]
- Navarrete, F.; García-Gutiérrez, M.S.; Manzanares, J. Pharmacological regulation of cannabinoid CB2 receptor modulates the reinforcing and motivational actions of ethanol. Biochem. Pharmacol. 2018, 157, 227–234. [Google Scholar] [CrossRef]
- Gomes, F.V.; Edelson, J.R.; Volk, D.W.; Grace, A.A. Altered brain cannabinoid 1 receptor mRNA expression across postnatal development in the MAM model of schizophrenia. Schizophr. Res. 2018, 201, 254–260. [Google Scholar] [CrossRef]
- Zhou, S.; Wu, Q.; Lin, X.; Ling, X.; Miao, J.; Liu, X.; Zhou, L.; Hu, C.; Zhang, Y.; Jia, N.; et al. Cannabinoid receptor type 2 promotes kidney fibrosis through orchestrating beta-catenin signaling. Kidney Int. 2021, 99, 364–381. [Google Scholar] [CrossRef]
- de Oliveira, H.U.; Dos Santos, R.S.; Malta, I.H.S.; Pinho, J.P.; Almeida, A.F.S.; Sorgi, C.A.; Galdino, G.; Ferranti Peti, A.P.; Xavier, G.S.; dos Reis, S.M.; et al. Investigation of the Involvement of the Endocannabinoid System in TENS-Induced Antinociception. J. Pain 2020, 21, 820–835. [Google Scholar] [CrossRef]
- Erdozain, A.M.; Rubio, M.; Meana, J.J.; Fernández-Ruiz, J.; Callado, L.F. Altered CB1 receptor coupling to G-proteins in the post-mortem caudate nucleus and cerebellum of alcoholic subjects. J. Psychopharmacol. 2015, 29, 1137–1145. [Google Scholar] [CrossRef]
- Espejo-Porras, F.; Fernández-Ruiz, J.; de Lago, E. Analysis of endocannabinoid receptors and enzymes in the post-mortem motor cortex and spinal cord of amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Jean-Gilles, L.; Braitch, M.; Latif, M.L.; Aram, J.; Fahey, A.J.; Edwards, L.J.; Robins, R.A.; Tanasescu, R.; Tighe, P.J.; Gran, B.; et al. Effects of pro-inflammatory cytokines on cannabinoid CB1and CB2receptors in immune cells. Acta Physiol. 2015, 214, 63–74. [Google Scholar] [CrossRef]
- Fitzgerald, M.L.; Mackie, K.; Pickel, V.M. Ultrastructural localization of cannabinoid CB1 and mGluR5 receptors in the prefrontal cortex and amygdala. J. Comp. Neurol. 2019, 527, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, K.; Li, C.; German, N.; Skobowiat, C.; Carrillo, M.; Kallem, R.R.; Larumbe, E.; Martinez, S.; Chuecos, M.; Ventolini, G.; et al. Effect of maternal high-fat diet on key components of the placental and hepatic endocannabinoid system. Am. J. Physiol. Metab. 2018, 314, E322–E333. [Google Scholar] [CrossRef]
- Tian, F.; Yang, H.; Huang, T.; Chen, F.; Xiong, F. Involvement of CB2 signalling pathway in the development of osteoporosis by regulating the proliferation and differentiation of hBMSCs. J. Cell. Mol. Med. 2021, 25, 2426–2435. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Lin, J.; Chen, Z.; Mao, Y.; Wu, X.; Xu, C.; Du, J.; Dong, Z.; Yang, H.; Zhou, F.; et al. CB2-mediated attenuation of nucleus pulposus degeneration via the amelioration of inflammation and oxidative stress in vivo and in vitro. Mol. Med. 2021, 27, 1–13. [Google Scholar] [CrossRef]
- Duerr, G.D.; Feißt, A.; Halbach, K.; Verfuerth, L.; Gestrich, C.; Wenzel, D.; Zimmer, A.; Breuer, J.; Dewald, O. CB2-deficiency is associated with a stronger hypertrophy and remodeling of the right ventricle in a murine model of left pulmonary artery occlusion. Life Sci. 2018, 215, 96–105. [Google Scholar] [CrossRef]
- Navia-Paldanius, D.; Aaltonen, N.; Lehtonen, M.; Savinainen, J.R.; Taschler, U.; Radner, F.P.; Zimmermann, R.; Laitinen, J.T. Increased tonic cannabinoid CB1R activity and brain region-specific desensitization of CB1R Gi/o signaling axis in mice with global genetic knockout of monoacylglycerol lipase. Eur. J. Pharm. Sci. 2015, 77, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Ginsburg, B.C.; Hensler, J.G. Age-related changes in CB1 receptor expression and function and the behavioral effects of cannabinoid receptor ligands. Pharmacol. Biochem. Behav. 2022, 213, 173339. [Google Scholar] [CrossRef]
- Pottier, G.; Gómez-Vallejo, V.; Padro, D.; Boisgard, R.; Dollé, F.; Llop, J.; Winkeler, A.; Martín, A. PET imaging of cannabinoid type 2 receptors with [11C]A-836339 did not evidence changes following neuroinflammation in rats. J. Cereb. Blood Flow Metab. 2017, 37, 1163–1178. [Google Scholar] [CrossRef] [Green Version]
- Ni, R.; Herde, A.M.; Haider, A.; Keller, C.; Louloudis, G.; Vaas, M.; Schibli, R.; Ametamey, S.M.; Klohs, J.; Mu, L. In vivo Imaging of Cannabinoid Type 2 Receptors: Functional and Structural Alterations in Mouse Model of Cerebral Ischemia by PET and MRI. Mol. Imaging Biol. 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Spindle, T.R.; Kuwabara, H.; Eversole, A.; Nandi, A.; Vandrey, R.; Antoine, D.G.; Umbricht, A.; Guarda, A.S.; Wong, D.F.; Weerts, E.M. Brain imaging of cannabinoid type I (CB1) receptors in women with cannabis use disorder and male and female healthy controls. Addict. Biol. 2021, 26, e13061. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Washington, DC, USA, 2014. [Google Scholar]
- Stein, D.J.; Scott, K.M.; de Jonge, P.; Kessler, R.C. Epidemiology of anxiety disorders: From surveys to nosology and back. Dialog- Clin. Neurosci. 2017, 19, 127–136. [Google Scholar] [CrossRef]
- Baxter, A.J.; Scott, K.M.; Vos, T.; Whiteford, H.A. Global prevalence of anxiety disorders: A systematic review and meta-regression. Psychol. Med. 2013, 43, 897–910. [Google Scholar] [CrossRef]
- Newman, M.G.; Llera, S.J.; Erickson, T.M.; Przeworski, A.; Castonguay, L.G. Worry and Generalized Anxiety Disorder: A Review and Theoretical Synthesis of Evidence on Nature, Etiology, Mechanisms, and Treatment. Annu. Rev. Clin. Psychol. 2013, 9, 275–297. [Google Scholar] [CrossRef] [Green Version]
- Navarria, A.; Tamburella, A.; Iannotti, F.A.; Micale, V.; Camillieri, G.; Gozzo, L.; Verde, R.; Imperatore, R.; Leggio, G.M.; Drago, F.; et al. The dual blocker of FAAH/TRPV1 N-arachidonoylserotonin reverses the behavioral despair induced by stress in rats and modulates the HPA-axis. Pharmacol. Res. 2014, 87, 151–159. [Google Scholar] [CrossRef]
- Demers, C.H.; Conley, E.D.; Bogdan, R.; Hariri, A.R. Interactions Between Anandamide and Corticotropin-Releasing Factor Signaling Modulate Human Amygdala Function and Risk for Anxiety Disorders: An Imaging Genetics Strategy for Modeling Molecular Interactions. Biol. Psychiatry 2016, 80, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Lazary, J.; Eszlari, N.; Juhasz, G.; Bagdy, G. Genetically reduced FAAH activity may be a risk for the development of anxiety and depression in persons with repetitive childhood trauma. Eur. Neuropsychopharmacol. 2016, 26, 1020–1028. [Google Scholar] [CrossRef]
- Harris, B.N.; Hohman, Z.P.; Campbell, C.M.; King, K.S.; Tucker, C.A. FAAH genotype, CRFR1 genotype, and cortisol interact to predict anxiety in an aging, rural Hispanic population: A Project FRONTIER study. Neurobiol. Stress 2019, 10, 100154. [Google Scholar] [CrossRef]
- Gärtner, A.; Dörfel, D.; Diers, K.; Witt, S.H.; Strobel, A.; Brocke, B. Impact of FAAH genetic variation on fronto-amygdala function during emotional processing. Eur. Arch. Psychiatry Clin. Neurosci. 2018, 269, 209–221. [Google Scholar] [CrossRef]
- Lazary, J.; Eszlari, N.; Juhasz, G.; Bagdy, G. A functional variant of CB2 receptor gene interacts with childhood trauma and FAAH gene on anxious and depressive phenotypes. J. Affect. Disord. 2019, 257, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Gonda, X.; Petschner, P.; Eszlari, N.; Sütöri, S.; Gál, Z.; Koncz, S.; Anderson, I.M.; Deakin, J.; Juhasz, G.; Bagdy, G. Effects of Different Stressors Are Modulated by Different Neurobiological Systems: The Role of GABA-A Versus CB1 Receptor Gene Variants in Anxiety and Depression. Front. Cell. Neurosci. 2019, 13, 138. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; McLaughlin, R.; Morrish, A.C.; Viau, V.; Floresco, S.; Hillard, C.J.; Gorzalka, B.B. Suppression of Amygdalar Endocannabinoid Signaling by Stress Contributes to Activation of the Hypothalamic–Pituitary–Adrenal Axis. Neuropsychopharmacology 2009, 34, 2733–2745. [Google Scholar] [CrossRef]
- McLaughlin, R.J.; Hill, M.N.; Bambico, F.R.; Stuhr, K.L.; Gobbi, G.; Hillard, C.J.; Gorzalka, B.B. Prefrontal cortical anandamide signaling coordinates coping responses to stress through a serotonergic pathway. Eur. Neuropsychopharmacol. 2012, 22, 664–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.N.; A Kumar, S.; Filipski, S.B.; Iverson, M.; Stuhr, K.L.; Keith, J.M.; Cravatt, B.F.; Hillard, C.J.; Chattarji, S.; McEwen, B.S. Disruption of fatty acid amide hydrolase activity prevents the effects of chronic stress on anxiety and amygdalar microstructure. Mol. Psychiatry 2013, 18, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Roelke, C.T.; Rademacher, D.J.; Hillard, C.J. Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling. Eur. J. Neurosci. 2005, 21, 1057–1069. [Google Scholar] [CrossRef]
- Patel, S.; Kingsley, P.J.; Mackie, K.; Marnett, L.J.; Winder, D.G. Repeated Homotypic Stress Elevates 2-Arachidonoylglycerol Levels and Enhances Short-Term Endocannabinoid Signaling at Inhibitory Synapses in Basolateral Amygdala. Neuropsychopharmacology 2009, 34, 2699–2709. [Google Scholar] [CrossRef] [Green Version]
- Surkin, P.N.; Gallino, S.L.; Luce, V.; Correa, F.; Fernandez-Solari, J.; De Laurentiis, A. Pharmacological augmentation of endocannabinoid signaling reduces the neuroendocrine response to stress. Psychoneuroendocrinology 2018, 87, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Evanson, N.K.; Tasker, J.G.; Hill, M.N.; Hillard, C.J.; Herman, J.P. Fast Feedback Inhibition of the HPA Axis by Glucocorticoids Is Mediated by Endocannabinoid Signaling. Endocrinology 2010, 151, 4811–4819. [Google Scholar] [CrossRef] [Green Version]
- Rademacher, D.J.; Meier, S.E.; Shi, L.; Ho, W.-S.V.; Jarrahian, A.; Hillard, C.J. Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum, and medial prefrontal cortex in mice. Neuropharmacology 2008, 54, 108–116. [Google Scholar] [CrossRef]
- Hill, M.N.; McLaughlin, R.; Pan, B.; Fitzgerald, M.L.; Roberts, C.; Lee, T.T.-Y.; Karatsoreos, I.N.; Mackie, K.; Viau, V.; Pickel, V.M.; et al. Recruitment of Prefrontal Cortical Endocannabinoid Signaling by Glucocorticoids Contributes to Termination of the Stress Response. J. Neurosci. 2011, 31, 10506–10515. [Google Scholar] [CrossRef] [PubMed]
- Tomas-Roig, J.; Piscitelli, F.; Gil, V.; Quintana, E.; Ramió-Torrentà, L.L.; del Río, J.A.; Moore, T.P.; Agbemenyah, H.; Salinas, G.; Pommerenke, C.; et al. Effects of repeated long-term psychosocial stress and acute cannabinoid exposure on mouse corticostriatal circuitries: Implications for neuropsychiatric disorders. CNS Neurosci. Ther. 2018, 24, 528–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.N.; McLaughlin, R.J.; Bingham, B.; Shrestha, L.; Lee, T.T.Y.; Gray, J.M.; Hillard, C.J.; Gorzalka, B.B.; Viau, V. Endogenous cannabinoid signaling is essential for stress adaptation. Proc. Natl. Acad. Sci. USA 2010, 107, 9406–9411. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.N.; Patel, S.; Carrier, E.J.; Rademacher, D.J.; Ormerod, B.; Hillard, C.J.; Gorzalka, B.B. Downregulation of Endocannabinoid Signaling in the Hippocampus Following Chronic Unpredictable Stress. Neuropsychopharmacology 2004, 30, 508–515. [Google Scholar] [CrossRef]
- Lee, T.T.; Hill, M.N. Age of stress exposure modulates the immediate and sustained effects of repeated stress on corticolimbic cannabinoid CB (1) receptor binding in male rats. Neuroscience 2013, 249, 106–114. [Google Scholar] [CrossRef]
- Benjet, C.; Bromet, E.; Karam, E.G.; Kessler, R.C.; McLaughlin, K.A.; Ruscio, A.M.; Shahly, V.; Stein, D.J.; Petukhova, M.; Hill, E.; et al. The epidemiology of traumatic event exposure worldwide: Results from the World Mental Health Survey Consortium. Psychol. Med. 2016, 46, 327–343. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.T.; Ogdie, M.N.; Jarvelin, M.-R.; Moilanen, I.K.; Loo, S.K.; McCracken, J.T.; McGough, J.J.; Yang, M.H.; Peltonen, L.; Nelson, S.F.; et al. Association of the cannabinoid receptor gene (CNR1) with ADHD and post-traumatic stress disorder. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2008, 147B, 1488–1494. [Google Scholar] [CrossRef] [Green Version]
- Kucukalic, S.; Bojic, E.F.; Babic, R.; Avdibegovic, E.; Babic, D.; Agani, F.; Jakovljevic, M.; Kucukalic, A.; Mehmedbasic, A.B.; Dzananovic, E.S.; et al. Genetic susceptibility to posttraumatic stress disorder: Analyses of the oxytocin receptor, retinoic acid receptor-related orphan receptor a and cannabinoid receptor 1 genes. Psychiatr. Danub. 2019, 31, 219–226. [Google Scholar] [CrossRef]
- Neumeister, A.; Normandin, M.; Pietrzak, R.H.; Piomelli, D.; Zheng, M.Q.; Anton, A.I.G.; Potenza, M.N.; Bailey, C.R.; Lin, S.F.; Najafzadeh, S.; et al. Elevated brain cannabinoid CB1 receptor availability in post-traumatic stress disorder: A positron emission tomography study. Mol. Psychiatry 2013, 18, 1034–1040. [Google Scholar] [CrossRef]
- Mota, N.; Sumner, J.A.; Lowe, S.R.; Neumeister, A.; Uddin, M.; Aiello, A.E.; Wildman, D.; Galea, S.; Koenen, K.C.; Pietrzak, R.H. The rs1049353 polymorphism in the CNR1 gene interacts with childhood abuse to predict posttraumatic threat symptoms. J. Clin. Psychiatry 2015, 76, 18765. [Google Scholar] [CrossRef] [Green Version]
- Ney, L.J.; Matthews, A.; Hsu, C.K.; Zuj, D.V.; Nicholson, E.; Steward, T.; Nichols, D.; Graham, B.; Harrison, B.; Bruno, R.; et al. Cannabinoid polymorphisms interact with plasma endocannabinoid levels to predict fear extinction learning. Depress. Anxiety 2021, 38, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.; Ferreira, F.R.; da Silva, W.A.; Guimarães, F.S. Predator threat stress promotes long lasting anxiety-like behaviors and modulates synaptophysin and CB1 receptors expression in brain areas associated with PTSD symptoms. Neurosci. Lett. 2013, 533, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Le, T.; McGuire, J.; Xing, G.; Zhang, L.; Li, H.; Parker, C.C.; Johnson, L.R.; Ursano, R.J. Expression pattern of the cannabinoid receptor genes in the frontal cortex of mood disorder patients and mice selectively bred for high and low fear. J. Psychiatr. Res. 2012, 46, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Chhatwal, J.P.; Davis, M.; A Maguschak, K.; Ressler, K. Enhancing Cannabinoid Neurotransmission Augments the Extinction of Conditioned Fear. Neuropsychopharmacology 2004, 30, 516–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparyan, A.; Navarrete, F.; Manzanares, J. Cannabidiol and Sertraline Regulate Behavioral and Brain Gene Expression Alterations in an Animal Model of PTSD. Front. Pharmacol. 2021, 12, 694510. [Google Scholar] [CrossRef]
- Maymon, N.; Zer-Aviv, T.M.; Sabban, E.L.; Akirav, I. Neuropeptide Y and cannabinoids interaction in the amygdala after exposure to shock and reminders model of PTSD. Neuropharmacology 2020, 162, 107804. [Google Scholar] [CrossRef] [PubMed]
- Xing, G.; Carlton, J.; Zhang, L.; Jiang, X.; Fullerton, C.; Li, H.; Ursano, R. Cannabinoid receptor expression and phosphorylation are differentially regulated between male and female cerebellum and brain stem after repeated stress: Implication for PTSD and drug abuse. Neurosci. Lett. 2011, 502, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Piggott, V.; Lloyd, S.; Matchynski, J.; Perrine, S.; Conti, A. Traumatic Stress, Chronic Ethanol Exposure, or the Combination, Alter Cannabinoid System Components in Reward and Limbic Regions of the Mouse Brain. Molecules 2021, 26, 2086. [Google Scholar] [CrossRef]
- Morena, M.; Berardi, A.; Colucci, P.; Palmery, M.; Trezza, V.; Hill, M.N.; Campolongo, P. Enhancing Endocannabinoid Neurotransmission Augments The Efficacy of Extinction Training and Ameliorates Traumatic Stress-Induced Behavioral Alterations in Rats. Neuropsychopharmacology 2017, 43, 1284–1296. [Google Scholar] [CrossRef]
- Fride, E.; Suris, R.; Weidenfeld, J.; Mechoulam, R. Differential response to acute and repeated stress in cannabinoid CB1 receptor knockout newborn and adult mice. Behav. Pharmacol. 2005, 16, 431–440. [Google Scholar] [CrossRef]
- A Varvel, S.; E Wise, L.; Niyuhire, F.; Cravatt, B.F.; Lichtman, A.H. Inhibition of Fatty-Acid Amide Hydrolase Accelerates Acquisition and Extinction Rates in a Spatial Memory Task. Neuropsychopharmacology 2006, 32, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- IHME. Global Health Data Exchange (GHDx); Institute of Health Metrics and Evaluation (IHME), 2019. Available online: http://ghdx.healthdata.org/gbd-results-tool?params=gbd-api-2019-permalink/27a7644e8ad28e739382d31e77589dd7 (accessed on 5 February 2022).
- World Health Organization. WHO, Depression and Other Common Mental Disorders; Global Health Estimates: Geneva, Switzerland, 2017.
- The American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM), 5th ed.; The American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Estrada, J.A.; Contreras, I. Endocannabinoid Receptors in the CNS: Potential Drug Targets for the Prevention and Treatment of Neurologic and Psychiatric Disorders. Curr. Neuropharmacol. 2020, 18, 769–787. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, S. Epidemiology of Suicide and the Psychiatric Perspective. Int. J. Environ. Res. Public Health 2018, 15, 1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jesulola, E.; Micalos, P.; Baguley, I.J. Understanding the pathophysiology of depression: From monoamines to the neurogenesis hypothesis model—Are we there yet? Behav. Brain Res. 2018, 341, 79–90. [Google Scholar] [CrossRef]
- Bassi, M.A.U.S.; Gilio, L.; Maffei, P.; Dolcetti, E.; Bruno, A.; Buttari, F.; Centonze, D.; Iezzi, E. Exploiting the Multifaceted Effects of Cannabinoids on Mood to Boost Their Therapeutic Use Against Anxiety and Depression. Front. Mol. Neurosci. 2018, 11, 424. [Google Scholar] [CrossRef] [Green Version]
- Nestler, E.J.; Barrot, M.; DiLeone, R.J.; Eisch, A.J.; Gold, S.J.; Monteggia, L.M. Neurobiology of depression. Neuron 2002, 34, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Langlois, C.; Potvin, S.; Khullar, A.; Tourjman, S.V. Down and High: Reflections Regarding Depression and Cannabis. Front. Psychiatry 2021, 12, 625158. [Google Scholar] [CrossRef]
- Torres-Berrío, A.; Issler, O.; Parise, E.M.; Nestler, E.J. Unraveling the epigenetic landscape of depression: Focus on early life stress. Dialog. Clin. Neurosci. 2019, 21, 341–357. [Google Scholar] [CrossRef]
- Nelson, C.A., 3rd; Gabard-Durnam, L.J. Early Adversity and Critical Periods: Neurodevelopmental Consequences of Violating the Expectable Environment. Trends Neurosci. 2020, 43, 133–143. [Google Scholar] [CrossRef]
- Kendler, K.S.; Gatz, M.; Gardner, C.O.; Pedersen, N.L. A Swedish National Twin Study of Lifetime Major Depression. Am. J. Psychiatry 2006, 163, 109–114. [Google Scholar] [CrossRef]
- Moreira, F.A.; Grieb, M.; Lutz, B. Central side-effects of therapies based on CB1 cannabinoid receptor agonists and antagonists: Focus on anxiety and depression. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713. [Google Scholar] [CrossRef]
- Després, J.-P.; Van Gaal, L.; Pi-Sunyer, X.; Scheen, A. Efficacy and safety of the weight-loss drug rimonabant. Lancet 2008, 371, 555–557. [Google Scholar] [CrossRef]
- Blüher, M. Efficacy and safety of the weight-loss drug rimonabant. Lancet 2008, 371, 555–556. [Google Scholar] [CrossRef]
- Elbatsh, M.M.; Moklas, M.A.A.; Marsden, C.A.; Kendall, D.A. Antidepressant-like effects of Delta(9)-tetrahydrocannabinol and rimonabant in the olfactory bulbectomised rat model of depression. Pharmacol. Biochem. Behav. 2012, 102, 357–365. [Google Scholar] [CrossRef]
- Griebel, G.; Stemmelin, J.; Scatton, B. Effects of the cannabinoid CB1 receptor antagonist rimonabant in models of emotional reactivity in rodents. Biol. Psychiatry 2005, 57, 261–267. [Google Scholar] [CrossRef]
- Beyer, C.E.; Dwyer, J.M.; Piesla, M.J.; Platt, B.J.; Shen, R.; Rahman, Z.; Chan, K.; Manners, M.T.; Samad, T.A.; Kennedy, J.D.; et al. Depression-like phenotype following chronic CB1 receptor antagonism. Neurobiol. Dis. 2010, 39, 148–155. [Google Scholar] [CrossRef]
- Lee, S.; Kim, D.H.; Yoon, S.-H.; Ryu, J.H. Sub-chronic administration of rimonabant causes loss of antidepressive activity and decreases doublecortin immunoreactivity in the mouse hippocampus. Neurosci. Lett. 2009, 467, 111–116. [Google Scholar] [CrossRef]
- Ettaro, R.; Laudermilk, L.; Clark, S.D.; Maitra, R. Behavioral assessment of rimonabant under acute and chronic conditions. Behav. Brain Res. 2020, 390, 112697. [Google Scholar] [CrossRef]
- Mondimore, F.; Zandi, P.; MacKinnon, D.; McInnis, M.; Miller, E.; Crowe, R.; Scheftner, W.; Marta, D.; Weissman, M.; Levinson, D.; et al. Familial Aggregation of Illness Chronicity in Recurrent, Early-Onset Major Depression Pedigrees. Am. J. Psychiatry 2006, 163, 1554. [Google Scholar] [CrossRef]
- Scherma, M.; Muntoni, A.L.; Riedel, G.; Fratta, W.; Fadda, P. Cannabinoids and their therapeutic applications in mental disorders. Dialog. Clin. Neurosci. 2020, 22, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Mitjans, M.; Serretti, A.; Fabbri, C.; Gastó, C.; Catalán, R.; Fañanás, L.; Arias, B. Screening genetic variability at the CNR1 gene in both major depression etiology and clinical response to citalopram treatment. Psychopharmacology 2013, 227, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Domschke, K.; Dannlowski, U.; Ohrmann, P.; Lawford, B.; Bauer, J.; Kugel, H.; Heindel, W.; Young, R.; Morris, P.; Arolt, V.; et al. Cannabinoid receptor 1 (CNR1) gene: Impact on antidepressant treatment response and emotion processing in Major Depression. Eur. Neuropsychopharmacol. 2008, 18, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Icick, R.; Peoc’H, K.; Karsinti, E.; Ksouda, K.; Hajj, A.; Bloch, V.; Prince, N.; Mouly, S.; Bellivier, F.; Lépine, J.-P.; et al. A cannabinoid receptor 1 polymorphism is protective against major depressive disorder in methadone-maintained outpatients. Am. J. Addict. 2015, 24, 613–620. [Google Scholar] [CrossRef]
- Kong, X.; Miao, Q.; Lu, X.; Zhang, Z.; Chen, M.; Zhang, J.; Zhai, J. The association of endocannabinoid receptor genes (CNR1 and CNR2) polymorphisms with depression: A meta-analysis. Medicine 2019, 98, e17403. [Google Scholar] [CrossRef]
- Yang, C.; Nolte, I.M.; Ma, Y.; An, X.; Bosker, F.J.; Li, J. The associations of CNR1 SNPs and haplotypes with vulnerability and treatment response phenotypes in Han Chinese with major depressive disorder: A case–control association study. Mol. Genet. Genom. Med. 2021, 9, e1752. [Google Scholar] [CrossRef]
- Koethe, D.; Llenos, I.C.; Dulay, J.R.; Hoyer, C.; Torrey, E.F.; Leweke, F.M.; Weis, S. Expression of CB1 cannabinoid receptor in the anterior cingulate cortex in schizophrenia, bipolar disorder, and major depression. J. Neural Transm. 2007, 114, 1055–1063. [Google Scholar] [CrossRef]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Callado, L.F.; Meana, J.J.; Garzón-Niño, J. Schizophrenia and depression, two poles of endocannabinoid system deregulation. Transl. Psychiatry 2017, 7, 1291. [Google Scholar] [CrossRef] [Green Version]
- Vinod, K.Y.; Arango, V.; Xie, S.; Kassir, S.A.; Mann, J.J.; Cooper, T.B.; Hungund, B.L. Elevated levels of endocannabinoids and CB1 receptor-mediated G-protein signaling in the prefrontal cortex of alcoholic suicide victims. Biol. Psychiatry 2005, 57, 480–486. [Google Scholar] [CrossRef]
- Mato, S.; Pilar-Cuéllar, F.; Valdizán, E.M.; González-Maeso, J.; Rodríguez-Puertas, R.; Meana, J.; Sallés, J.; Crespo-Facorro, B.; Pazos, Á. Selective up-regulation of cannabinoid CB1 receptor coupling to Go-proteins in suicide victims with mood disorders. Biochem. Pharmacol. 2018, 157, 258–265. [Google Scholar] [CrossRef]
- Hungund, B.L.; Vinod, K.Y.; Kassir, S.A.; Basavarajappa, B.S.; Yalamanchili, R.; Cooper, T.B.; Arango, V.; Mann, J.J. Upregulation of CB1 receptors and agonist-stimulated [35S] GTPgammaS binding in the prefrontal cortex of depressed suicide victims. Mol. Psychiatry 2004, 9, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.; Miller, G.; Ho, W.-S.; Gorzalka, B.; Hillard, C. Serum Endocannabinoid Content is Altered in Females with Depressive Disorders: A Preliminary Report. Pharmacopsychiatry 2008, 41, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.N.; Miller, G.E.; Carrier, E.J.; Gorzalka, B.B.; Hillard, C.J. Circulating endocannabinoids and N-acyl ethanolamines are differentially regulated in major depression and following exposure to social stress. Psychoneuroendocrinology 2009, 34, 1257–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Sanchiz, P.; Nogueira-Arjona, R.; Pastor, A.; Araos, P.; Serrano, A.; Boronat, A.; Garcia-Marchena, N.; Mayoral-Cleries, F.; Bordallo, A.; Alen, F.; et al. Plasma concentrations of oleoylethanolamide in a primary care sample of depressed patients are increased in those treated with selective serotonin reuptake inhibitor-type antidepressants. Neuropharmacology 2019, 149, 212–220. [Google Scholar] [CrossRef]
- Heyman, E.; Gamelin, F.-X.; Goekint, M.; Piscitelli, F.; Roelands, B.; Leclair, E.; Di Marzo, V.; Meeusen, R. Intense exercise increases circulating endocannabinoid and BDNF levels in humans—Possible implications for reward and depression. Psychoneuroendocrinology 2012, 37, 844–851. [Google Scholar] [CrossRef]
- Meyer, J.D.; Crombie, K.M.; Cook, D.B.; Hillard, C.J.; Koltyn, K.F. Serum Endocannabinoid and Mood Changes after Exercise in Major Depressive Disorder. Med. Sci. Sports Exerc. 2019, 51, 1909–1917. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.S.V.; Hill, M.N.; E Miller, G.; Gorzalka, B.B.; Hillard, C.J. Serum contents of endocannabinoids are correlated with blood pressure in depressed women. Lipids Health Dis. 2012, 11, 32. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, J.M.; Chesney, S.A.; Lee, T.S.; Brasel, K.; Larson, C.L.; Hillard, C.J.; Deroon-Cassini, T.A. Circulating endocannabinoids and prospective risk for depression in trauma-injury survivors. Neurobiol. Stress 2021, 14, 100304. [Google Scholar] [CrossRef]
- Hill, M.N.; Ho, W.-S.V.; Hillard, C.J.; Gorzalka, B.B. Differential effects of the antidepressants tranylcypromine and fluoxetine on limbic cannabinoid receptor binding and endocannabinoid contents. J. Neural Transm. 2008, 115, 1673–1679. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.N.; Carrier, E.J.; McLaughlin, R.J.; Morrish, A.C.; Meier, S.E.; Hillard, C.J.; Gorzalka, B.B. Regional alterations in the endocannabinoid system in an animal model of depression: Effects of concurrent antidepressant treatment. J. Neurochem. 2008, 106, 2322–2336. [Google Scholar] [CrossRef] [Green Version]
- Reich, C.G.; Taylor, M.E.; McCarthy, M.M. Differential effects of chronic unpredictable stress on hippocampal CB1 receptors in male and female rats. Behav. Brain Res. 2009, 203, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Sun, D.; Pan, B.; Roberts, C.; Sun, X.; Hillard, C.J.; Liu, Q.-S. Deficiency in Endocannabinoid Signaling in the Nucleus Accumbens Induced by Chronic Unpredictable Stress. Neuropsychopharmacology 2010, 35, 2249–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Rhee, J.; Lee, S.; Chung, C. Selectively Impaired Endocannabinoid-Dependent Long-Term Depression in the Lateral Habenula in an Animal Model of Depression. Cell Rep. 2017, 20, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Vinod, K.Y.; Xie, S.; Psychoyos, D.; Hungund, B.L.; Cooper, T.B.; Tejani-Butt, S.M. Dysfunction in Fatty Acid Amide Hydrolase Is Associated with Depressive-Like Behavior in Wistar Kyoto Rats. PLoS ONE 2012, 7, e36743. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.-J.; Gao, M.; Gao, F.-F.; Su, Q.-X.; Wu, J. Brain cannabinoid receptor 2: Expression, function and modulation. Acta Pharmacol. Sin. 2017, 38, 312–316. [Google Scholar] [CrossRef]
- Rafiei, D.; Kolla, N.J. Elevated Brain Fatty Acid Amide Hydrolase Induces Depressive-Like Phenotypes in Rodent Models: A Review. Int. J. Mol. Sci. 2021, 22, 1047. [Google Scholar] [CrossRef]
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Marder, S.R.; Cannon, T.D. Schizophrenia. N. Engl. J. Med. 2019, 381, 1753–1761. [Google Scholar] [CrossRef]
- Kahn, R.S.; Sommer, I.E.; Murray, R.M.; Meyer-Lindenberg, A.; Weinberg, D.R.; Cannon, T.D.; O’Donovan, M.; Correll, C.U.; Kane, J.M.; van OS, J.; et al. Schizophrenia. Nat. Rev. Dis. Primers 2015, 1, 15067. [Google Scholar] [CrossRef]
- Laursen, T.M.; Nordentoft, M.; Mortensen, P.B. Excess Early Mortality in Schizophrenia. Annu. Rev. Clin. Psychol. 2014, 10, 425–448. [Google Scholar] [CrossRef]
- McGrath, J.; Saha, S.; Chant, D.; Welham, J. Schizophrenia: A Concise Overview of Incidence, Prevalence, and Mortality. Epidemiol. Rev. 2008, 30, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jääskeläinen, E.; Juola, P.; Hirvonen, N.; McGrath, J.J.; Saha, S.; Isohanni, M.; Veijola, J.; Miettunen, J. A Systematic Review and Meta-Analysis of Recovery in Schizophrenia. Schizophr. Bull. 2013, 39, 1296–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, J.M.; Kishimoto, T.; Correll, C.U. Non-adherence to medication in patients with psychotic disorders: Epidemiology, contributing factors and management strategies. World Psychiatry 2013, 12, 216–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickard, B.S. Schizophrenia biomarkers: Translating the descriptive into the diagnostic. J. Psychopharmacol. 2015, 29, 138–143. [Google Scholar] [CrossRef] [Green Version]
- Sherif, M.; Radhakrishnan, R.; D’Souza, D.C.; Ranganathan, M. Human Laboratory Studies on Cannabinoids and Psychosis. Biol. Psychiatry 2016, 79, 526–538. [Google Scholar] [CrossRef]
- Minichino, A.; Senior, M.; Brondino, N.; Zhang, S.H.; Godlewska, B.R.; Burnet, P.W.; Lennox, B.R.; Cipriani, A. Measuring Disturbance of the Endocannabinoid System in Psychosis: A Systematic Review and Meta-analysis. JAMA Psychiatry 2019, 76, 914–923. [Google Scholar] [CrossRef]
- Leroy, S.; Griffon, N.; Bourdel, M.; Olié, J.; Poirier, M.; Krebs, M. Schizophrenia and the cannabinoid receptor type 1 (CB1): Association study using a single-base polymorphism in coding exon 1. Am. J. Med Genet. 2001, 105, 749–752. [Google Scholar] [CrossRef]
- Ujike, H.; Takaki, M.; Nakata, K.; Tanaka, Y.; Takeda, T.; Kodama, M.; Fujiwara, Y.; Sakai, A.; Kuroda, S. CNR1, central cannabinoid receptor gene, associated with susceptibility to hebephrenic schizophrenia. Mol. Psychiatry 2002, 7, 515–518. [Google Scholar] [CrossRef] [Green Version]
- Zammit, S.; Spurlock, G.; Williams, H.; Norton, N.; Williams, N.; O’Donovan, M.C.; Owen, M.J. Genotype effects of CHRNA7, CNR1 and COMT in schizophrenia: Interactions with tobacco and cannabis use. Br. J. Psychiatry 2007, 191, 402–407. [Google Scholar] [CrossRef] [Green Version]
- Seifert, J.; Ossege, S.; Emrich, H.M.; Schneider, U.; Stuhrmann, M. No association of CNR1 gene variations with susceptibility to schizophrenia. Neurosci. Lett. 2007, 426, 29–33. [Google Scholar] [CrossRef]
- Hamdani, N.; Tabeze, J.-P.; Ramoz, N.; Ades, J.; Hamon, M.; Sarfati, Y.; Boni, C.; Gorwood, P. The CNR1 gene as a pharmacogenetic factor for antipsychotics rather than a susceptibility gene for schizophrenia. Eur. Neuropsychopharmacol. 2008, 18, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Kim, J.Y.; Park, B.-L.; Kim, J.-H.; Kim, B.; Park, C.S.; Kim, B.-J.; Lee, C.-S.; Lee, M.; Choi, W.H.; et al. Genetic association analysis of CNR1 and CNR2 polymorphisms with schizophrenia in a Korean population. Psychiatr. Genet. 2014, 24, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Squassina, A.; Congiu, D.; Chillotti, C.; Niola, P.; Galderisi, S.; Pistis, M.; Del Zompo, M. Investigation of endocannabinoid system genes suggests association between peroxisome proliferator activator receptor-α gene (PPARA) and schizophrenia. Eur. Neuropsychopharmacol. 2013, 23, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.-J.; Wang, Y.-C.; Hong, C.-J. Association study of a cannabinoid receptor gene (CNR1) polymorphism and schizophrenia. Psychiatr. Genet. 2000, 10, 149–151. [Google Scholar] [CrossRef]
- Ferretjans, R.; de Souza, R.P.; Panizzutti, B.; Ferrari, P.; Mantovani, L.; de Campos-Carli, S.M.; Santos, R.R.; Guimarães, F.C.; Teixeira, A.L.; Gama, C.S.; et al. Cannabinoid receptor gene polymorphisms and cognitive performance in patients with schizophrenia and controls. Rev. Bras. de Psiquiatr. 2022, 44, 26–34. [Google Scholar] [CrossRef]
- Suárez-Pinilla, P.; Roiz-Santiañez, R.; de la Foz, V.O.G.; Guest, P.C.; Ayesa-Arriola, R.; Córdova-Palomera, A.; Tordesillas-Gutierrez, D.; Crespo-Facorro, B. Brain structural and clinical changes after first episode psychosis: Focus on cannabinoid receptor 1 polymorphisms. Psychiatry Res. Neuroimaging 2015, 233, 112–119. [Google Scholar] [CrossRef]
- Kuzman, M.R.; Kuharic, D.B.; Ganoci, L.; Makaric, P.; Kekin, I.; Gajsak, L.R.; Prpic, N.; Bozina, T.; Bajić, Ž.; Bozina, N. Association of CNR1 genotypes with changes in neurocognitive performance after eighteen-month treatment in patients with first-episode psychosis. Eur. Psychiatry 2019, 61, 88–96. [Google Scholar] [CrossRef]
- Schennach, R.; Zill, P.; Obermeier, M.; Hauer, D.; Dehning, S.; Cerovecki, A.; Opgen-Rhein, M.; Musil, R.; Spellmann, I.; Matz, J.; et al. The CNR1 gene in depression and schizophrenia—Is there an association with early improvement and response? Psychiatry Res. 2012, 196, 160. [Google Scholar] [CrossRef]
- Yu, W.; De Hert, M.; Moons, T.; Claes, S.J.; Correll, C.U.; van Winkel, R. CNR1 gene and risk of the metabolic syndrome in patients with schizophrenia. J. Clin. Psychopharmacol. 2013, 33, 186–192. [Google Scholar] [CrossRef]
- Dalton, V.S.; Long, L.; Weickert, C.S.; Zavitsanou, K. Paranoid Schizophrenia is Characterized by Increased CB1 Receptor Binding in the Dorsolateral Prefrontal Cortex. Neuropsychopharmacology 2011, 36, 1620–1630. [Google Scholar] [CrossRef]
- Zavitsanou, K.; Garrick, T.; Huang, X.F. Selective antagonist [3H]SR141716A binding to cannabinoid CB1 receptors is increased in the anterior cingulate cortex in schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2004, 28, 355–360. [Google Scholar] [CrossRef]
- Newell, K.A.; Deng, C.; Huang, X.-F. Increased cannabinoid receptor density in the posterior cingulate cortex in schizophrenia. Exp. Brain Res. 2006, 172, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Liu, X.; Liu, Y.; Deng, Z.; Nie, X.; Wang, X.; Jin, Y. Enrichment of epidermal stem cells by rapid adherence and analysis of the reciprocal interaction of epidermal stem cells with neighboring cells using an organotypic system. Cell Biol. Int. 2007, 31, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Eggan, S.M.; Hashimoto, T.; Lewis, D.A. Reduced Cortical Cannabinoid 1 Receptor Messenger RNA and Protein Expression in Schizophrenia. Arch. Gen. Psychiatry 2008, 65, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Urigüen, L.; García-Fuster, M.J.; Callado, L.F.; Morentin, B.; La Harpe, R.; Casadó, V.; Lluis, C.; Franco, R.; García-Sevilla, J.A.; Meana, J.J. Immunodensity and mRNA expression of A2A adenosine, D2 dopamine, and CB1 cannabinoid receptors in postmortem frontal cortex of subjects with schizophrenia: Effect of antipsychotic treatment. Psychopharmacology 2009, 206, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Guillozet-Bongaarts, A.L.; Hyde, T.M.; Dalley, R.A.; Hawrylycz, M.J.; Henry, A.; Hof, P.R.; Hohmann, J.; Jones, A.R.; Kuan, C.L.; Royall, J.; et al. Altered gene expression in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol. Psychiatry 2014, 19, 478–485. [Google Scholar] [CrossRef] [Green Version]
- Volk, D.W.; Eggan, S.M.; Horti, A.G.; Wong, D.F.; Lewis, D. Reciprocal alterations in cortical cannabinoid receptor 1 binding relative to protein immunoreactivity and transcript levels in schizophrenia. Schizophr. Res. 2014, 159, 124–129. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, N.; De Petrocellis, L.; Orlando, P.; Daniele, F.; Fezza, F.; Di Marzo, V. Endocannabinoid signalling in the blood of patients with schizophrenia. Lipids Health Dis. 2003, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Ferretjans, R.; de Campos, S.M.; Ribeiro-Santos, R.; Guimarães, F.C.; de Oliveira, K.; Cardoso, A.C.A.; Araújo, M.S.; Teixeira-Carvalho, A.; Martins-Filho, O.A.; Teixeira, A.L.; et al. Cognitive performance and peripheral endocannabinoid system receptor expression in schizophrenia. Schizophr. Res. 2014, 156, 254–260. [Google Scholar] [CrossRef]
- de Campos-Carli, S.M.; Sobreira Araujo, M.; Cardoso de Oliveira Silveira, A.; Bortolo de Rezende, V.; Pessoa Rocha, N.; Feretjans, R.; Ribeiro-Santos, R.; Texeira-Carvalho, A.; Martins-Filho, O.A.; Berk, M.; et al. Lucio-Teixeira Cannabinoid receptors on peripheral leukocytes from patients with schizophrenia: Evidence for defective immunomodulatory mechanisms. J. Psychiatry Res. 2017, 87, 44–52. [Google Scholar] [CrossRef]
- Chase, K.A.; Feiner, B.; Rosen, C.; Gavin, D.P.; Sharma, R.P. Characterization of peripheral cannabinoid receptor expression and clinical correlates in schizophrenia. Psychiatry Res. 2016, 245, 346–353. [Google Scholar] [CrossRef] [PubMed]
- D’Addario, C.; Micale, V.; Di Bartolomeo, M.; Stark, T.; Pucci, M.; Sulcova, A.; Palazzo, M.; Babinska, Z.; Cremaschi, L.; Drago, F.; et al. A preliminary study of endocannabinoid system regulation in psychosis: Distinct alterations of CNR1 promoter DNA methylation in patients with schizophrenia. Schizophr. Res. 2017, 188, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Guinart, D.; Moreno, E.; Galindo, L.; Cuenca-Royo, A.; Barrera-Conde, M.; Pérez, E.J.; Fernández-Avilés, C.; Correll, C.U.; I Canela, E.; Casadó, V.; et al. Altered Signaling in CB1R-5-HT2AR Heteromers in Olfactory Neuroepithelium Cells of Schizophrenia Patients is Modulated by Cannabis Use. Schizophr. Bull. 2020, 46, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.F.; Kuwabara, H.; Horti, A.G.; Raymont, V.; Brasic, J.; Guevara, M.; Ye, W.; Dannals, R.F.; Ravert, H.T.; Nandi, A.; et al. Quantification of cerebral cannabinoid receptors subtype 1 (CB1) in healthy subjects and schizophrenia by the novel PET radioligand [11C]OMAR. NeuroImage 2010, 52, 1505–1513. [Google Scholar] [CrossRef]
- Ceccarini, J.; De Hert, M.; Van Winkel, R.; Peuskens, J.; Bormans, G.; Kranaster, L.; Enning, F.; Koethe, D.; Leweke, F.M.; Van Laere, K. Increased ventral striatal CB1 receptor binding is related to negative symptoms in drug-free patients with schizophrenia. NeuroImage 2013, 79, 304–312. [Google Scholar] [CrossRef]
- Ranganathan, M.; Cortes-Briones, J.; Radhakrishnan, R.; Thurnauer, H.; Planeta, B.; Skosnik, P.; Gao, H.; Labaree, D.; Neumeister, A.; Pittman, B.; et al. Reduced Brain Cannabinoid Receptor Availability in Schizophrenia. Biol. Psychiatry 2016, 79, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- Mihov, Y. Positron Emission Tomography Studies on Cannabinoid Receptor Type 1 in Schizophrenia. Biol. Psychiatry 2016, 79, e97–e99. [Google Scholar] [CrossRef]
- Borgan, F.; Laurikainen, H.; Veronese, M.; Marques, T.R.; Haaparanta-Solin, M.; Solin, O.; Dahoun, T.; Rogdaki, M.; Salokangas, R.K.; Karukivi, M.; et al. In Vivo Availability of Cannabinoid 1 Receptor Levels in Patients with First-Episode Psychosis. JAMA Psychiatry 2019, 76, 1074–1084. [Google Scholar] [CrossRef] [Green Version]
- Borgan, F.; Veronese, M.; Marques, T.R.; Lythgoe, D.J.; Howes, O. Association between cannabinoid 1 receptor availability and glutamate levels in healthy controls and drug-free patients with first episode psychosis: A multi-modal PET and 1H-MRS study. Eur. Arch. Psychiatry Neurol. Sci. 2021, 271, 677–687. [Google Scholar] [CrossRef]
- Borgan, F.; O’Daly, O.; Veronese, M.; Reis Marques, T.; Laurikainen, H.; Hietala, J.; Howes, O. The neural and molecular basis of working memory function in psychosis: A multimodal PET-fMRI study. Mol. Psychiatry 2021, 26, 4464–4474. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, H.; Horiuchi, Y.; Ishikawa, M.; Koga, M.; Imai, K.; Suzuki, Y.; Morikawa, M.; Inada, T.; Watanabe, Y.; Takahashi, M.; et al. Brain Cannabinoid CB2 Receptor in Schizophrenia. Biol. Psychiatry 2010, 67, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.; He, S.; Wang, L.; Jin, L.; Si, P.; Cheng, X. Association of Single-Nucleotide Polymorphisms in the Cannabinoid Receptor 2 Gene with Schizophrenia in the Han Chinese Population. J. Mol. Neurosci. 2013, 51, 454–460. [Google Scholar] [CrossRef]
- Potvin, S.; Mahrouche, L.; Assaf, R.; Chicoine, M.; Giguère, C.-É.; Furtos, A.; Godbout, R. Peripheral Endogenous Cannabinoid Levels Are Increased in Schizophrenia Patients Evaluated in a Psychiatric Emergency Setting. Front. Psychiatry 2020, 11, 628. [Google Scholar] [CrossRef] [PubMed]
- Leweke, F.M.; Giuffrida, A.; Koethe, D.; Schreiber, D.; Nolden, B.M.; Kranaster, L.; Neatby, M.A.; Schneider, M.; Gerth, C.W.; Hellmich, M.; et al. Anandamide levels in cerebrospinal fluid of first-episode schizophrenic patients: Impact of cannabis use. Schizophr. Res. 2007, 94, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Koethe, D.; Pahlisch, F.; Hellmich, M.; Rohleder, C.; Mueller, J.K.; Meyer-Lindenberg, A.; Torrey, E.F.; Piomelli, D.; Leweke, F.M. Familial abnormalities of endocannabinoid signaling in schizophrenia. World J. Biol. Psychiatry 2018, 20, 117–125. [Google Scholar] [CrossRef]
- Potvin, S.; Kouassi, E.; Lipp, O.; Bouchard, R.-H.; Roy, M.-A.; Demers, M.-F.; Gendron, A.; Astarita, G.; Piomelli, D.; Stip, E. Endogenous cannabinoids in patients with schizophrenia and substance use disorder during quetiapine therapy. J. Psychopharmacol. 2008, 22, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Desfossés, J.; Stip, E.; Bentaleb, L.A.; Lipp, O.; Chiasson, J.-P.; Furtos, A.; Venne, K.; Kouassi, E.; Potvin, S. Plasma Endocannabinoid Alterations in Individuals with Substance Use Disorder are Dependent on the “Mirror Effect” of Schizophrenia. Front. Psychiatry 2012, 3, 85. [Google Scholar] [CrossRef] [Green Version]
- Muguruza, C.; Lehtonen, M.; Aaltonen, N.; Morentin, B.; Meana, J.J.; Callado, L.F. Quantification of endocannabinoids in postmortem brain of schizophrenic subjects. Schizophr. Res. 2013, 148, 145–150. [Google Scholar] [CrossRef]
- Joaquim, H.P.G.; Costa, A.C.; Pereira, C.A.C.; Talib, L.L.; Bilt, M.M.V.; Loch, A.A.; Gattaz, W.F. Plasmatic endocannabinoids are decreased in subjects with ultra-high risk of psychosis. Eur. J. Neurosci. 2021, 55, 1079–1087. [Google Scholar] [CrossRef]
- Morita, Y.; Ujike, H.; Tanaka, Y.; Uchida, N.; Nomura, A.; Ohtani, K.; Kishimoto, M.; Morio, A.; Imamura, T.; Sakai, A.; et al. A nonsynonymous polymorphism in the human fatty acid amide hydrolase gene did not associate with either methamphetamine dependence or schizophrenia. Neurosci. Lett. 2005, 376, 182–187. [Google Scholar] [CrossRef]
- Monteleone, P.; Milano, W.; Petrella, C.; Canestrelli, B.; Maj, M. Endocannabinoid Pro129Thr FAAH Functional Polymorphism But Not 1359G/A CNR1 Polymorphism Is Associated With Antipsychotic-Induced Weight Gain. J. Clin. Psychopharmacol. 2010, 30, 441–445. [Google Scholar] [CrossRef]
- Martínez-Magaña, J.J.; Genis-Mendoza, A.D.; González-Covarrubias, V.; Juárez-Rojop, I.E.; Tovilla-Zárate, C.A.; Soberón, X.; Lanzagorta, N.; Nicolini, H. Association of FAAH p.Pro129Thr and COMT p.Ala72Ser with schizophrenia and comorbid substance use through next-generation sequencing: An exploratory analysis. Rev. Bras. de Psiquiatr. 2021. [Google Scholar] [CrossRef] [PubMed]
- Volk, D.W.; Siegel, B.I.; Verrico, C.D.; Lewis, D.A. Endocannabinoid metabolism in the prefrontal cortex in schizophrenia. Schizophr. Res. 2013, 147, 53–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bioque, M.; Cabrera, B.; García-Bueno, B.; Mac-Dowell, K.S.; Torrent, C.; Saiz, P.A.; Parellada, M.; González-Pinto, A.; Lobo, A.; Leza, J.C.; et al. Dysregulated peripheral endocannabinoid system signaling is associated with cognitive deficits in first-episode psychosis. J. Psychiatr. Res. 2016, 75, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.B.; Zhou, X.; Geyer, M.A. Prepulse inhibition and genetic mouse models of schizophrenia. Behav. Brain Res. 2009, 204, 282–294. [Google Scholar] [CrossRef] [Green Version]
- Braff, D.L. Prepulse Inhibition of the Startle Reflex: A Window on the Brain in Schizophrenia. Behav. Neurobiol. Alcohol Addict. 2010, 4, 349–371. [Google Scholar] [CrossRef]
- Mansbach, R.S.; Rovetti, C.C.; Winston, E.N.; A Lowe, J. Effects of the cannabinoid CB1 receptor antagonist SR141716A on the behavior of pigeons and rats. Psychopharmacology 1996, 124, 315–322. [Google Scholar] [CrossRef]
- Martin, R.S.; Secchi, R.L.; Sung, E.; Lemaire, M.; Bonhaus, D.W.; Hedley, L.R.; Lowe, D.A. Effects of cannabinoid receptor ligands on psychosis-relevant behavior models in the rat. Psychopharmacology 2003, 165, 128–135. [Google Scholar] [CrossRef]
- Hajós, M.; Hoffmann, W.E.; Kocsis, B. Activation of Cannabinoid-1 Receptors Disrupts Sensory Gating and Neuronal Oscillation: Relevance to Schizophrenia. Biol. Psychiatry 2008, 63, 1075–1083. [Google Scholar] [CrossRef]
- Lee, G.; Zhou, Y. NMDAR Hypofunction Animal Models of Schizophrenia. Front. Mol. Neurosci. 2019, 12, 185. [Google Scholar] [CrossRef] [Green Version]
- Ballmaier, M.; Bortolato, M.; Rizzetti, C.; Zoli, M.; Gessa, G.; Heinz, A.; Spano, P. Cannabinoid Receptor Antagonists Counteract Sensorimotor Gating Deficits in the Phencyclidine Model of Psychosis. Neuropsychopharmacology 2007, 32, 2098–2107. [Google Scholar] [CrossRef] [PubMed]
- Guidali, C.; Viganò, D.; Petrosino, S.; Zamberletti, E.; Realini, N.; Binelli, G.; Rubino, T.; Di Marzo, V.; Parolaro, D. Cannabinoid CB1 receptor antagonism prevents neurochemical and behavioural deficits induced by chronic phencyclidine. Int. J. Neuropsychopharmacol. 2011, 14, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, M.D.; Stevens, R.J.; Rogacki, N.; Featherstone, R.E.; Senyah, Y.; Giardino, O.; Borowsky, B.; Stemmelin, J.; Cohen, C.; Pichat, P.; et al. AVE1625, a cannabinoid CB1 receptor antagonist, as a co-treatment with antipsychotics for schizophrenia: Improvement in cognitive function and reduction of antipsychotic-side effects in rodents. Psychopharmacology 2010, 215, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Kruk-Slomka, M.; Budzynska, B.; Słomka, T.; Banaszkiewicz, I.; Biala, G. The Influence of the CB1 Receptor Ligands on the Schizophrenia-Like Effects in Mice Induced by MK-801. Neurotox. Res. 2016, 30, 658–676. [Google Scholar] [CrossRef] [Green Version]
- Neary, J.L.; Perez, S.M.; Peterson, K.; Lodge, D.J.; Carless, M.A. Comparative analysis of MBD-seq and MeDIP-seq and estimation of gene expression changes in a rodent model of schizophrenia. Genomics 2017, 109, 204–213. [Google Scholar] [CrossRef]
- Perez, S.M.; Aguilar, D.D.; Neary, J.L.; A Carless, M.; Giuffrida, A.; Lodge, D.J. Schizophrenia-Like Phenotype Inherited by the F2 Generation of a Gestational Disruption Model of Schizophrenia. Neuropsychopharmacology 2015, 41, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.M.; Donegan, J.; Boley, A.M.; Aguilar, D.D.; Giuffrida, A.; Lodge, D.J. Ventral hippocampal overexpression of Cannabinoid Receptor Interacting Protein 1 (CNRIP1) produces a schizophrenia-like phenotype in the rat. Schizophr. Res. 2019, 206, 263–270. [Google Scholar] [CrossRef]
- Szűcs, E.; Dvorácskó, S.; Tömböly, C.; Büki, A.; Kékesi, G.; Horváth, G.; Benyhe, S. Decreased CB receptor binding and cannabinoid signaling in three brain regions of a rat model of schizophrenia. Neurosci. Lett. 2016, 633, 87–93. [Google Scholar] [CrossRef]
- Almeida, V.; Levin, R.; Peres, F.F.; Suiama, M.A.; Vendramini, A.M.; Santos, C.M.; Silva, N.D.; Zuardi, A.W.; Hallak, J.E.C.; Crippa, J.A.; et al. Role of the endocannabinoid and endovanilloid systems in an animal model of schizophrenia-related emotional processing/cognitive deficit. Neuropharmacology 2019, 155, 44–53. [Google Scholar] [CrossRef]
- Röpke, J.; Ferreira-Vieira, T.H.; Iglesias, L.P.; Asth, L.; Ribeiro, F.M.; Moreira, F.A. Protective role of endocannabinoid signaling in an animal model of haloperidol-induced tardive dyskinesia. Pharmacol. Biochem. Behav. 2021, 206, 173193. [Google Scholar] [CrossRef]
- Banaszkiewicz, I.; Biala, G.; Kruk-Slomka, M. Contribution of CB2 receptors in schizophrenia-related symptoms in various animal models: Short review. Neurosci. Biobehav. Rev. 2020, 114, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Cortez, I.L.; Da Silva, N.R.; Guimarães, F.S.; Gomes, F.V. Are CB2 Receptors a New Target for Schizophrenia Treatment? Front. Psychiatry 2020, 11, 587154. [Google Scholar] [CrossRef] [PubMed]
- Ortega Álvaro, A.; Aracil-Fernández, A.; García-Gutiérrez, M.S.; Navarrete, F.; Manzanares, J. Deletion of CB2 Cannabinoid Receptor Induces Schizophrenia-Related Behaviors in Mice. Neuropsychopharmacology 2011, 36, 1489–1504. [Google Scholar] [CrossRef] [Green Version]
- Khella, R.; Short, J.L.; Malone, D.T. CB2 receptor agonism reverses MK-801-induced disruptions of prepulse inhibition in mice. Psychopharmacology 2014, 231, 3071–3087. [Google Scholar] [CrossRef]
- Kruk-Slomka, M.; Banaszkiewicz, I.; Biala, G. The Impact of CB2 Receptor Ligands on the MK-801-Induced Hyperactivity in Mice. Neurotox. Res. 2017, 31, 410–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenstein, S.A.; Clapper, J.R.; Holmes, P.V.; Piomelli, D.; Hohmann, A.G. A role for 2-arachidonoylglycerol and endocannabinoid signaling in the locomotor response to novelty induced by olfactory bulbectomy. Pharmacol. Res. 2010, 61, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Karl, T. Neuregulin 1: A prime candidate for research into gene-environment interactions in schizophrenia? Insights from genetic rodent models. Front. Behav. Neurosci. 2013, 7, 106. [Google Scholar] [CrossRef] [Green Version]
- Clarke, D.; Stuart, J.; Mcgregor, I.; Arnold, J.C. Endocannabinoid dysregulation in cognitive and stress-related brain regions in the Nrg1 mouse model of schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 72, 9–15. [Google Scholar] [CrossRef]
- Baxter, A.J.; Brugha, T.S.; Erskine, H.; Scheurer, R.W.; Vos, T.; Scott, J. The epidemiology and global burden of autism spectrum disorders. Psychol. Med. 2015, 45, 601–613. [Google Scholar] [CrossRef]
- Hill, A.P.; Zuckerman, K.E.; Hagen, A.D.; Kriz, D.J.; Duvall, S.W.; van Santen, J.; Nigg, J.; Fair, D.; Fombonne, E. Aggressive behavior problems in children with autism spectrum disorders: Prevalence and correlates in a large clinical sample. Res. Autism Spectr. Disord. 2014, 8, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Steenfeldt-Kristensen, C.; Jones, C.A.; Richards, C. The Prevalence of Self-injurious Behaviour in Autism: A Meta-analytic Study. J. Autism Dev. Disord. 2020, 50, 3857–3873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2016. Morb. Mortal. Wkly. Rep. Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef]
- Lai, M.-C.; Kassee, C.; Besney, R.; Bonato, S.; Hull, L.; Mandy, W.; Szatmari, P.; Ameis, S.H. Prevalence of co-occurring mental health diagnoses in the autism population: A systematic review and meta-analysis. Lancet Psychiatry 2019, 6, 819–829. [Google Scholar] [CrossRef]
- Muskens, J.B.; Velders, F.P.; Staal, W.G. Medical comorbidities in children and adolescents with autism spectrum disorders and attention deficit hyperactivity disorders: A systematic review. Eur. Child Adolesc. Psychiatry 2017, 26, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Geng, H.; Liu, W.; Zhang, G. Prenatal, perinatal, and postnatal factors associated with autism: A meta-analysis. Medicine 2017, 96, e6696. [Google Scholar] [CrossRef]
- Kim, J.Y.; Son, M.J.; Son, C.Y.; Radua, J.; Eisenhut, M.; Gressier, F.; Koyanagi, A.; Carvalho, A.F.; Stubbs, B.; Solmi, M.; et al. Environmental risk factors and biomarkers for autism spectrum disorder: An umbrella review of the evidence. Lancet Psychiatry 2019, 6, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Marotta, R.; Risoleo, M.C.; Messina, G.; Parisi, L.; Carotenuto, M.; Vetri, L.; Roccella, M. The Neurochemistry of Autism. Brain Sci. 2020, 10, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, C.; Anacker, A.; Veenstra-VanderWeele, J. The serotonin system in autism spectrum disorder: From biomarker to animal models. Neuroscience 2016, 321, 24–41. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Lee, J.; Kim, E. Excitation/inhibition imbalance in animal models of autism spectrum disorders. Biol. Psychiatry 2017, 81, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Zamberletti, E.; Gabaglio, M.; Parolaro, D. The Endocannabinoid System and Autism Spectrum Disorders: Insights from Animal Models. Int. J. Mol. Sci. 2017, 18, 1916. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.; Siniscalco, D. Endocannabinoid system involvement in autism spectrum disorder: An overview with potential therapeutic applications. AIMS Mol. Sci. 2019, 6, 27–37. [Google Scholar] [CrossRef]
- Purcell, A.E.; Jeon, O.H.; Zimmerman, A.W.; Blue, M.E.; Pevsner, J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 2001, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, M.; Liu, Y.; Xie, S.; Wang, L.; Li, D.; Li, L.; Wang, F.; Zhang, Y.; Xia, W.; Sun, C.; et al. Alterations of the endocannabinoid system and its therapeutic potential in autism spectrum disorder. Open Biol. 2021, 11, 200306. [Google Scholar] [CrossRef]
- Siniscalco, D.; Sapone, A.; Giordano, C.; Cirillo, A.; de Magistris, L.; Rossi, F.; Fasano, A.; Bradstreet, J.J.; Maione, S.; Antonucci, N. Cannabinoid Receptor Type 2, but not Type 1, is Up-Regulated in Peripheral Blood Mononuclear Cells of Children Affected by Autistic Disorders. J. Autism Dev. Disord. 2013, 43, 2686–2695. [Google Scholar] [CrossRef]
- Aran, A.; Eylon, M.; Harel, M.; Polianski, L.; Nemirovski, A.; Tepper, S.; Schnapp, A.; Cassuto, H.; Wattad, N.; Tam, J. Lower circulating endocannabinoid levels in children with autism spectrum disorder. Mol. Autism 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Karhson, D.S.; Krasinska, K.M.; Dallaire, J.A.; Libove, R.A.; Phillips, J.M.; Chien, A.S.; Garner, J.P.; Hardan, A.Y.; Parker, K.J. Plasma anandamide concentrations are lower in children with autism spectrum disorder. Mol. Autism 2018, 9, 1–6. [Google Scholar] [CrossRef]
- Kerr, D.; Downey, L.; Conboy, M.; Finn, D.; Roche, M. Alterations in the endocannabinoid system in the rat valproic acid model of autism. Behav. Brain Res. 2013, 249, 124–132. [Google Scholar] [CrossRef]
- Wu, H.-F.; Lu, T.-Y.; Chu, M.-C.; Chen, P.S.; Lee, C.-W.; Lin, H.-C. Targeting the inhibition of fatty acid amide hydrolase ameliorate the endocannabinoid-mediated synaptic dysfunction in a valproic acid-induced rat model of Autism. Neuropharmacology 2020, 162, 107736. [Google Scholar] [CrossRef]
- Kerr, D.M.; Gilmartin, A.; Roche, M. Pharmacological inhibition of fatty acid amide hydrolase attenuates social behavioural deficits in male rats prenatally exposed to valproic acid. Pharmacol. Res. 2016, 113, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Hosie, S.; Malone, D.T.; Liu, S.; Glass, M.; Adlard, P.A.; Hannan, A.; Hill-Yardin, E.L. Altered Amygdala Excitation and CB1 Receptor Modulation of Aggressive Behavior in the Neuroligin-3R451C Mouse Model of Autism. Front. Cell. Neurosci. 2018, 12, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martella, G.; Meringolo, M.; Trobiani, L.; De Jaco, A.; Pisani, A.; Bonsi, P. The neurobiological bases of autism spectrum disorders: The R451C-neuroligin 3 mutation hampers the expression of long-term synaptic depression in the dorsal striatum. Eur. J. Neurosci. 2017, 47, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, A.; Eubig, P.; Schantz, S.L. Attention Deficit/Hyperactivity Disorder: A Focused Overview for Children’s Environmental Health Researchers. Environ. Health Perspect. 2010, 118, 1646–1653. [Google Scholar] [CrossRef] [Green Version]
- Banaschewski, T.; Becker, K.; Dofner, M.; HoLtman, M.; Rosler, M.; Romanos, M. Attention-Deficit/Hyperactivity Disorder. Dtsch. Arztebl. Int. 2017, 114, 149–159. [Google Scholar]
- Brown, R.T.; Freeman, W.S.; Perrin, J.M.; Stein, M.T.; Amler, R.W.; Feldman, H.M.; Pierce, K.; Wolraich, M.L. Prevalence and Assessment of Attention-Deficit/Hyperactivity Disorder in Primary Care Settings. Pediatrics 2001, 107, e43. [Google Scholar] [CrossRef] [Green Version]
- Faraone, S.V.; Biederman, J. What Is the Prevalence of Adult ADHD? Results of a Population Screen of 966 Adults. J. Atten. Disord. 2005, 9, 384–391. [Google Scholar] [CrossRef]
- Kessler, R.C.; Adler, L.; Barkley, R.; Biederman, J.; Conners, C.K.; Demler, O.; Faraone, S.V.; Greenhill, L.L.; Howes, M.J.; Secnik, K.; et al. The Prevalence and Correlates of Adult ADHD in the United States: Results From the National Comorbidity Survey Replication. Am. J. Psychiatry 2006, 163, 716–723. [Google Scholar] [CrossRef]
- Luo, Y.; Weibman, D.; Halperin, J.M.; Li, X. A Review of Heterogeneity in Attention Deficit/Hyperactivity Disorder (ADHD). Front. Hum. Neurosci. 2019, 13, 42. [Google Scholar] [CrossRef] [Green Version]
- Polanczyk, G. The Worldwide Prevalence of ADHD: A Systematic Review and Metaregression Analysis. Am. J. Psychiatry 2007, 164, 942. [Google Scholar] [CrossRef]
- Simon, V.; Czobor, P.; Bálint, S.; Mészáros, Á.; Bitter, I. Prevalence and correlates of adult attention-deficit hyperactivity disorder: Meta-analysis. Br. J. Psychiatry 2009, 194, 204–211. [Google Scholar] [CrossRef]
- Thapar, A.; Cooper, M. Attention deficit hyperactivity disorder. Lancet 2016, 387, 1240–1250. [Google Scholar] [CrossRef]
- Nikolas, M.A.; Burt, S.A. Genetic and environmental influences on ADHD symptom dimensions of inattention and hyperactivity: A meta-analysis. J. Abnorm. Psychol. 2010, 119, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Thapar, A.; Cooper, M.; Eyre, O.; Langley, K. Practitioner Review: What have we learnt about the causes of ADHD? J. Child Psychol. Psychiatry 2012, 54, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, J.M.; Kinsbourne, M.; Nigg, J.; Lanphear, B.; Stefanatos, G.A.; Volkow, N.; Taylor, E.; Casey, B.J.; Castellanos, F.; Wadhwa, P.D. Etiologic Subtypes of Attention-Deficit/Hyperactivity Disorder: Brain Imaging, Molecular Genetic and Environmental Factors and the Dopamine Hypothesis. Neuropsychol. Rev. 2007, 17, 39–59. [Google Scholar] [CrossRef]
- Brennan, A.R.; Arnsten, A.F. Neuronal mechanisms underlying attention deficit hyperactivity disorder: The influence of arousal on prefrontal cortical function. Ann. N. Y. Acad. Sci. 2008, 1129, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Ponce, G.; Hoenicka, J.; Rubio, G.; Ampuero, I.; A Jiménez-Arriero, M.; Rodríguez-Jiménez, R.; Palomo, T.; A Ramos, J. Association between cannabinoid receptor gene (CNR1) and childhood attention deficit/hyperactivity disorder in Spanish male alcoholic patients. Mol. Psychiatry 2003, 8, 466–467. [Google Scholar] [CrossRef]
- Ehlers, C.L.; Slutske, W.S.; Lind, P.A.; Wilhelmsen, K.C. Association Between Single Nucleotide Polymorphisms in the Cannabinoid Receptor Gene (CNR1) and Impulsivity in Southwest California Indians. Twin Res. Hum. Genet. 2007, 10, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Buchmann, A.F.; Hohm, E.; Witt, S.H.; Blomeyer, D.; Jennen-Steinmetz, C.; Schmidt, M.H.; Esser, G.; Banaschewski, T.; Brandeis, D.; Laucht, M. Role of CNR1 polymorphisms in moderating the effects of psychosocial adversity on impulsivity in adolescents. J. Neural Transm. 2015, 122, 455–463. [Google Scholar] [CrossRef]
- Ahmadalipour, A.; Fanid, L.M.; Zeinalzadeh, N.; Alizadeh, M.; Vaezi, H.; Aydinlou, Z.H.; Noorazar, S.G. The first evidence of an association between a polymorphism in the endocannabinoid-degrading enzyme FAAH (FAAH rs2295633) with attention deficit hyperactivity disorder. Genomics 2020, 112, 1330–1334. [Google Scholar] [CrossRef]
- Centonze, D.; Bari, M.; Di Michele, B.; Rossi, S.; Gasperi, V.; Pasini, A.; Battista, N.; Bernardi, G.; Curatolo, P.; Maccarrone, M. Altered anandamide degradation in attention-deficit/hyperactivity disorder. Neurology 2009, 72, 1526–1527. [Google Scholar] [CrossRef]
- Brunkhorst-Kanaan, N.; Trautmann, S.; Schreiber, Y.; Thomas, D.; Kittel-Schneider, S.; Gurke, R.; Geisslinger, G.; Reif, A.; Tegeder, I. Sphingolipid and Endocannabinoid Profiles in Adult Attention Deficit Hyperactivity Disorder. Biomedicines 2021, 9, 1173. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.E.; Williams, E.; Seegobin, S.; Tye, C.; Kuntsi, J.; Asherson, P. Cannabinoids in attention-deficit/hyperactivity disorder: A randomised-controlled trial. Eur. Neuropsychopharmacol. 2017, 27, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Adriani, W.; Laviola, G. Windows of vulnerability to psychopathology and therapeutic strategy in the adolescent rodent model. Behav. Pharmacol. 2004, 15, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Tomizawa, M.; Suzuki, K.; Shirakawa, Y.; Ono, H.; Adachi, K.; Suzuki, H.; Shimomura, K.; Nabeshima, T.; Kamijima, M. Organophosphate Agent Induces ADHD-Like Behaviors via Inhibition of Brain Endocannabinoid-Hydrolyzing Enzyme(s) in Adolescent Male Rats. J. Agric. Food Chem. 2020, 68, 2547–2553. [Google Scholar] [CrossRef] [PubMed]
- Pattij, T.; Janssen, M.C.W.; Schepers, I.; González-Cuevas, G.; de Vries, T.J.; Schoffelmeer, A.N.M. Effects of the cannabinoid CB1 receptor antagonist rimonabant on distinct measures of impulsive behavior in rats. Psychopharmacology 2007, 193, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Leffa, D.T.; Ferreira, S.G.; Machado, N.J.; Souza, C.M.; da Rosa, F.; de Carvalho, C.; Kincheski, G.C.; Takahashi, R.N.; Porciúncula, L.O.; Souza, D.O.; et al. Caffeine and cannabinoid receptors modulate impulsive behavior in an animal model of attentional deficit and hyperactivity disorder. Eur. J. Neurosci. 2019, 49, 1673–1683. [Google Scholar] [CrossRef]
- Beltramo, M.; de Fonseca, F.R.; Navarro, M.; Calignano, A.; Gorriti, M.A.; Grammatikopoulos, G.; Sadile, A.G.; Giuffrida, A.; Piomelli, D. Reversal of Dopamine D2 Receptor Responses by an Anandamide Transport Inhibitor. J. Neurosci. 2000, 20, 3401–3407. [Google Scholar] [CrossRef] [Green Version]
- Castelli, M.; Federici, M.; Rossi, S.; de Chiara, V.; Napolitano, F.; Studer, V.; Motta, C.; Sacchetti, L.; Romano, R.; Musella, A.; et al. Loss of striatal cannabinoid CB1 receptor function in attention-deficit / hyperactivity disorder mice with point-mutation of the dopamine transporter. Eur. J. Neurosci. 2011, 34, 1369–1377. [Google Scholar] [CrossRef]
- Bracci, E. The endocannabinoid system misfires in ADHD mice (Commentary on Castelli et al.). Eur. J. Neurosci. 2011, 34, 1368. [Google Scholar] [CrossRef]
- Rossi, M.A.; Stuber, G.D. Overlapping Brain Circuits for Homeostatic and Hedonic Feeding. Cell Metab. 2018, 27, 42–56. [Google Scholar] [CrossRef]
- Di Marzo, V.; Petrosino, S. Endocannabinoids and the regulation of their levels in health and disease. Curr. Opin. Lipidol. 2007, 18, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Milano, W.; Cauli, O. Changes in the Peripheral Endocannabinoid System as a Risk Factor for the Development of Eating Disorders. Endocr. Metab. Immune Disord.—Drug Targets 2018, 18, 325–332. [Google Scholar] [CrossRef]
- Keski-Rahkonen, A.; Mustelin, L. Epidemiology of eating disorders in Europe: Prevalence, incidence, comorbidity, course, consequences, and risk factors. Curr. Opin. Psychiatry 2016, 29, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, P.; Maj, M. Dysfunctions of leptin, ghrelin, BDNF and endocannabinoids in eating disorders: Beyond the homeostatic control of food intake. Psychoneuroendocrinology 2013, 38, 312–330. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, Z.; Kanyas, K.; Latzer, Y.; Karni, O.; Bloch, M.; Lerer, B.; Berry, E. Association study of cannabinoid receptor gene (CNR1) alleles and anorexia nervosa: Differences between restricting and bingeing/purging subtypes. Am. J. Med Genet. 2004, 125B, 126–130. [Google Scholar] [CrossRef]
- Müller, T.D.; Reichwald, K.; Brönner, G.; Kirschner, J.; Nguyen, T.T.; Scherag, A.; Herzog, W.; Herpertz-Dahlmann, B.; Lichtner, P.; Meitinger, T.; et al. Lack of association of genetic variants in genes of the endocannabinoid system with anorexia nervosa. Child Adolesc. Psychiatry Ment. Health 2008, 2, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Paolacci, S.; Kiani, A.K.; Manara, E.; Beccari, T.; Ceccarini, M.R.; Stuppia, L.; Chiurazzi, P.; Ragione, L.D.; Bertelli, M. Genetic contributions to the etiology of anorexia nervosa: New perspectives in molecular diagnosis and treatment. Mol. Genet. Genom. Med. 2020, 8, e1244. [Google Scholar] [CrossRef]
- Monteleone, P.; Bifulco, M.; Di Fillipo, C.; Gazzerro, P.; Monteleone, F.; Proto, M.C.; Di Genio, M.; Grimaldi, M.; Maj, M. Association of CNR1 and FAAH endocannabinoid gene polymorphisms with anorexia nervosa and bulimia nervosa: Evidence for synergistic effects. Genes Brain Behav. 2009, 8, 728–732. [Google Scholar] [CrossRef]
- González, L.M.; García-Herráiz, A.; Mota-Zamorano, S.; Flores, I.; Albuquerque, D.; Gervasini, G. Variability in cannabinoid receptor genes is associated with psychiatric comorbidities in anorexia nervosa. Eat. Weight Disord.—Stud. Anorex. Bulim. Obes. 2021, 26, 2597–2606. [Google Scholar] [CrossRef]
- Frieling, H.; Albrecht, H.; Jedtberg, S.; Gozner, A.; Lenz, B.; Wilhelm, J.; Hillemacher, T.; de Zwaan, M.; Kornhuber, J.; Bleich, S. Elevated cannabinoid 1 receptor mRNA is linked to eating disorder related behavior and attitudes in females with eating disorders. Psychoneuroendocrinology 2009, 34, 620–624. [Google Scholar] [CrossRef]
- Schroeder, M.; Eberlein, C.; De Zwaan, M.; Kornhuber, J.; Bleich, S.; Frieling, H. Lower levels of cannabinoid 1 receptor mRNA in female eating disorder patients: Association with wrist cutting as impulsive self-injurious behavior. Psychoneuroendocrinology 2012, 37, 2032–2036. [Google Scholar] [CrossRef] [PubMed]
- Gérard, N.; Pieters, G.; Goffin, K.; Bormans, G.; Van Laere, K. Brain Type 1 Cannabinoid Receptor Availability in Patients with Anorexia and Bulimia Nervosa. Biol. Psychiatry 2011, 70, 777–784. [Google Scholar] [CrossRef] [Green Version]
- Ceccarini, J.; Weltens, N.; Ly, H.G.; Tack, J.; Van Oudenhove, L.; Van Laere, K. Association between cerebral cannabinoid 1 receptor availability and body mass index in patients with food intake disorders and healthy subjects: A [(18)F]MK-9470 PET study. Transl. Psychiatry 2016, 6, e853. [Google Scholar] [CrossRef] [Green Version]
- Monteleone, A.M.; Di Marzo, V.; Aveta, T.; Piscitelli, F.; Grave, R.D.; Scognamiglio, P.; El Ghoch, M.; Calugi, S.; Monteleone, P.; Maj, M. Deranged endocannabinoid responses to hedonic eating in underweight and recently weight-restored patients with anorexia nervosa. Am. J. Clin. Nutr. 2014, 101, 262–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccolo, M.; Claussen, M.C.; Bluemel, S.; Schumacher, S.; Cronin, A.; Fried, M.; Goetze, O.; Martin-Soelch, C.; Milos, G. Altered circulating endocannabinoids in anorexia nervosa during acute and weight-restored phases: A pilot study. Eur. Eat. Disord. Rev. 2019, 28, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, A.M.; Piscitelli, F.; Grave, R.D.; El Ghoch, M.; Di Marzo, V.; Maj, M.; Monteleone, P. Peripheral Endocannabinoid Responses to Hedonic Eating in Binge-Eating Disorder. Nutrients 2017, 9, 1377. [Google Scholar] [CrossRef] [Green Version]
- Yagin, N.L.; Aliasgari, F.; Alizadeh, M.; Aliasgharzadeh, S.; Mahdavi, R. Comparison of endocannabinoids levels, FAAH gene polymorphisms, and appetite regulatory substances in women with and without binge eating disorder: A cross- sectional study. Nutr. Res. 2020, 83, 86–93. [Google Scholar] [CrossRef]
- Monteleone, P.; Matias, I.; Martiadis, V.; De Petrocellis, L.; Maj, M.; Di Marzo, V. Blood Levels of the Endocannabinoid Anandamide are Increased in Anorexia Nervosa and in Binge-Eating Disorder, but not in Bulimia Nervosa. Neuropsychopharmacology 2005, 30, 1216–1221. [Google Scholar] [CrossRef] [Green Version]
- Sipe, J.C.; Waalen, J.; Gerber, A.; Beutler, E. Overweight and obesity associated with a missense polymorphism in fatty acid amide hydrolase (FAAH). Int. J. Obes. 2005, 29, 755–759. [Google Scholar] [CrossRef] [Green Version]
- Monteleone, P.; Tortorella, A.; Martiadis, V.; Di Filippo, C.; Canestrelli, B.; Maj, M. The cDNA 385C to A missense polymorphism of the endocannabinoid degrading enzyme fatty acid amide hydrolase (FAAH) is associated with overweight/obesity but not with binge eating disorder in overweight/obese women. Psychoneuroendocrinology 2008, 33, 546–550. [Google Scholar] [CrossRef]
- Bulik, C.M.; Yilmaz, Z.; Hardaway, J.A. Genetics and epigenetics of eating disorders. Adv. Genom. Genet. 2015, 5, 131–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casteels, C.; Gérard, N.; van Kuyck, K.; Pottel, L.; Nuttin, B.; Bormans, G.; Van Laere, K. Small animal PET imaging of the type 1 cannabinoid receptor in a rodent model for anorexia nervosa. Eur. J. Pediatr. 2014, 41, 308–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collu, R.; Scherma, M.; Piscitelli, F.; Giunti, E.; Satta, V.; Castelli, M.P.; Verde, R.; Fratta, W.; Bisogno, T.; Fadda, P. Impaired brain endocannabinoid tone in the activity-based model of anorexia nervosa. Int. J. Eat. Disord. 2019, 52, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Gandia, M.; Aracil-Fernández, A.; Romero, S.M.; Aguilar, M.; Manzanares, J.; Miñarro, J.; Rodríguez-Arias, M. Changes in gene expression and sensitivity of cocaine reward produced by a continuous fat diet. Psychopharmacology 2017, 234, 2337–2352. [Google Scholar] [CrossRef]
- Satta, V.; Scherma, M.; Piscitelli, F.; Usai, P.; Castelli, M.P.; Bisogno, T.; Fratta, W.; Fadda, P. Limited Access to a High Fat Diet Alters Endocannabinoid Tone in Female Rats. Front. Neurosci. 2018, 12, 40. [Google Scholar] [CrossRef]
- Berland, C.; Castel, J.; Terrasi, R.; Montalban, E.; Foppen, E.; Martin, C.; Muccioli, G.G.; Luquet, S.; Gangarossa, G. Identification of an endocannabinoid gut-brain vagal mechanism controlling food reward and energy homeostasis. Mol. Psychiatry 2022, 1–15. [Google Scholar] [CrossRef]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773. [Google Scholar] [CrossRef]
- Galaj, E.; Xi, Z.-X. Potential of Cannabinoid Receptor Ligands as Treatment for Substance Use Disorders. CNS Drugs 2019, 33, 1001–1030. [Google Scholar] [CrossRef]
- Jansma, J.M.; Van Hell, H.H.; Vanderschuren, L.; Bossong, M.G.; Jager, G.; Kahn, R.S.; Ramsey, N.F. THC reduces the anticipatory nucleus accumbens response to reward in subjects with a nicotine addiction. Transl. Psychiatry 2013, 3, e234. [Google Scholar] [CrossRef] [Green Version]
- Hirvonen, J.; Zanotti-Fregonara, P.; Gorelick, D.A.; Lyoo, C.H.; Rallis-Frutos, D.; Morse, C.; Zoghbi, S.S.; Pike, V.W.; Volkow, N.D.; Huestis, M.A.; et al. Decreased Cannabinoid CB1 Receptors in Male Tobacco Smokers Examined With Positron Emission Tomography. Biol. Psychiatry 2018, 84, 715–721. [Google Scholar] [CrossRef]
- Evans, D.E.; Sutton, S.; Jentink, K.G.; Lin, H.-Y.; Park, J.Y.; Drobes, D.J. Cannabinoid receptor 1 (CNR1) gene variant moderates neural index of cognitive disruption during nicotine withdrawal. Genes Brain Behav. 2016, 15, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherma, M.; Muntoni, A.L.; Melis, M.; Fattore, L.; Fadda, P.; Fratta, W.; Pistis, M. Interactions between the endocannabinoid and nicotinic cholinergic systems: Preclinical evidence and therapeutic perspectives. Psychopharmacology 2016, 233, 1765–1777. [Google Scholar] [CrossRef] [PubMed]
- Saravia, R.; Ten-Blanco, M.; Pereda-Pérez, I.; Berrendero, F. New Insights in the Involvement of the Endocannabinoid System and Natural Cannabinoids in Nicotine Dependence. Int. J. Mol. Sci. 2021, 22, 13316. [Google Scholar] [CrossRef]
- Castañé, A.; Valjent, E.; Ledent, C.; Parmentier, M.; Maldonado, R.; Valverde, O. Lack of CB1 cannabinoid receptors modifies nicotine behavioural responses, but not nicotine abstinence. Neuropharmacology 2002, 43, 857–867. [Google Scholar] [CrossRef]
- Merritt, L.L.; Martin, B.R.; Walters, C.; Lichtman, A.H.; Damaj, M.I. The Endogenous Cannabinoid System Modulates Nicotine Reward and Dependence. J. Pharmacol. Exp. Ther. 2008, 326, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Muldoon, P.P.; Akinola, L.S.; Schlosburg, J.E.; Lichtman, A.H.; Sim-Selley, L.J.; Mahadevan, A.; Cravatt, B.F.; Damaj, M.I. Inhibition of monoacylglycerol lipase reduces nicotine reward in the conditioned place preference test in male mice. Neuropharmacology 2020, 176, 108170. [Google Scholar] [CrossRef]
- Navarrete, F.; Rodriguez-Arias, M.; Martin-García, E.; Navarro, D.; García-Gutiérrez, M.S.; Aguilar, M.A.; Aracil-Fernández, A.; Berbel, P.; Miñarro, J.; Maldonado, R.; et al. Role of CB2 Cannabinoid Receptors in the Rewarding, Reinforcing, and Physical Effects of Nicotine. Neuropsychopharmacology 2013, 38, 2515–2524. [Google Scholar] [CrossRef] [Green Version]
- Ignatowska-Jankowska, B.M.; Muldoon, P.P.; Lichtman, A.H.; Damaj, M.I. The cannabinoid CB2 receptor is necessary for nicotine-conditioned place preference, but not other behavioral effects of nicotine in mice. Psychopharmacology 2013, 229, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Canseco-Alba, A.; Schanz, N.; Sanabria, B.; Zhao, J.; Lin, Z.; Liu, Q.-R.; Onaivi, E.S. Behavioral effects of psychostimulants in mutant mice with cell-type specific deletion of CB2 cannabinoid receptors in dopamine neurons. Behav. Brain Res. 2019, 360, 286–297. [Google Scholar] [CrossRef]
- Neumeister, A.; Normandin, M.D.; Murrough, J.W.; Henry, S.; Bailey, C.R.; Luckenbaugh, D.A.; Tuit, K.; Zheng, M.-Q.; Galatzer-Levy, I.R.; Sinha, R.; et al. Positron Emission Tomography Shows Elevated Cannabinoid CB1 Receptor Binding in Men with Alcohol Dependence. Alcohol. Clin. Exp. Res. 2012, 36, 2104–2109. [Google Scholar] [CrossRef] [Green Version]
- Ceccarini, J.; Hompes, T.; Verhaeghen, A.; Casteels, C.; Peuskens, H.; Bormans, G.; Claes, S.; Van Laere, K. Changes in Cerebral CB1 Receptor Availability after Acute and Chronic Alcohol Abuse and Monitored Abstinence. J. Neurosci. 2014, 34, 2822–2831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcos, M.; Pastor, I.; de la Calle, C.; Barrio-Real, L.; Laso, F.-J.; González-Sarmiento, R. Cannabinoid Receptor 1 Gene is Associated with Alcohol Dependence. Alcohol. Clin. Exp. Res. 2011, 36, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.G.; Samochowiec, J.; Finckh, U.; Fiszer-Piosik, E.; Horodnicki, J.; Wendel, B.; Rommelspacher, H.; Hoehe, M.R. Association of a CB1 Cannabinoid Receptor Gene (CNR1) polymorphism with severe alcohol dependence. Drug Alcohol Depend. 2002, 65, 221–224. [Google Scholar] [CrossRef]
- Wildenberg, E.V.D.; Janssen, R.G.J.H.; Hutchison, K.E.; van Breukelen, G.J.P.; Wiers, R.W. Polymorphisms of the dopamine D4 receptor gene (DRD4 VNTR) and cannabinoid CB1 receptor gene (CNR1) are not strongly related to cue-reactivity after alcohol exposure. Addict. Biol. 2007, 12, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Hirvonen, J.; Zanotti-Fregonara, P.; Umhau, J.C.; George, D.T.; Rallis-Frutos, D.; Lyoo, C.H.; Li, C.-T.; Hines, C.S.; Sun, H.; Terry, G.E.; et al. Reduced cannabinoid CB1 receptor binding in alcohol dependence measured with positron emission tomography. Mol. Psychiatry 2012, 18, 916–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soundararajan, S.; Kazmi, N.; Brooks, A.T.; Krumlauf, M.; Schwandt, M.L.; George, D.T.; Hodgkinson, C.A.; Wallen, G.R.; Ramchandani, V.A. FAAH and CNR1 Polymorphisms in the Endocannabinoid System and Alcohol-Related Sleep Quality. Front. Psychiatry 2021, 12, 712178. [Google Scholar] [CrossRef]
- Pabalan, N.; Chaweeborisuit, P.; Tharabenjasin, P.; Tasanarong, A.; Jarjanazi, H.; Eiamsitrakoon, T.; Tapanadechopone, P. Associations of CB1 cannabinoid receptor (CNR1) gene polymorphisms with risk for alcohol dependence: Evidence from meta-analyses of genetic and genome-wide association studies. Medicine 2021, 100, e27343. [Google Scholar] [CrossRef]
- Ishiguro, H.; Iwasaki, S.; Teasenfitz, L.; Higuchi, S.; Horiuchi, Y.; Saito, T.; Arinami, T.; Onaivi, E.S. Involvement of cannabinoid CB2 receptor in alcohol preference in mice and alcoholism in humans. Pharm. J. 2006, 7, 380–385. [Google Scholar] [CrossRef] [Green Version]
- Kärkkäinen, O.; Lehtonen, M.; Laukkanen, V.; Tupala, E.; Hyytiä, P.; Kautiainen, H.; Tiihonen, J.; Callaway, J.; Storvik, M. Endogenous cannabinoids in amygdala and hippocampus in post-mortem brains of Cloninger type 1 and 2 alcoholics. Alcohol 2013, 47, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Erdozain, A.M.; Rubio, M.; Valdizán, E.M.; Pazos, A.; Meana, J.J.; Fernández-Ruiz, J.; Alexander, S.P.H.; Callado, L.F. The endocannabinoid system is altered in the post-mortem prefrontal cortex of alcoholic subjects. Addict. Biol. 2014, 20, 773–783. [Google Scholar] [CrossRef] [Green Version]
- Hungund, B.L.; Szakall, I.; Adam, A.; Basavarajappa, B.; Vadasz, C. Cannabinoid CB1 receptor knockout mice exhibit markedly reduced voluntary alcohol consumption and lack alcohol-induced dopamine release in the nucleus accumbens. J. Neurochem. 2003, 84, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Houchi, H.; Babovic, D.; Pierrefiche, O.; Ledent, C.; Daoust, M.; Naassila, M. CB1 Receptor Knockout Mice Display Reduced Ethanol-Induced Conditioned Place Preference and Increased Striatal Dopamine D2 Receptors. Neuropsychopharmacology 2004, 30, 339–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naassila, M.; Pierrefiche, O.; Ledent, C.; Daoust, M. Decreased alcohol self-administration and increased alcohol sensitivity and withdrawal in CB1 receptor knockout mice. Neuropharmacology 2004, 46, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Álvaro, A.; Ternianov, A.; Aracil-Fernández, A.; Navarrete, F.; García-Gutiérrez, M.S.; Manzanares, J. Role of cannabinoid CB2receptor in the reinforcing actions of ethanol. Addict. Biol. 2015, 20, 43–55. [Google Scholar] [CrossRef] [PubMed]
- A Blednov, Y.; Cravatt, B.F.; Boehm, S.L.; Walker, D.; Harris, R.A. Role of Endocannabinoids in Alcohol Consumption and Intoxication: Studies of Mice Lacking Fatty Acid Amide Hydrolase. Neuropsychopharmacology 2006, 32, 1570–1582. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Yalamanchili, R.; Cravatt, B.F.; Cooper, T.B.; Hungund, B.L. Increased ethanol consumption and preference and decreased ethanol sensitivity in female FAAH knockout mice. Neuropharmacology 2006, 50, 834–844. [Google Scholar] [CrossRef]
- Serrano, A.; Rivera, P.; Pavon, F.J.; Decara, J.; Suárez, J.; de Fonseca, F.R.; Parsons, L.H. Differential Effects of Single Versus Repeated Alcohol Withdrawal on the Expression of Endocannabinoid System-Related Genes in the Rat Amygdala. Alcohol. Clin. Exp. Res. 2012, 36, 984–994. [Google Scholar] [CrossRef] [Green Version]
- Marín, L.S.; Pavon, F.J.; Decara, J.; Suarez, J.; Gavito, A.; Castilla-Ortega, E.; De Fonseca, F.R.; Serrano, A. Effects of Intermittent Alcohol Exposure on Emotion and Cognition: A Potential Role for the Endogenous Cannabinoid System and Neuroinflammation. Front. Behav. Neurosci. 2017, 11, 15. [Google Scholar] [CrossRef] [Green Version]
- Hirvonen, J.; Goodwin, R.S.; Li, C.-T.; Terry, G.E.; Zoghbi, S.S.; Morse, C.; Pike, V.W.; Volkow, N.D.; Huestis, M.A.; Innis, R.B. Reversible and regionally selective downregulation of brain cannabinoid CB1 receptors in chronic daily cannabis smokers. Mol. Psychiatry 2012, 17, 642–649. [Google Scholar] [CrossRef] [Green Version]
- Ceccarini, J.; Kuepper, R.; Kemels, D.; van Os, J.; Henquet, C.; Van Laere, K. [18F]MK-9470 PET measurement of cannabinoid CB1receptor availability in chronic cannabis users. Addict. Biol. 2015, 20, 357–367. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, D.; Cortes-Briones, J.; Ranganathan, M.; Thurnauer, H.; Creatura, G.; Surti, T.; Skosnik, P. Rapid Changes in CB1 Receptor Availability in Cannabis Dependent Males after Abstinence from Cannabis. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2016, 1, 60–67. [Google Scholar] [PubMed]
- Hopfer, C.J.; Young, S.E.; Purcell, S.; Crowley, T.J.; Stallings, M.C.; Corley, R.P.; Rhee, S.H.; Smolen, A.; Krauter, K.; Hewitt, J.K.; et al. Cannabis receptor haplotype associated with fewer cannabis dependence symptoms in adolescents. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2006, 141B, 895–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, A.; Wetherill, L.; Dick, D.M.; Xuei, X.; Hinrichs, A.; Hesselbrock, V.; Kramer, J.; Nurnberger, J.I., Jr.; Schuckit, M.; Bierut, L.J.; et al. Evidence for association between polymorphisms in the cannabinoid receptor 1 (CNR1) gene and cannabis dependence. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2009, 150B, 736–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schacht, J.P.; E Hutchison, K.; Filbey, F.M. Associations between Cannabinoid Receptor-1 (CNR1) Variation and Hippocampus and Amygdala Volumes in Heavy Cannabis Users. Neuropsychopharmacology 2012, 37, 2368–2376. [Google Scholar] [CrossRef] [Green Version]
- Haughey, H.M.; Marshall, E.; Schacht, J.P.; Louis, A.; Hutchison, K.E. Marijuana withdrawal and craving: Influence of the cannabinoid receptor 1 (CNR1) and fatty acid amide hydrolase (FAAH) genes. Addiction 2008, 103, 1678–1686. [Google Scholar] [CrossRef]
- Pehlivan, S.; Aytac, H.M.; Kurnaz, S.; Pehlivan, M.; Aydin, P.C. Evaluation of COMT (rs4680), CNR2 (rs2501432), CNR2 (rs2229579), UCP2 (rs659366), and IL-17 (rs763780) gene variants in synthetic cannabinoid use disorder patients. J. Addict. Dis. 2020, 38, 495–505. [Google Scholar] [CrossRef]
- Villares, J. Chronic use of marijuana decreases cannabinoid receptor binding and mRNA expression in the human brain. Neuroscience 2007, 145, 323–334. [Google Scholar] [CrossRef]
- Manza, P.; Yuan, K.; Shokri-Kojori, E.; Tomasi, D.; Volkow, N.D. Brain structural changes in cannabis dependence: Association with MAGL. Mol. Psychiatry 2020, 25, 3256–3266. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, S.; Kittler, J.; Kirby, M.T.; Sim, L.J.; Hampson, R.E.; Childers, S.R.; Deadwyler, S.A. Effects of long-term exposure to delta9-THC on expression of cannabinoid receptor (CB1) mRNA in different rat brain regions. Brain Res. Mol. Brain Res. 1998, 62, 141–149. [Google Scholar] [CrossRef]
- Di Marzo, V.; Berrendero, F.; Bisogno, T.; González, S.; Cavaliere, P.; Romero, J.; Cebeira, M.; Ramos, J.A.; Fernández-Ruiz, J.J. Enhancement of anandamide formation in the limbic forebrain and reduction of endocannabinoid contents in the striatum of delta9-tetrahydrocannabinol-tolerant rats. J. Neurochem. 2000, 74, 1627–1635. [Google Scholar] [CrossRef]
- Friend, L.; Weed, J.; Sandoval, P.; Nufer, T.; Ostlund, I.; Edwards, J.G. CB1-Dependent Long-Term Depression in Ventral Tegmental Area GABA Neurons: A Novel Target for Marijuana. J. Neurosci. 2017, 37, 10943–10954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, L.; Kranzler, H.R.; Luo, X.; Yang, B.-Z.; Weiss, R.; Brady, K.; Poling, J.; Farrer, L.; Gelernter, J. Interaction between Two Independent CNR1 Variants Increases Risk for Cocaine Dependence in European Americans: A Replication Study in Family-Based Sample and Population-Based Sample. Neuropsychopharmacology 2009, 34, 1504–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.M.; Nielsen, D.A.; Kosten, T.R.; De La Garza, R.; Newton, T.F.; Verrico, C.D. FAAH variant Pro129Thr modulates subjective effects produced by cocaine administration. Am. J. Addict. 2018, 27, 567–573. [Google Scholar] [CrossRef]
- Álvaro-Bartolomé, M.; García-Sevilla, J. Dysregulation of cannabinoid CB1 receptor and associated signaling networks in brains of cocaine addicts and cocaine-treated rodents. Neuroscience 2013, 247, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Mendizábal, V.; Touriño, C.; Robledo, P.; Ledent, C.; Parmentier, M.; Maldonado, R.; Valverde, O. Lack of CB1 Cannabinoid Receptor Impairs Cocaine Self-Administration. Neuropsychopharmacology 2005, 30, 1670–1680. [Google Scholar] [CrossRef]
- Martín-García, E.; Bourgoin, L.; Cathala, A.; Kasanetz, F.; Mondesir, M.; Gutiérrez-Rodríguez, A.; Reguero, L.; Fiancette, J.-F.; Grandes, P.; Spampinato, U.; et al. Differential Control of Cocaine Self-Administration by GABAergic and Glutamatergic CB1 Cannabinoid Receptors. Neuropsychopharmacology 2015, 41, 2192–2205. [Google Scholar] [CrossRef] [Green Version]
- Turner, B.D.; Smith, N.K.; Manz, K.M.; Chang, B.T.; Delpire, E.; Grueter, C.A.; Grueter, B.A. Cannabinoid type 1 receptors in A2a neurons contribute to cocaine-environment association. Psychopharmacology 2021, 238, 1121–1131. [Google Scholar] [CrossRef]
- Zapata, A.; Lupica, C.R. Lateral habenula cannabinoid CB1 receptor involvement in drug-associated impulsive behavior. Neuropharmacology 2021, 192, 108604. [Google Scholar] [CrossRef]
- Aracil-Fernández, A.; Trigo, J.M.; García-Gutiérrez, M.S.; Álvaro, A.O.; Ternianov, A.; Navarro, D.; Robledo, P.; Berbel, P.; Maldonado, R.; Manzanares, J. Decreased Cocaine Motor Sensitization and Self-Administration in Mice Overexpressing Cannabinoid CB2 Receptors. Neuropsychopharmacology 2012, 37, 1749–1763. [Google Scholar] [CrossRef] [Green Version]
- Bystrowska, B.; Frankowska, M.; Smaga, I.; Pomierny-Chamioło, L.; Filip, M. Effects of Cocaine Self-Administration and Its Extinction on the Rat Brain Cannabinoid CB1 and CB2 Receptors. Neurotox. Res. 2018, 34, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.-Y.; Zhang, M.; Cao, Y. Exposure to morphine affects the expression of endocannabinoid receptors and immune functions. J. Neuroimmunol. 2012, 247, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-W.; Ho, W.-C.; Huang, C.-L.; Wang, R.-Y. Precision therapeutic opioid dosing implications from genetic biomarkers and craving score. Medicine 2020, 99, e20429. [Google Scholar] [CrossRef] [PubMed]
- Ledent, C.; Valverde, O.; Cossu, G.; Petitet, F.; Aubert, J.-F.; Beslot, F.; Böhme, G.A.; Imperato, A.; Pedrazzini, T.; Roques, B.P.; et al. Unresponsiveness to Cannabinoids and Reduced Addictive Effects of Opiates in CB1 Receptor Knockout Mice. Science 1999, 283, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Ledent, C.; Parmentier, M.; Maldonado, R.; Valverde, O. Cocaine, but not morphine, induces conditioned place preference and sensitization to locomotor responses in CB1 knockout mice. Eur. J. Neurosci. 2000, 12, 4038–4046. [Google Scholar] [CrossRef] [PubMed]
- Iyer, V.; Slivicki, R.A.; Thomaz, A.C.; Crystal, J.D.; Mackie, K.; Hohmann, A.G. The cannabinoid CB2 receptor agonist LY2828360 synergizes with morphine to suppress neuropathic nociception and attenuates morphine reward and physical dependence. Eur. J. Pharmacol. 2020, 886, 173544. [Google Scholar] [CrossRef] [PubMed]
- Naudon, L.; Piscitelli, F.; Giros, B.; Di Marzo, V.; Daugé, V. Possible involvement of endocannabinoids in the increase of morphine consumption in maternally deprived rat. Neuropharmacology 2013, 65, 193–199. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navarro, D.; Gasparyan, A.; Navarrete, F.; Torregrosa, A.B.; Rubio, G.; Marín-Mayor, M.; Acosta, G.B.; Garcia-Gutiérrez, M.S.; Manzanares, J. Molecular Alterations of the Endocannabinoid System in Psychiatric Disorders. Int. J. Mol. Sci. 2022, 23, 4764. https://doi.org/10.3390/ijms23094764
Navarro D, Gasparyan A, Navarrete F, Torregrosa AB, Rubio G, Marín-Mayor M, Acosta GB, Garcia-Gutiérrez MS, Manzanares J. Molecular Alterations of the Endocannabinoid System in Psychiatric Disorders. International Journal of Molecular Sciences. 2022; 23(9):4764. https://doi.org/10.3390/ijms23094764
Chicago/Turabian StyleNavarro, Daniela, Ani Gasparyan, Francisco Navarrete, Abraham B. Torregrosa, Gabriel Rubio, Marta Marín-Mayor, Gabriela B. Acosta, Maria Salud Garcia-Gutiérrez, and Jorge Manzanares. 2022. "Molecular Alterations of the Endocannabinoid System in Psychiatric Disorders" International Journal of Molecular Sciences 23, no. 9: 4764. https://doi.org/10.3390/ijms23094764
APA StyleNavarro, D., Gasparyan, A., Navarrete, F., Torregrosa, A. B., Rubio, G., Marín-Mayor, M., Acosta, G. B., Garcia-Gutiérrez, M. S., & Manzanares, J. (2022). Molecular Alterations of the Endocannabinoid System in Psychiatric Disorders. International Journal of Molecular Sciences, 23(9), 4764. https://doi.org/10.3390/ijms23094764