
Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies


Abstract
:1. Introduction
2. Introductory Eye Physiology
3. Pathogenesis of RP
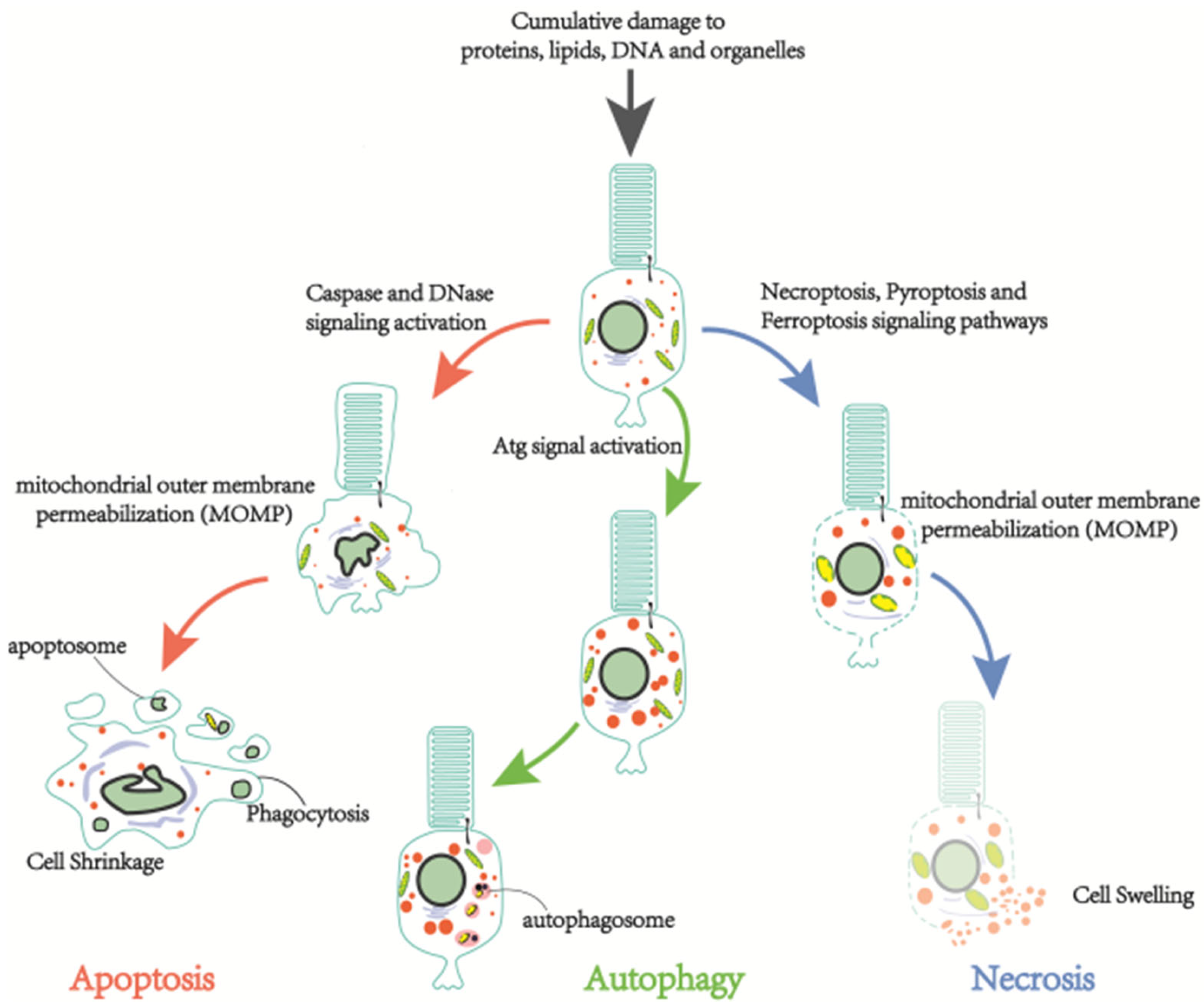
3.1. Cell Death in RP
3.1.1. Apoptosis
3.1.2. Necrosis
3.1.3. Autophagy-Dependent Cell Death
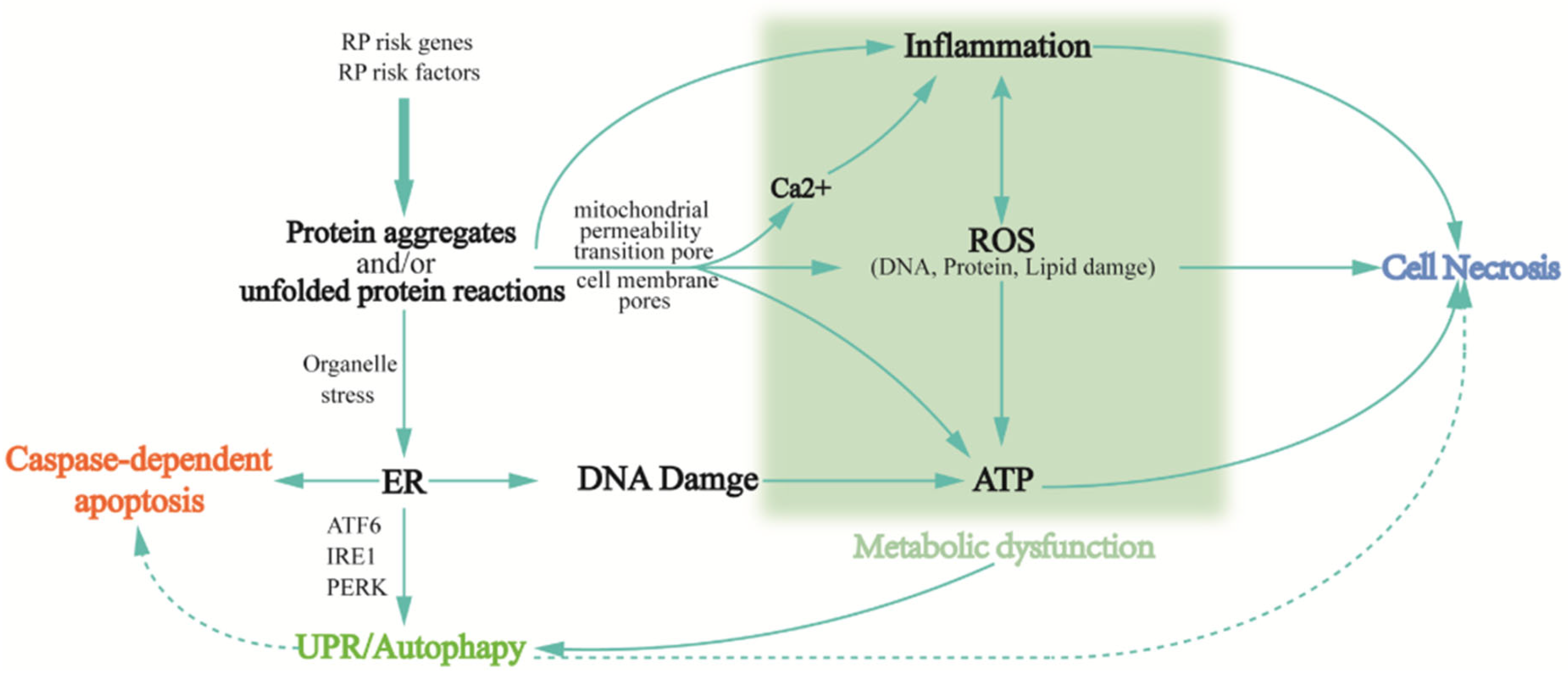
3.2. Phenotypic Switch in RP
3.2.1. Protein Aggregation and/or Unfolded Protein Reactions
3.2.2. Inflammatory Response
3.2.3. Oxidative Stress
3.2.4. Autophagy
3.2.5. Metabolic Dysfunction
3.3. Aberrant Biochemical Reaction in RP
3.3.1. Phototransduction Cascade Reaction
3.3.2. RNA Splicing
3.3.3. Transcription Factor Regulation
3.3.4. Cellular Structure and Function Regulation
3.3.5. Retinal Metabolism
4. Clinical Manifestations and Diagnosis of RP
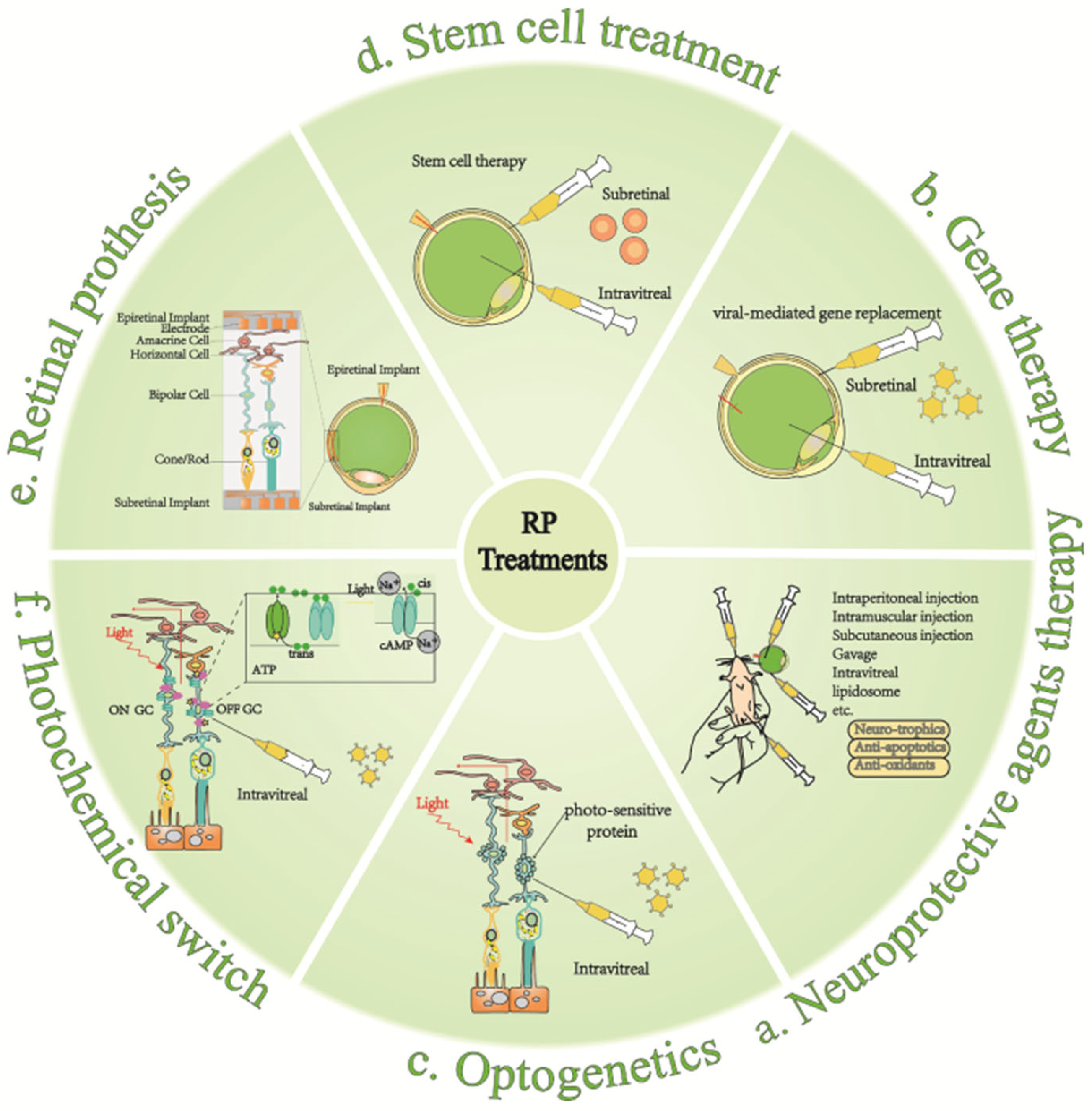
5. Therapeutic Approaches to RP
5.1. Neuroprotective Agent
5.2. Gene Therapy
5.3. Stem Cell Therapy
5.4. Optogenetics
5.5. Artificial Retina
5.6. Chemical Photoswitches
6. Summary and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Dias, M.F.; Joo, K.; Kemp, J.A.; Fialho, S.L.; da Silva Cunha, A., Jr.; Woo, S.J.; Kwon, Y.J. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye Res. 2018, 63, 107–131. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Sharma, T. Retinitis Pigmentosa (Non-syndromic). Adv. Exp. Med. Biol. 2018, 1085, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Koch, S.; Sothilingam, V.; Garcia Garrido, M.; Tanimoto, N.; Schulze, E.; Becirovic, E.; Koch, F.; Seide, C.; Beck, S.C.; et al. Gene therapy restores vision and delays degeneration in the CNGB1(-/-) mouse model of retinitis pigmentosa. Adv. Exp. Med. Biol. 2014, 801, 733–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, S.A.; Farrar, G.J.; Kenna, P.; Humphries, M.M.; Sheils, D.M.; Kumar-Singh, R.; Sharp, E.M.; Soriano, N.; Ayuso, C.; Benitez, J.; et al. Localization of an autosomal dominant retinitis pigmentosa gene to chromosome 7q. Nat. Genet. 1993, 4, 54–58. [Google Scholar] [CrossRef]
- Banerjee, P.; Kleyn, P.W.; Knowles, J.A.; Lewis, C.A.; Ross, B.M.; Parano, E.; Kovats, S.G.; Lee, J.J.; Penchaszadeh, G.K.; Ott, J.; et al. TULP1 mutation in two extended Dominican kindreds with autosomal recessive retinitis pigmentosa. Nat. Genet. 1998, 18, 177–179. [Google Scholar] [CrossRef]
- Maw, M.A.; Kennedy, B.; Knight, A.; Bridges, R.; Roth, K.E.; Mani, E.J.; Mukkadan, J.K.; Nancarrow, D.; Crabb, J.W.; Denton, M.J. Mutation of the gene encoding cellular retinaldehyde-binding protein in autosomal recessive retinitis pigmentosa. Nat. Genet. 1997, 17, 198–200. [Google Scholar] [CrossRef]
- Vervoort, R.; Lennon, A.; Bird, A.C.; Tulloch, B.; Axton, R.; Miano, M.G.; Meindl, A.; Meitinger, T.; Ciccodicola, A.; Wright, A.F. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 2000, 25, 462–466. [Google Scholar] [CrossRef]
- Bunker, C.H.; Berson, E.L.; Bromley, W.C.; Hayes, R.P.; Roderick, T.H. Prevalence of retinitis pigmentosa in Maine. Am. J. Ophthalmol. 1984, 97, 357–365. [Google Scholar] [CrossRef]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef]
- Tam, B.M.; Moritz, O.L. Characterization of rhodopsin P23H-induced retinal degeneration in a Xenopus laevis model of retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3234–3241. [Google Scholar] [CrossRef] [Green Version]
- Mathur, P.; Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 2015, 1852, 406–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearring, J.N.; Salinas, R.Y.; Baker, S.A.; Arshavsky, V.Y. Protein sorting, targeting and trafficking in photoreceptor cells. Prog. Retin. Eye Res. 2013, 36, 24–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaroop, A.; Kim, D.; Forrest, D. Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat. Rev. Neurosci. 2010, 11, 563–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krol, J.; Roska, B. Rods Feed Cones to Keep them Alive. Cell 2015, 161, 706–708. [Google Scholar] [CrossRef] [Green Version]
- van Soest, S.; Westerveld, A.; de Jong, P.T.; Bleeker-Wagemakers, E.M.; Bergen, A.A. Retinitis pigmentosa: Defined from a molecular point of view. Surv. Ophthalmol. 1999, 43, 321–334. [Google Scholar] [CrossRef]
- Slijkerman, R.W.; Song, F.; Astuti, G.D.; Huynen, M.A.; van Wijk, E.; Stieger, K.; Collin, R.W. The pros and cons of vertebrate animal models for functional and therapeutic research on inherited retinal dystrophies. Prog. Retin. Eye Res. 2015, 48, 137–159. [Google Scholar] [CrossRef]
- Lakkaraju, A.; Umapathy, A.; Tan, L.X.; Daniele, L.; Philp, N.J.; Boesze-Battaglia, K.; Williams, D.S. The cell biology of the retinal pigment epithelium. Prog. Retin. Eye Res. 2020, 78, 100846. [Google Scholar] [CrossRef]
- Jones, B.W.; Pfeiffer, R.L.; Ferrell, W.D.; Watt, C.B.; Marmor, M.; Marc, R.E. Retinal remodeling in human retinitis pigmentosa. Exp. Eye Res. 2016, 150, 149–165. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Notomi, S.; Hisatomi, T.; Nakazawa, T.; Ishibashi, T.; Miller, J.W.; Vavvas, D.G. Photoreceptor cell death and rescue in retinal detachment and degenerations. Prog. Retin. Eye Res. 2013, 37, 114–140. [Google Scholar] [CrossRef] [Green Version]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Ramirez, M.L.G.; Salvesen, G.S. A primer on caspase mechanisms. Semin. Cell Dev. Biol. 2018, 82, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.E.; Krammer, P.H. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003, 10, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.; Hughes, M.A.; Schilling, R.; Sticht, C.; Tenev, T.; Ploesser, M.; Meier, P.; Sprick, M.R.; MacFarlane, M.; Leverkus, M. Caspase-10 Negatively Regulates Caspase-8-Mediated Cell Death, Switching the Response to CD95L in Favor of NF-κB Activation and Cell Survival. Cell Rep. 2017, 19, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Allen, H.; Banerjee, S.; Franklin, S.; Herzog, L.; Johnston, C.; McDowell, J.; Paskind, M.; Rodman, L.; Salfeld, J.; et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 1995, 80, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Ma, W.; Benchimol, S. Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat. Genet. 2000, 26, 122–127. [Google Scholar] [CrossRef]
- Modjtahedi, N.; Giordanetto, F.; Madeo, F.; Kroemer, G. Apoptosis-inducing factor: Vital and lethal. Trends Cell Biol. 2006, 16, 264–272. [Google Scholar] [CrossRef]
- Polster, B.M.; Basañez, G.; Etxebarria, A.; Hardwick, J.M.; Nicholls, D.G. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J. Biol. Chem. 2005, 280, 6447–6454. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef] [Green Version]
- Churbanova, I.Y.; Sevrioukova, I.F. Redox-dependent changes in molecular properties of mitochondrial apoptosis-inducing factor. J. Biol. Chem. 2008, 283, 5622–5631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanges, D.; Comitato, A.; Tammaro, R.; Marigo, V. Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 17366–17371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Szamier, R.B.; Berson, E.L. Retinal ultrastructure in advanced retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 1977, 16, 947–962. [Google Scholar]
- Murakami, Y.; Matsumoto, H.; Roh, M.; Suzuki, J.; Hisatomi, T.; Ikeda, Y.; Miller, J.W.; Vavvas, D.G. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 14598–14603. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Shu, D.Y.; Butcher, E.R.; Saint-Geniez, M. Suppression of PGC-1α Drives Metabolic Dysfunction in TGFβ2-Induced EMT of Retinal Pigment Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 4701. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Liang, J.J.; Zhuang, X.; Chen, S.W.; Ng, T.K.; Chen, H. Intravitreal Injection of Hydrogen Peroxide Induces Acute Retinal Degeneration, Apoptosis, and Oxidative Stress in Mice. Oxidative Med. Cell. Longev. 2018, 2018, 5489476. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Oveson, B.C.; Jo, Y.J.; Lauer, T.W.; Usui, S.; Komeima, K.; Xie, B.; Campochiaro, P.A. Increased expression of glutathione peroxidase 4 strongly protects retina from oxidative damage. Antioxid. Redox Signal. 2009, 11, 715–724. [Google Scholar] [CrossRef]
- Ueta, T.; Inoue, T.; Furukawa, T.; Tamaki, Y.; Nakagawa, Y.; Imai, H.; Yanagi, Y. Glutathione peroxidase 4 is required for maturation of photoreceptor cells. J. Biol. Chem. 2012, 287, 7675–7682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhang, L. Regulation of ATG and Autophagy Initiation. Adv. Exp. Med. Biol. 2019, 1206, 41–65. [Google Scholar] [CrossRef]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef]
- Kirisako, T.; Ichimura, Y.; Okada, H.; Kabeya, Y.; Mizushima, N.; Yoshimori, T.; Ohsumi, M.; Takao, T.; Noda, T.; Ohsumi, Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 2000, 151, 263–276. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Kunchithapautham, K.; Rohrer, B. Apoptosis and autophagy in photoreceptors exposed to oxidative stress. Autophagy 2007, 3, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Sethi, C.S.; Lewis, G.P.; Fisher, S.K.; Leitner, W.P.; Mann, D.L.; Luthert, P.J.; Charteris, D.G. Glial remodeling and neural plasticity in human retinal detachment with proliferative vitreoretinopathy. Investig. Ophthalmol. Vis. Sci. 2005, 46, 329–342. [Google Scholar] [CrossRef]
- Roesch, K.; Stadler, M.B.; Cepko, C.L. Gene expression changes within Müller glial cells in retinitis pigmentosa. Mol. Vis. 2012, 18, 1197–1214. [Google Scholar] [PubMed]
- Pfeiffer, R.L.; Marc, R.E.; Jones, B.W. Persistent remodeling and neurodegeneration in late-stage retinal degeneration. Prog. Retin. Eye Res. 2020, 74, 100771. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, M.S.; Knox, T.; LaVail, M.M.; Gorbatyuk, O.S.; Noorwez, S.M.; Hauswirth, W.W.; Lin, J.H.; Muzyczka, N.; Lewin, A.S. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. USA 2010, 107, 5961–5966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasheva, V.I.; Domingos, P.M. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis 2009, 14, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [Green Version]
- Illing, M.E.; Rajan, R.S.; Bence, N.F.; Kopito, R.R. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J. Biol. Chem. 2002, 277, 34150–34160. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Sharma, M.; Südhof, T.C. Definition of a molecular pathway mediating α-synuclein neurotoxicity. J. Neurosci. 2015, 35, 5221–5232. [Google Scholar] [CrossRef] [Green Version]
- Majd, S.; Power, J.H.; Grantham, H.J. Neuronal response in Alzheimer’s and Parkinson’s disease: The effect of toxic proteins on intracellular pathways. BMC Neurosci. 2015, 16, 69. [Google Scholar] [CrossRef] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Alpha-synuclein and beta-amyloid—Different targets, same players: Calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem. Biophys. Res. Commun. 2017, 483, 1110–1115. [Google Scholar] [CrossRef]
- Murakami, Y.; Ishikawa, K.; Nakao, S.; Sonoda, K.H. Innate immune response in retinal homeostasis and inflammatory disorders. Prog. Retin. Eye Res. 2020, 74, 100778. [Google Scholar] [CrossRef]
- O’Koren, E.G.; Mathew, R.; Saban, D.R. Fate mapping reveals that microglia and recruited monocyte-derived macrophages are definitively distinguishable by phenotype in the retina. Sci. Rep. 2016, 6, 20636. [Google Scholar] [CrossRef] [PubMed]
- McMenamin, P.G.; Saban, D.R.; Dando, S.J. Immune cells in the retina and choroid: Two different tissue environments that require different defenses and surveillance. Prog. Retin. Eye Res. 2019, 70, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Karlstetter, M.; Scholz, R.; Rutar, M.; Wong, W.T.; Provis, J.M.; Langmann, T. Retinal microglia: Just bystander or target for therapy? Prog. Retin. Eye Res. 2015, 45, 30–57. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Nakabeppu, Y.; Sonoda, K.H. Oxidative Stress and Microglial Response in Retinitis Pigmentosa. Int. J. Mol. Sci. 2020, 21, 7170. [Google Scholar] [CrossRef]
- Silverman, S.M.; Ma, W.; Wang, X.; Zhao, L.; Wong, W.T. C3- and CR3-dependent microglial clearance protects photoreceptors in retinitis pigmentosa. J. Exp. Med. 2019, 216, 1925–1943. [Google Scholar] [CrossRef] [Green Version]
- Micera, A.; Balzamino, B.O.; Di Zazzo, A.; Dinice, L.; Bonini, S.; Coassin, M. Biomarkers of Neurodegeneration and Precision Therapy in Retinal Disease. Front. Pharmacol. 2020, 11, 601647. [Google Scholar] [CrossRef]
- Scimone, C.; Donato, L.; Alibrandi, S.; Vadalà, M.; Giglia, G.; Sidoti, A.; D’Angelo, R. N-retinylidene-N-retinylethanolamine adduct induces expression of chronic inflammation cytokines in retinal pigment epithelium cells. Exp. Eye Res. 2021, 209, 108641. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial cytopathies. Cell Calcium 2016, 60, 199–206. [Google Scholar] [CrossRef]
- Rinaldi, C.; Donato, L.; Alibrandi, S.; Scimone, C.; D’Angelo, R.; Sidoti, A. Oxidative Stress and the Neurovascular Unit. Life 2021, 11, 767. [Google Scholar] [CrossRef]
- Jang, J.Y.; Blum, A.; Liu, J.; Finkel, T. The role of mitochondria in aging. J. Clin. Investig. 2018, 128, 3662–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.L.; Mérida, S.; Bosch-Morell, F.; Miranda, M.; Villar, V.M. Autophagy Dysfunction and Oxidative Stress, Two Related Mechanisms Implicated in Retinitis Pigmentosa. Front. Physiol. 2018, 9, 1008. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.T.; Gauss, K.A. Structure and regulation of the neutrophil respiratory burst oxidase: Comparison with nonphagocyte oxidases. J. Leukoc. Biol. 2004, 76, 760–781. [Google Scholar] [CrossRef]
- Usui, S.; Oveson, B.C.; Lee, S.Y.; Jo, Y.J.; Yoshida, T.; Miki, A.; Miki, K.; Iwase, T.; Lu, L.; Campochiaro, P.A. NADPH oxidase plays a central role in cone cell death in retinitis pigmentosa. J. Neurochem. 2009, 110, 1028–1037. [Google Scholar] [CrossRef] [Green Version]
- Nishiguchi, K.M.; Carvalho, L.S.; Rizzi, M.; Powell, K.; Holthaus, S.M.; Azam, S.A.; Duran, Y.; Ribeiro, J.; Luhmann, U.F.; Bainbridge, J.W.; et al. Gene therapy restores vision in rd1 mice after removal of a confounding mutation in Gpr179. Nat. Commun. 2015, 6, 6006. [Google Scholar] [CrossRef]
- Yamada, H.; Yamada, E.; Hackett, S.F.; Ozaki, H.; Okamoto, N.; Campochiaro, P.A. Hyperoxia causes decreased expression of vascular endothelial growth factor and endothelial cell apoptosis in adult retina. J. Cell Physiol. 1999, 179, 149–156. [Google Scholar] [CrossRef]
- Lee, S.Y.; Usui, S.; Zafar, A.B.; Oveson, B.C.; Jo, Y.J.; Lu, L.; Masoudi, S.; Campochiaro, P.A. N-Acetylcysteine promotes long-term survival of cones in a model of retinitis pigmentosa. J. Cell Physiol. 2011, 226, 1843–1849. [Google Scholar] [CrossRef]
- Yoshida, N.; Ikeda, Y.; Notomi, S.; Ishikawa, K.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Laboratory evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology 2013, 120, e5–e12. [Google Scholar] [CrossRef]
- Xiong, W.; MacColl Garfinkel, A.E.; Li, Y.; Benowitz, L.I.; Cepko, C.L. NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J. Clin. Investig. 2015, 125, 1433–1445. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Ikeda, Y.; Yoshida, N.; Notomi, S.; Hisatomi, T.; Oka, S.; De Luca, G.; Yonemitsu, Y.; Bignami, M.; Nakabeppu, Y.; et al. MutT homolog-1 attenuates oxidative DNA damage and delays photoreceptor cell death in inherited retinal degeneration. Am. J. Pathol. 2012, 181, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández de la Cámara, C.; Salom, D.; Sequedo, M.D.; Hervás, D.; Marín-Lambíes, C.; Aller, E.; Jaijo, T.; Díaz-Llopis, M.; Millán, J.M.; Rodrigo, R. Altered antioxidant-oxidant status in the aqueous humor and peripheral blood of patients with retinitis pigmentosa. PLoS ONE 2013, 8, e74223. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa-Ishimoto, Y.; Hwang, S.; Cadwell, K. Autophagy and Inflammation. Annu. Rev. Immunol. 2018, 36, 73–101. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q. Oxidative Stress and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 179–198. [Google Scholar] [CrossRef]
- Vázquez, M.C.; Balboa, E.; Alvarez, A.R.; Zanlungo, S. Oxidative stress: A pathogenic mechanism for Niemann-Pick type C disease. Oxidative Med. Cell. Longev. 2012, 2012, 205713. [Google Scholar] [CrossRef] [Green Version]
- Chai, P.; Ni, H.; Zhang, H.; Fan, X. The Evolving Functions of Autophagy in Ocular Health: A Double-edged Sword. Int. J. Biol. Sci. 2016, 12, 1332–1340. [Google Scholar] [CrossRef] [Green Version]
- Cadwell, K. Crosstalk between autophagy and inflammatory signalling pathways: Balancing defence and homeostasis. Nat. Rev. Immunol. 2016, 16, 661–675. [Google Scholar] [CrossRef]
- Indo, H.P.; Hawkins, C.L.; Nakanishi, I.; Matsumoto, K.I.; Matsui, H.; Suenaga, S.; Davies, M.J.; St Clair, D.K.; Ozawa, T.; Majima, H.J. Role of Mitochondrial Reactive Oxygen Species in the Activation of Cellular Signals, Molecules, and Function. Handb. Exp. Pharmacol. 2017, 240, 439–456. [Google Scholar] [CrossRef]
- Intartaglia, D.; Giamundo, G.; Conte, I. Autophagy in the retinal pigment epithelium: A new vision and future challenges. FEBS J. 2021, 10. [Google Scholar] [CrossRef]
- Venkatesh, A.; Ma, S.; Le, Y.Z.; Hall, M.N.; Rüegg, M.A.; Punzo, C. Activated mTORC1 promotes long-term cone survival in retinitis pigmentosa mice. J. Clin. Investig. 2015, 125, 1446–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, M.; Yin, Y.; Wang, X.; Wang, Q.; Zhang, L.; Hu, H.; Wang, C. Mice deficient in UXT exhibit retinitis pigmentosa-like features via aberrant autophagy activation. Autophagy 2021, 17, 1873–1888. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Butcher, E.; Saint-Geniez, M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2020, 21, 4271. [Google Scholar] [CrossRef]
- Rosales, M.A.B.; Shu, D.Y.; Iacovelli, J.; Saint-Geniez, M. Loss of PGC-1α in RPE induces mesenchymal transition and promotes retinal degeneration. Life Sci. Alliance 2019, 2, e201800212. [Google Scholar] [CrossRef]
- Chan, P.; Stolz, J.; Kohl, S.; Chiang, W.C.; Lin, J.H. Endoplasmic reticulum stress in human photoreceptor diseases. Brain Res. 2016, 1648, 538–541. [Google Scholar] [CrossRef] [Green Version]
- Athanasiou, D.; Aguilà, M.; Bevilacqua, D.; Novoselov, S.S.; Parfitt, D.A.; Cheetham, M.E. The cell stress machinery and retinal degeneration. FEBS Lett. 2013, 587, 2008–2017. [Google Scholar] [CrossRef] [Green Version]
- Cideciyan, A.V.; Sudharsan, R.; Dufour, V.L.; Massengill, M.T.; Iwabe, S.; Swider, M.; Lisi, B.; Sumaroka, A.; Marinho, L.F.; Appelbaum, T.; et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc. Natl. Acad. Sci. USA 2018, 115, E8547–E8556. [Google Scholar] [CrossRef] [Green Version]
- Deisseroth, K.; Hegemann, P. The form and function of channelrhodopsin. Science 2017, 357, aan5544. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Kawada, M.; Havlioglu, N.; Tang, H.; Wu, J.Y. Mutations in PRPF31 inhibit pre-mRNA splicing of rhodopsin gene and cause apoptosis of retinal cells. J. Neurosci. 2005, 25, 748–757. [Google Scholar] [CrossRef]
- Schaffert, N.; Hossbach, M.; Heintzmann, R.; Achsel, T.; Lührmann, R. RNAi knockdown of hPrp31 leads to an accumulation of U4/U6 di-snRNPs in Cajal bodies. EMBO J. 2004, 23, 3000–3009. [Google Scholar] [CrossRef] [Green Version]
- Azizzadeh Pormehr, L.; Ahmadian, S.; Daftarian, N.; Mousavi, S.A.; Shafiezadeh, M. PRPF31 reduction causes mis-splicing of the phototransduction genes in human organotypic retinal culture. Eur. J. Hum. Genet. 2020, 28, 491–498. [Google Scholar] [CrossRef]
- Azizzadeh Pormehr, L.; Daftarian, N.; Ahmadian, S.; Rezaei Kanavi, M.; Ahmadieh, H.; Shafiezadeh, M. Human organotypic retinal flat-mount culture (HORFC) as a model for retinitis pigmentosa11. J. Cell. Biochem. 2018, 119, 6775–6783. [Google Scholar] [CrossRef] [PubMed]
- Ajmal, M.; Khan, M.I.; Neveling, K.; Khan, Y.M.; Azam, M.; Waheed, N.K.; Hamel, C.P.; Ben-Yosef, T.; De Baere, E.; Koenekoop, R.K.; et al. A missense mutation in the splicing factor gene DHX38 is associated with early-onset retinitis pigmentosa with macular coloboma. J. Med. Genet. 2014, 51, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Xie, Y.A.; Abouzeid, H.; Gordon, C.T.; Fiorentino, A.; Sun, Z.; Lehman, A.; Osman, I.S.; Dharmat, R.; Riveiro-Alvarez, R.; et al. Mutations in the Spliceosome Component CWC27 Cause Retinal Degeneration with or without Additional Developmental Anomalies. Am. J. Hum. Genet. 2017, 100, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busetto, V.; Barbosa, I.; Basquin, J.; Marquenet, É.; Hocq, R.; Hennion, M.; Paternina, J.A.; Namane, A.; Conti, E.; Bensaude, O.; et al. Structural and functional insights into CWC27/CWC22 heterodimer linking the exon junction complex to spliceosomes. Nucleic Acids Res. 2020, 48, 5670–5683. [Google Scholar] [CrossRef]
- Garancher, A.; Lin, C.Y.; Morabito, M.; Richer, W.; Rocques, N.; Larcher, M.; Bihannic, L.; Smith, K.; Miquel, C.; Leboucher, S.; et al. NRL and CRX Define Photoreceptor Identity and Reveal Subgroup-Specific Dependencies in Medulloblastoma. Cancer Cell 2018, 33, 435–449.e436. [Google Scholar] [CrossRef] [Green Version]
- Ng, L.; Lu, A.; Swaroop, A.; Sharlin, D.S.; Swaroop, A.; Forrest, D. Two transcription factors can direct three photoreceptor outcomes from rod precursor cells in mouse retinal development. J. Neurosci. 2011, 31, 11118–11125. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.M.; Skowronska-Krawczyk, D.; Chao, D.L. Targeting of the NRL Pathway as a Therapeutic Strategy to Treat Retinitis Pigmentosa. J. Clin. Med. 2020, 9, 2224. [Google Scholar] [CrossRef]
- Han, S.; Chen, J.; Hua, J.; Hu, X.; Jian, S.; Zheng, G.; Wang, J.; Li, H.; Yang, J.; Hejtmancik, J.F.; et al. MITF protects against oxidative damage-induced retinal degeneration by regulating the NRF2 pathway in the retinal pigment epithelium. Redox Biol. 2020, 34, 101537. [Google Scholar] [CrossRef]
- Westfall, J.E.; Hoyt, C.; Liu, Q.; Hsiao, Y.C.; Pierce, E.A.; Page-McCaw, P.S.; Ferland, R.J. Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the Joubert syndrome gene, Ahi1. J. Neurosci. 2010, 30, 8759–8768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhogaraju, S.; Cajanek, L.; Fort, C.; Blisnick, T.; Weber, K.; Taschner, M.; Mizuno, N.; Lamla, S.; Bastin, P.; Nigg, E.A.; et al. Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science 2013, 341, 1009–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, T.; Brown, J.M.; Bellve, K.; Craige, B.; Craft, J.M.; Fogarty, K.; Lechtreck, K.F.; Witman, G.B. Together, the IFT81 and IFT74 N-termini form the main module for intraflagellar transport of tubulin. J. Cell Sci. 2016, 129, 2106–2119. [Google Scholar] [CrossRef] [Green Version]
- Tebbe, L.; Kakakhel, M.; Makia, M.S.; Al-Ubaidi, M.R.; Naash, M.I. The Interplay between Peripherin 2 Complex Formation and Degenerative Retinal Diseases. Cells 2020, 9, 784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, L.; Abdalla, E.M.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Nabil, K.M.; D’Angelo, R.; Sidoti, A. Impairments of Photoreceptor Outer Segments Renewal and Phototransduction Due to a Peripherin Rare Haplotype Variant: Insights from Molecular Modeling. Int. J. Mol. Sci. 2021, 22, 3484. [Google Scholar] [CrossRef]
- Clarke, G.; Goldberg, A.F.; Vidgen, D.; Collins, L.; Ploder, L.; Schwarz, L.; Molday, L.L.; Rossant, J.; Szél, A.; Molday, R.S.; et al. Rom-1 is required for rod photoreceptor viability and the regulation of disk morphogenesis. Nat. Genet. 2000, 25, 67–73. [Google Scholar] [CrossRef]
- Kevany, B.M.; Tsybovsky, Y.; Campuzano, I.D.; Schnier, P.D.; Engel, A.; Palczewski, K. Structural and functional analysis of the native peripherin-ROM1 complex isolated from photoreceptor cells. J. Biol. Chem. 2013, 288, 36272–36284. [Google Scholar] [CrossRef] [Green Version]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. PRPH2/RDS and ROM-1: Historical context, current views and future considerations. Prog. Retin. Eye Res. 2016, 52, 47–63. [Google Scholar] [CrossRef] [Green Version]
- Zulliger, R.; Conley, S.M.; Mwoyosvi, M.L.; Al-Ubaidi, M.R.; Naash, M.I. Oligomerization of Prph2 and Rom1 is essential for photoreceptor outer segment formation. Hum. Mol. Genet. 2018, 27, 3507–3518. [Google Scholar] [CrossRef] [Green Version]
- Strayve, D.; Makia, M.S.; Kakakhel, M.; Sakthivel, H.; Conley, S.M.; Al-Ubaidi, M.R.; Naash, M.I. ROM1 contributes to phenotypic heterogeneity in PRPH2-associated retinal disease. Hum. Mol. Genet. 2020, 29, 2708–2722. [Google Scholar] [CrossRef]
- Pellikka, M.; Tanentzapf, G.; Pinto, M.; Smith, C.; McGlade, C.J.; Ready, D.F.; Tepass, U. Crumbs, the Drosophila homologue of human CRB1/RP12, is essential for photoreceptor morphogenesis. Nature 2002, 416, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Izaddoost, S.; Nam, S.C.; Bhat, M.A.; Bellen, H.J.; Choi, K.W. Drosophila Crumbs is a positional cue in photoreceptor adherens junctions and rhabdomeres. Nature 2002, 416, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.H.; Pellissier, L.P.; Wijnholds, J. The CRB1 and adherens junction complex proteins in retinal development and maintenance. Prog. Retin. Eye Res. 2014, 40, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Hollyfield, J.G. Hyaluronan and the functional organization of the interphotoreceptor matrix. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2767–2769. [Google Scholar]
- Kanan, Y.; Hoffhines, A.; Rauhauser, A.; Murray, A.; Al-Ubaidi, M.R. Protein tyrosine-O-sulfation in the retina. Exp. Eye Res. 2009, 89, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Kam, J.H.; Weinrich, T.W.; Shinhmar, H.; Powner, M.B.; Roberts, N.W.; Aboelnour, A.; Jeffery, G. Fundamental differences in patterns of retinal ageing between primates and mice. Sci. Rep. 2019, 9, 12574. [Google Scholar] [CrossRef] [Green Version]
- Ala-Laurila, P.; Kolesnikov, A.V.; Crouch, R.K.; Tsina, E.; Shukolyukov, S.A.; Govardovskii, V.I.; Koutalos, Y.; Wiggert, B.; Estevez, M.E.; Cornwall, M.C. Visual cycle: Dependence of retinol production and removal on photoproduct decay and cell morphology. J. Gen. Physiol. 2006, 128, 153–169. [Google Scholar] [CrossRef]
- Bermúdez, V.; Tenconi, P.E.; Giusto, N.M.; Mateos, M.V. Lipid Signaling in Retinal Pigment Epithelium Cells Exposed to Inflammatory and Oxidative Stress Conditions. Molecular Mechanisms Underlying Degenerative Retinal Diseases. Adv. Exp. Med. Biol. 2019, 1185, 289–293. [Google Scholar] [CrossRef]
- Isken, A.; Golczak, M.; Oberhauser, V.; Hunzelmann, S.; Driever, W.; Imanishi, Y.; Palczewski, K.; von Lintig, J. RBP4 disrupts vitamin A uptake homeostasis in a STRA6-deficient animal model for Matthew-Wood syndrome. Cell Metab. 2008, 7, 258–268. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, M.E.; Roy, S.; Cantin, L.; Salesse, C. Comparison between the enzymatic activity, structure and substrate binding of mouse and human lecithin retinol acyltransferase. Biochem. Biophys. Res. Commun. 2019, 519, 832–837. [Google Scholar] [CrossRef]
- Branham, K.; Yashar, B.M. Providing comprehensive genetic-based ophthalmic care. Clin. Genet. 2013, 84, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergott, R.C. Retinal segmentation using multicolor laser imaging. J. Neuroophthalmol. 2014, 34, S24–S28. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Possin, D.E.; Milam, A.H. Histopathology of bone spicule pigmentation in retinitis pigmentosa. Ophthalmology 1995, 102, 805–816. [Google Scholar] [CrossRef]
- Cellini, M.; Strobbe, E.; Gizzi, C.; Campos, E.C. ET-1 plasma levels and ocular blood flow in retinitis pigmentosa. Can. J. Physiol. Pharmacol. 2010, 88, 630–635. [Google Scholar] [CrossRef]
- Hwang, Y.H.; Kim, S.W.; Kim, Y.Y.; Na, J.H.; Kim, H.K.; Sohn, Y.H. Optic nerve head, retinal nerve fiber layer, and macular thickness measurements in young patients with retinitis pigmentosa. Curr. Eye Res. 2012, 37, 914–920. [Google Scholar] [CrossRef]
- Strong, S.; Liew, G.; Michaelides, M. Retinitis pigmentosa-associated cystoid macular oedema: Pathogenesis and avenues of intervention. Br. J. Ophthalmol. 2017, 101, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Chebil, A.; Touati, S.; Maamouri, R.; Kort, F.; El Matri, L. Spectral Domain optical coherence tomography findings in patients with retinitis pigmentosa. Tunis. Med. 2016, 94, 265–271. [Google Scholar]
- Yoshida, N.; Ikeda, Y.; Murakami, Y.; Nakatake, S.; Tachibana, T.; Notomi, S.; Hisatomi, T.; Ishibashi, T. Vitreous cysts in patients with retinitis pigmentosa. Jpn. J. Ophthalmol. 2015, 59, 373–377. [Google Scholar] [CrossRef]
- Grover, S.; Fishman, G.A.; Brown, J., Jr. Frequency of optic disc or parapapillary nerve fiber layer drusen in retinitis pigmentosa. Ophthalmology 1997, 104, 295–298. [Google Scholar] [CrossRef]
- Liu, G.; Du, Q.; Keyal, K.; Wang, F. Morphologic characteristics and clinical significance of the macular-sparing area in patients with retinitis pigmentosa as revealed by multicolor imaging. Exp. Ther. Med. 2017, 14, 5387–5394. [Google Scholar] [CrossRef] [Green Version]
- Hood, D.C.; Lazow, M.A.; Locke, K.G.; Greenstein, V.C.; Birch, D.G. The transition zone between healthy and diseased retina in patients with retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2011, 52, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, N.R.; Greenberg, J.P.; Laud, K.; Tsang, S.; Freund, K.B. Outer retinal tubulation in degenerative retinal disorders. Retina 2013, 33, 1871–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, M.; Hirami, Y.; Hata, M.; Mandai, M.; Takahashi, M.; Kurimoto, Y. Intraretinal hyperreflective foci on spectral-domain optical coherence tomographic images of patients with retinitis pigmentosa. Clin. Ophthalmol. 2014, 8, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delori, F.C.; Dorey, C.K.; Staurenghi, G.; Arend, O.; Goger, D.G.; Weiter, J.J. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Investig. Ophthalmol. Vis. Sci. 1995, 36, 718–729. [Google Scholar]
- Keilhauer, C.N.; Delori, F.C. Near-infrared autofluorescence imaging of the fundus: Visualization of ocular melanin. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3556–3564. [Google Scholar] [CrossRef]
- Teussink, M.M.; Lambertus, S.; de Mul, F.F.; Rozanowska, M.B.; Hoyng, C.B.; Klevering, B.J.; Theelen, T. Lipofuscin-associated photo-oxidative stress during fundus autofluorescence imaging. PLoS ONE 2017, 12, e0172635. [Google Scholar] [CrossRef]
- Tsang, S.H.; Sharma, T. Fundus Autofluorescence. Adv. Exp. Med. Biol. 2018, 1085, 15–16. [Google Scholar] [CrossRef]
- Kashani, A.H.; Chen, C.L.; Gahm, J.K.; Zheng, F.; Richter, G.M.; Rosenfeld, P.J.; Shi, Y.; Wang, R.K. Optical coherence tomography angiography: A comprehensive review of current methods and clinical applications. Prog. Retin. Eye Res. 2017, 60, 66–100. [Google Scholar] [CrossRef]
- Georgiou, M.; Kalitzeos, A.; Patterson, E.J.; Dubra, A.; Carroll, J.; Michaelides, M. Adaptive optics imaging of inherited retinal diseases. Br. J. Ophthalmol. 2018, 102, 1028–1035. [Google Scholar] [CrossRef]
- Ratnam, K.; Carroll, J.; Porco, T.C.; Duncan, J.L.; Roorda, A. Relationship between foveal cone structure and clinical measures of visual function in patients with inherited retinal degenerations. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5836–5847. [Google Scholar] [CrossRef]
- Grover, S.; Fishman, G.A.; Brown, J., Jr. Patterns of visual field progression in patients with retinitis pigmentosa. Ophthalmology 1998, 105, 1069–1075. [Google Scholar] [CrossRef]
- Talib, M.; van Schooneveld, M.J.; van Genderen, M.M.; Wijnholds, J.; Florijn, R.J.; Ten Brink, J.B.; Schalij-Delfos, N.E.; Dagnelie, G.; Cremers, F.P.M.; Wolterbeek, R.; et al. Genotypic and Phenotypic Characteristics of CRB1-Associated Retinal Dystrophies: A Long-Term Follow-up Study. Ophthalmology 2017, 124, 884–895. [Google Scholar] [CrossRef] [Green Version]
- Berson, E.L.; Rosner, B.; Weigel-DiFranco, C.; Dryja, T.P.; Sandberg, M.A. Disease progression in patients with dominant retinitis pigmentosa and rhodopsin mutations. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3027–3036. [Google Scholar]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Berson, E.L. Retinitis pigmentosa and allied diseases: Applications of electroretinographic testing. Int. Ophthalmol. 1981, 4, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Berson, E.L. Retinitis pigmentosa. The Friedenwald Lecture. Investig. Ophthalmol. Vis. Sci. 1993, 34, 1659–1676. [Google Scholar]
- Messias, K.; Jägle, H.; Saran, R.; Ruppert, A.D.; Siqueira, R.; Jorge, R.; Messias, A. Psychophysically determined full-field stimulus thresholds (FST) in retinitis pigmentosa: Relationships with electroretinography and visual field outcomes. Doc. Ophthalmol. 2013, 127, 123–129. [Google Scholar] [CrossRef]
- Branham, K.; Schlegel, D.; Fahim, A.T.; Jayasundera, K.T. Genetic testing for inherited retinal degenerations: Triumphs and tribulations. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 571–577. [Google Scholar] [CrossRef]
- Buch, P.K.; MacLaren, R.E.; Ali, R.R. Neuroprotective gene therapy for the treatment of inherited retinal degeneration. Curr. Gene Ther. 2007, 7, 434–445. [Google Scholar] [CrossRef]
- Delplace, V.; Ortin-Martinez, A.; Tsai, E.L.S.; Amin, A.N.; Wallace, V.; Shoichet, M.S. Controlled release strategy designed for intravitreal protein delivery to the retina. J. Control. Release 2019, 293, 10–20. [Google Scholar] [CrossRef]
- Gupta, V.K.; You, Y.; Gupta, V.B.; Klistorner, A.; Graham, S.L. TrkB receptor signalling: Implications in neurodegenerative, psychiatric and proliferative disorders. Int. J. Mol. Sci. 2013, 14, 10122–10142. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.C.; Xiang, J.J.; Wu, H.H.; Zhang, T.Y.; Zhang, D.P.; Xu, Q.H.; Huang, X.L.; Kong, X.L.; Sun, J.H.; Hu, Y.L.; et al. Neural Stem Cells Transfected with Reactive Oxygen Species-Responsive Polyplexes for Effective Treatment of Ischemic Stroke. Adv. Mater. 2019, 31, e1807591. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [Green Version]
- Hecht, R.; Li, Y.S.; Sun, J.; Belouski, E.; Hall, M.; Hager, T.; Yie, J.; Wang, W.; Winters, D.; Smith, S.; et al. Rationale-Based Engineering of a Potent Long-Acting FGF21 Analog for the Treatment of Type 2 Diabetes. PLoS ONE 2012, 7, e49345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, D.M.; Barnard, A.R.; Singh, M.S.; Martin, C.; Lee, E.J.; Davies, W.I.L.; MacLaren, R.E. CNTF Gene Therapy Confers Lifelong Neuroprotection in a Mouse Model of Human Retinitis Pigmentosa. Mol. Ther. 2015, 23, 1308–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omura, T.; Asari, M.; Yamamoto, J.; Oka, K.; Hoshina, C.; Maseda, C.; Awaya, T.; Tasaki, Y.; Shiono, H.; Yonezawa, A.; et al. Sodium tauroursodeoxycholate prevents paraquat-induced cell death by suppressing endoplasmic reticulum stress responses in human lung epithelial A549 cells. Biochem. Biophys. Res. Commun. 2013, 432, 689–694. [Google Scholar] [CrossRef]
- Fernández-Sánchez, L.; Lax, P.; Pinilla, I.; Martín-Nieto, J.; Cuenca, N. Tauroursodeoxycholic acid prevents retinal degeneration in transgenic P23H rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4998–5008. [Google Scholar] [CrossRef]
- Guadagni, V.; Novelli, E.; Piano, I.; Gargini, C.; Strettoi, E. Pharmacological approaches to retinitis pigmentosa: A laboratory perspective. Prog. Retin. Eye Res. 2015, 48, 62–81. [Google Scholar] [CrossRef]
- Morshedian, A.; Kaylor, J.J.; Ng, S.Y.; Tsan, A.; Frederiksen, R.; Xu, T.; Yuan, L.; Sampath, A.P.; Radu, R.A.; Fain, G.L.; et al. Light-Driven Regeneration of Cone Visual Pigments through a Mechanism Involving RGR Opsin in Müller Glial Cells. Neuron 2019, 102, 1172–1183.e1175. [Google Scholar] [CrossRef]
- Choi, E.H.; Daruwalla, A.; Suh, S.; Leinonen, H.; Palczewski, K. Retinoids in the visual cycle: Role of the retinal G protein-coupled receptor. J. Lipid Res. 2021, 62, 100040. [Google Scholar] [CrossRef]
- Johansson, I.; Monsen, V.T.; Pettersen, K.; Mildenberger, J.; Misund, K.; Kaarniranta, K.; Schønberg, S.; Bjørkøy, G. The marine n-3 PUFA DHA evokes cytoprotection against oxidative stress and protein misfolding by inducing autophagy and NFE2L2 in human retinal pigment epithelial cells. Autophagy 2015, 11, 1636–1651. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.S.; Calandria, J.M.; Gordon, W.C.; Jun, B.; Zhou, Y.; Gelfman, C.M.; Li, S.; Jin, M.; Knott, E.J.; Chang, B.; et al. Adiponectin receptor 1 conserves docosahexaenoic acid and promotes photoreceptor cell survival. Nat. Commun. 2015, 6, 6228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kijlstra, A.; Tian, Y.; Kelly, E.R.; Berendschot, T.T. Lutein: More than just a filter for blue light. Prog. Retin. Eye Res. 2012, 31, 303–315. [Google Scholar] [CrossRef]
- Shyam, R.; Gorusupudi, A.; Nelson, K.; Horvath, M.P.; Bernstein, P.S. RPE65 has an additional function as the lutein to meso-zeaxanthin isomerase in the vertebrate eye. Proc. Natl. Acad. Sci. USA 2017, 114, 10882–10887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Lau, A.G.; Sarti, F. Synaptic retinoic acid signaling and homeostatic synaptic plasticity. Neuropharmacology 2014, 78, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozaki, T.; Nakazawa, M.; Kudo, T.; Hirano, S.; Suzuki, K.; Ishiguro, S. Protection of cone photoreceptor M-opsin degradation with 9-cis-β-carotene-rich alga Dunaliella bardawil in Rpe65(-/-) mouse retinal explant culture. Curr. Eye Res. 2014, 39, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Pawlyk, B.S.; Li, T.; Scimeca, M.S.; Sandberg, M.A.; Berson, E.L. Absence of photoreceptor rescue with D-cis-diltiazem in the rd mouse. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1912–1915. [Google Scholar]
- Yang, J.; Weimer, R.M.; Kallop, D.; Olsen, O.; Wu, Z.; Renier, N.; Uryu, K.; Tessier-Lavigne, M. Regulation of axon degeneration after injury and in development by the endogenous calpain inhibitor calpastatin. Neuron 2013, 80, 1175–1189. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Sugawara, T.; Murakami, A.; Nakazawa, M.; Nao, I.N.; Machida, S.; Wada, Y.; Mashima, Y.; Myake, Y. Topical isopropyl unoprostone for retinitis pigmentosa: Microperimetric results of the phase 2 clinical study. Ophthalmol. Ther. 2012, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Jayakody, S.A.; Gonzalez-Cordero, A.; Ali, R.R.; Pearson, R.A. Cellular strategies for retinal repair by photoreceptor replacement. Prog. Retin. Eye Res. 2015, 46, 31–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, R.; Tao, W.; Li, Y.; Sieving, P.A. CNTF and retina. Prog. Retin. Eye Res. 2012, 31, 136–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bei, F.; Lee, H.H.C.; Liu, X.; Gunner, G.; Jin, H.; Ma, L.; Wang, C.; Hou, L.; Hensch, T.K.; Frank, E.; et al. Restoration of Visual Function by Enhancing Conduction in Regenerated Axons. Cell 2016, 164, 219–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksen, A.Z.; Eliasen, R.; Oswald, J.; Kempen, P.J.; Melander, F.; Andresen, T.L.; Young, M.; Baranov, P.; Urquhart, A.J. Multifarious Biologic Loaded Liposomes that Stimulate the Mammalian Target of Rapamycin Signaling Pathway Show Retina Neuroprotection after Retina Damage. ACS Nano 2018, 12, 7497–7508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, R.; Yang, M.; Fan, W.; Lan, J.; Zhou, Y.G. Paired Immunoglobulin-like Receptor B Inhibition in Müller Cells Promotes Neurite Regeneration After Retinal Ganglion Cell Injury in vitro. Neurosci. Bull. 2020, 36, 972–984. [Google Scholar] [CrossRef]
- Hu, Z.; Deng, N.; Liu, K.; Zhou, N.; Sun, Y.; Zeng, W. CNTF-STAT3-IL-6 Axis Mediates Neuroinflammatory Cascade across Schwann Cell-Neuron-Microglia. Cell Rep. 2020, 31, 107657. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Sidiropoulou, K.; Kallergi, E.; Dalezios, Y.; Tavernarakis, N. Modulation of Autophagy by BDNF Underlies Synaptic Plasticity. Cell Metab. 2017, 26, 230–242.e235. [Google Scholar] [CrossRef] [Green Version]
- Colgan, L.A.; Hu, M.; Misler, J.A.; Parra-Bueno, P.; Moran, C.M.; Leitges, M.; Yasuda, R. PKCα integrates spatiotemporally distinct Ca2+ and autocrine BDNF signaling to facilitate synaptic plasticity. Nat. Neurosci. 2018, 21, 1027–1037. [Google Scholar] [CrossRef]
- Barbereau, C.; Yehya, A.; Silhol, M.; Cubedo, N.; Verdier, J.M.; Maurice, T.; Rossel, M. Neuroprotective brain-derived neurotrophic factor signaling in the TAU-P301L tauopathy zebrafish model. Pharmacol. Res. 2020, 158, 104865. [Google Scholar] [CrossRef]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef]
- Timper, K.; Del Río-Martín, A.; Cremer, A.L.; Bremser, S.; Alber, J.; Giavalisco, P.; Varela, L.; Heilinger, C.; Nolte, H.; Trifunovic, A.; et al. GLP-1 Receptor Signaling in Astrocytes Regulates Fatty Acid Oxidation, Mitochondrial Integrity, and Function. Cell Metab 2020, 31, 1189–1205.e1113. [Google Scholar] [CrossRef] [PubMed]
- Froger, N.; Moutsimilli, L.; Cadetti, L.; Jammoul, F.; Wang, Q.P.; Fan, Y.; Gaucher, D.; Rosolen, S.G.; Neveux, N.; Cynober, L.; et al. Taurine: The comeback of a neutraceutical in the prevention of retinal degenerations. Prog. Retin. Eye Res. 2014, 41, 44–63. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.M.; Yoo, Y.M.; Park, S.Y.; Ahn, C.; Jeon, B.H.; Hong, E.J.; Kim, W.Y.; Jeung, E.B. Calbindin-D(9k) is a Novel Risk Gene for Neurodegenerative Disease. Cell Physiol. Biochem. 2020, 54, 438–456. [Google Scholar] [CrossRef] [PubMed]
- Afşar, E.; Kırımlıoglu, E.; Çeker, T.; Yılmaz, Ç.; Demir, N.; Aslan, M. Effect of ER stress on sphingolipid levels and apoptotic pathways in retinal pigment epithelial cells. Redox Biol. 2020, 30, 101430. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.M.; Lee, J.H.; Yun, S.P.; Han, Y.S.; Yun, C.W.; Lee, H.J.; Noh, H.; Lee, S.J.; Han, H.J.; Lee, S.H. Tauroursodeoxycholic acid reduces ER stress by regulating of Akt-dependent cellular prion protein. Sci. Rep. 2016, 6, 39838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, R.; Ribeiro, F.F.; Xapelli, S.; Genebra, T.; Ribeiro, M.F.; Sebastião, A.M.; Rodrigues, C.M.P.; Solá, S. Tauroursodeoxycholic Acid Enhances Mitochondrial Biogenesis, Neural Stem Cell Pool, and Early Neurogenesis in Adult Rats. Mol. Neurobiol. 2018, 55, 3725–3738. [Google Scholar] [CrossRef]
- Zhang, Y.; Qu, P.; Ma, X.; Qiao, F.; Ma, Y.; Qing, S.; Zhang, Y.; Wang, Y.; Cui, W. Tauroursodeoxycholic acid (TUDCA) alleviates endoplasmic reticulum stress of nuclear donor cells under serum starvation. PLoS ONE 2018, 13, e0196785. [Google Scholar] [CrossRef] [Green Version]
- Junghans, A.; Sies, H.; Stahl, W. Macular pigments lutein and zeaxanthin as blue light filters studied in liposomes. Arch. Biochem. Biophys. 2001, 391, 160–164. [Google Scholar] [CrossRef]
- Chucair, A.J.; Rotstein, N.P.; Sangiovanni, J.P.; During, A.; Chew, E.Y.; Politi, L.E. Lutein and zeaxanthin protect photoreceptors from apoptosis induced by oxidative stress: Relation with docosahexaenoic acid. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5168–5177. [Google Scholar] [CrossRef]
- Saddala, M.S.; Lennikov, A.; Mukwaya, A.; Yang, Y.; Hill, M.A.; Lagali, N.; Huang, H. Discovery of novel L-type voltage-gated calcium channel blockers and application for the prevention of inflammation and angiogenesis. J. Neuroinflamm. 2020, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A.; Lenaeus, M.J.; Gamal El-Din, T.M. Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 133–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.W.; Rohrer, B.; Wheless, L.; Samantaray, S.; Ray, S.K.; Inoue, J.; Azuma, M.; Banik, N.L. Calpain inhibition reduces structural and functional impairment of retinal ganglion cells in experimental optic neuritis. J. Neurochem. 2016, 139, 270–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birch, D.G.; Bernstein, P.S.; Iannacone, A.; Pennesi, M.E.; Lam, B.L.; Heckenlively, J.; Csaky, K.; Hartnett, M.E.; Winthrop, K.L.; Jayasundera, T.; et al. Effect of Oral Valproic Acid vs Placebo for Vision Loss in Patients With Autosomal Dominant Retinitis Pigmentosa: A Randomized Phase 2 Multicenter Placebo-Controlled Clinical Trial. JAMA Ophthalmol. 2018, 136, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Ding, C.; Yuan, S.; Pan, T.; Li, D.; Li, H.; Huang, B.; Liu, Q. Vitamin C- and Valproic Acid-Induced Fetal RPE Stem-like Cells Recover Retinal Degeneration via Regulating SOX2. Mol. Ther. 2020, 28, 1645–1657. [Google Scholar] [CrossRef]
- Samardzija, M.; Masarini, K.; Ueffing, M.; Trifunović, D. HDAC Inhibition Prevents Primary Cone Degeneration Even After the Onset of Degeneration. Adv. Exp. Med. Biol. 2019, 1185, 383–387. [Google Scholar] [CrossRef]
- Ducloyer, J.B.; Le Meur, G.; Cronin, T.; Adjali, O.; Weber, M. Gene therapy for retinitis pigmentosa. Med. Sci. 2020, 36, 607–615. [Google Scholar] [CrossRef]
- Dalkara, D.; Goureau, O.; Marazova, K.; Sahel, J.A. Let There Be Light: Gene and Cell Therapy for Blindness. Hum. Gene Ther. 2016, 27, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Dalkara, D.; Sahel, J.A. Gene therapy for inherited retinal degenerations. Comptes Rendus. Biol. 2014, 337, 185–192. [Google Scholar] [CrossRef]
- Fahim, A. Retinitis pigmentosa: Recent advances and future directions in diagnosis and management. Curr. Opin. Pediatr. 2018, 30, 725–733. [Google Scholar] [CrossRef]
- Vu, K.T.; Hulleman, J.D. An inducible form of Nrf2 confers enhanced protection against acute oxidative stresses in RPE cells. Exp. Eye Res. 2017, 164, 31–36. [Google Scholar] [CrossRef]
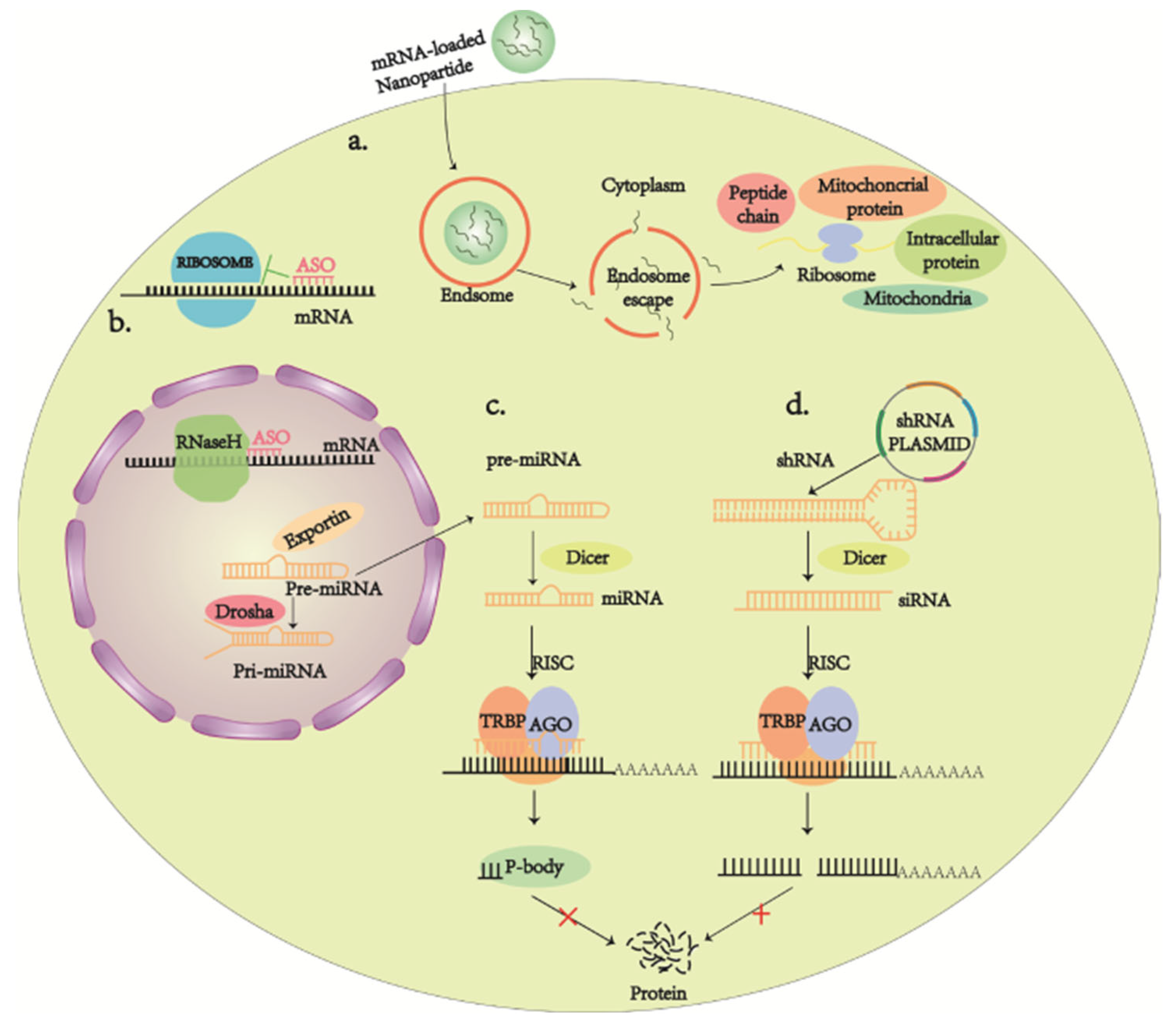
- Yau, E.H.; Butler, M.C.; Sullivan, J.M. A cellular high-throughput screening approach for therapeutic trans-cleaving ribozymes and RNAi against arbitrary mRNA disease targets. Exp. Eye Res. 2016, 151, 236–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, A.S.; Drenser, K.A.; Hauswirth, W.W.; Nishikawa, S.; Yasumura, D.; Flannery, J.G.; LaVail, M.M. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat. Med. 1998, 4, 967–971. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Rossor, A.M.; Reilly, M.M.; Sleigh, J.N. Antisense oligonucleotides and other genetic therapies made simple. Pract. Neurol. 2018, 18, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Schlake, T.; Thran, M.; Fiedler, K.; Heidenreich, R.; Petsch, B.; Fotin-Mleczek, M. mRNA: A Novel Avenue to Antibody Therapy? Mol. Ther. 2019, 27, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Wang, J.H.; Chen, J.; Li, F.; Edwards, T.L.; Hewitt, A.W.; Liu, G.S. Gene therapy for visual loss: Opportunities and concerns. Prog. Retin. Eye Res. 2019, 68, 31–53. [Google Scholar] [CrossRef]
- Carvalho, L.S.; Vandenberghe, L.H. Promising and delivering gene therapies for vision loss. Vision Res. 2015, 111, 124–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Collin, R.W.; Cremers, F.P.; den Hollander, A.I.; van den Born, L.I.; Pierce, E.A. Expression of wild-type Rp1 protein in Rp1 knock-in mice rescues the retinal degeneration phenotype. PLoS ONE 2012, 7, e43251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.J. Therapeutic siRNA: State of the art. Signal Transduct. Target. Ther. 2020, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Li, C.; Yang, T.; Hu, B.; Zhang, M.; Guo, S.; Xiao, H.; Liang, X.J.; Huang, Y. The challenge and prospect of mRNA therapeutics landscape. Biotechnol. Adv. 2020, 40, 107534. [Google Scholar] [CrossRef]
- Hasanzadeh Kafshgari, M.; Agiotis, L.; Largillière, I.; Patskovsky, S.; Meunier, M. Antibody-Functionalized Gold Nanostar-Mediated On-Resonance Picosecond Laser Optoporation for Targeted Delivery of RNA Therapeutics. Small 2021, 17, e2007577. [Google Scholar] [CrossRef] [PubMed]
- Trapani, I.; Auricchio, A. Seeing the Light after 25 Years of Retinal Gene Therapy. Trends Mol. Med. 2018, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Barnea-Cramer, A.O.; Singh, M.; Fischer, D.; De Silva, S.; McClements, M.E.; Barnard, A.R.; MacLaren, R.E. Repair of Retinal Degeneration following Ex Vivo Minicircle DNA Gene Therapy and Transplantation of Corrected Photoreceptor Progenitors. Mol. Ther. 2020, 28, 830–844. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.K.; Lu, B.; Girman, S.; Wang, S. Cell-based therapeutic strategies for replacement and preservation in retinal degenerative diseases. Prog. Retin. Eye Res. 2017, 58, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Mead, B.; Berry, M.; Logan, A.; Scott, R.A.; Leadbeater, W.; Scheven, B.A. Stem cell treatment of degenerative eye disease. Stem. Cell Res. 2015, 14, 243–257. [Google Scholar] [CrossRef] [Green Version]
- Goureau, O.; Orieux, G. [Photoreceptor cell transplantation for future treatment of retinitis pigmentosa]. Med. Sci. 2020, 36, 600–606. [Google Scholar] [CrossRef]
- Meyer, J.S.; Shearer, R.L.; Capowski, E.E.; Wright, L.S.; Wallace, K.A.; McMillan, E.L.; Zhang, S.C.; Gamm, D.M. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 16698–16703. [Google Scholar] [CrossRef] [Green Version]
- Zarbin, M. Cell-Based Therapy for Retinal Disease: The New Frontier. Methods Mol. Biol. 2019, 1834, 367–381. [Google Scholar] [CrossRef]
- Lee, I.K.; Ludwig, A.L.; Phillips, M.J.; Lee, J.; Xie, R.; Sajdak, B.S.; Jager, L.D.; Gong, S.; Gamm, D.M.; Ma, Z. Ultrathin micromolded 3D scaffolds for high-density photoreceptor layer reconstruction. Sci. Adv. 2021, 7, eabf0344. [Google Scholar] [CrossRef]
- Park, S.S.; Bauer, G.; Abedi, M.; Pontow, S.; Panorgias, A.; Jonnal, R.; Zawadzki, R.J.; Werner, J.S.; Nolta, J. Intravitreal autologous bone marrow CD34+ cell therapy for ischemic and degenerative retinal disorders: Preliminary phase 1 clinical trial findings. Investig. Ophthalmol. Vis. Sci. 2014, 56, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, R.C.; Messias, A.; Messias, K.; Arcieri, R.S.; Ruiz, M.A.; Souza, N.F.; Martins, L.C.; Jorge, R. Quality of life in patients with retinitis pigmentosa submitted to intravitreal use of bone marrow-derived stem cells (Reticell-clinical trial). Stem Cell Res. Ther. 2015, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriyan, A.E.; Albini, T.A.; Townsend, J.H.; Rodriguez, M.; Pandya, H.K.; Leonard, R.E., 2nd; Parrott, M.B.; Rosenfeld, P.J.; Flynn, H.W., Jr.; Goldberg, J.L. Vision Loss after Intravitreal Injection of Autologous “Stem Cells” for AMD. N. Engl. J. Med. 2017, 376, 1047–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, H.Y.; Watanabe, T.; Shirai, H.; Yamasaki, S.; Kinoshita, M.; Matsushita, K.; Hashiguchi, T.; Onoe, H.; Matsuyama, T.; Kuwahara, A.; et al. Medium- to long-term survival and functional examination of human iPSC-derived retinas in rat and primate models of retinal degeneration. EBioMedicine 2019, 39, 562–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; La, H.; Wei, X.; Zhou, Y.; Ou, Q.; Chen, Z.; Zhu, X.; Xu, J.Y.; Jin, C.; Gao, F.; et al. Transplantation Site Affects the Outcomes of Adipose-Derived Stem Cell-Based Therapy for Retinal Degeneration. Stem Cells Int. 2020, 2020, 9625798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuenca, N.; Fernández-Sánchez, L.; McGill, T.J.; Lu, B.; Wang, S.; Lund, R.; Huhn, S.; Capela, A. Phagocytosis of photoreceptor outer segments by transplanted human neural stem cells as a neuroprotective mechanism in retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6745–6756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugita, S.; Mandai, M.; Kamao, H.; Takahashi, M. Immunological aspects of RPE cell transplantation. Prog. Retin. Eye Res. 2021, 84, 100950. [Google Scholar] [CrossRef]
- Ha, T.W.; Jeong, J.H.; Shin, H.; Kim, H.K.; Im, J.S.; Song, B.H.; Hanna, J.; Oh, J.S.; Woo, D.H.; Han, J.; et al. Characterization of Endoplasmic Reticulum (ER) in Human Pluripotent Stem Cells Revealed Increased Susceptibility to Cell Death upon ER Stress. Cells 2020, 9, 1078. [Google Scholar] [CrossRef]
- Stern, J.H.; Tian, Y.; Funderburgh, J.; Pellegrini, G.; Zhang, K.; Goldberg, J.L.; Ali, R.R.; Young, M.; Xie, Y.; Temple, S. Regenerating Eye Tissues to Preserve and Restore Vision. Cell Stem Cell 2018, 22, 834–849. [Google Scholar] [CrossRef] [Green Version]
- Cehajic-Kapetanovic, J.; Eleftheriou, C.; Allen, A.E.; Milosavljevic, N.; Pienaar, A.; Bedford, R.; Davis, K.E.; Bishop, P.N.; Lucas, R.J. Restoration of Vision with Ectopic Expression of Human Rod Opsin. Curr. Biol. 2015, 25, 2111–2122. [Google Scholar] [CrossRef] [Green Version]
- Simunovic, M.P.; Shen, W.; Lin, J.Y.; Protti, D.A.; Lisowski, L.; Gillies, M.C. Optogenetic approaches to vision restoration. Exp. Eye Res. 2019, 178, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Nagel, G.; Szellas, T.; Huhn, W.; Kateriya, S.; Adeishvili, N.; Berthold, P.; Ollig, D.; Hegemann, P.; Bamberg, E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. USA 2003, 100, 13940–13945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gushchin, I.; Shevchenko, V.; Polovinkin, V.; Borshchevskiy, V.; Buslaev, P.; Bamberg, E.; Gordeliy, V. Structure of the light-driven sodium pump KR2 and its implications for optogenetics. FEBS J. 2016, 283, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Kandori, H.; Inoue, K.; Tsunoda, S.P. Light-Driven Sodium-Pumping Rhodopsin: A New Concept of Active Transport. Chem. Rev. 2018, 118, 10646–10658. [Google Scholar] [CrossRef] [PubMed]
- Garita-Hernandez, M.; Lampič, M.; Chaffiol, A.; Guibbal, L.; Routet, F.; Santos-Ferreira, T.; Gasparini, S.; Borsch, O.; Gagliardi, G.; Reichman, S.; et al. Restoration of visual function by transplantation of optogenetically engineered photoreceptors. Nat. Commun. 2019, 10, 4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordaz, J.D.; Wu, W.; Xu, X.M. Optogenetics and its application in neural degeneration and regeneration. Neural. Regen Res. 2017, 12, 1197–1209. [Google Scholar] [CrossRef]
- Lin, J.Y.; Knutsen, P.M.; Muller, A.; Kleinfeld, D.; Tsien, R.Y. ReaChR: A red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat. Neurosci. 2013, 16, 1499–1508. [Google Scholar] [CrossRef] [Green Version]
- Klapoetke, N.C.; Murata, Y.; Kim, S.S.; Pulver, S.R.; Birdsey-Benson, A.; Cho, Y.K.; Morimoto, T.K.; Chuong, A.S.; Carpenter, E.J.; Tian, Z.; et al. Independent optical excitation of distinct neural populations. Nat. Methods 2014, 11, 338–346. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.K.; Yang, S.J.; Pichamoorthy, N.; Young, N.P.; Kauvar, I.; Jennings, J.H.; Lerner, T.N.; Berndt, A.; Lee, S.Y.; Ramakrishnan, C.; et al. Simultaneous fast measurement of circuit dynamics at multiple sites across the mammalian brain. Nat. Methods 2016, 13, 325–328. [Google Scholar] [CrossRef]
- Park, D.W.; Schendel, A.A.; Mikael, S.; Brodnick, S.K.; Richner, T.J.; Ness, J.P.; Hayat, M.R.; Atry, F.; Frye, S.T.; Pashaie, R.; et al. Graphene-based carbon-layered electrode array technology for neural imaging and optogenetic applications. Nat. Commun. 2014, 5, 5258. [Google Scholar] [CrossRef]
- Renault, R.; Sukenik, N.; Descroix, S.; Malaquin, L.; Viovy, J.L.; Peyrin, J.M.; Bottani, S.; Monceau, P.; Moses, E.; Vignes, M. Combining microfluidics, optogenetics and calcium imaging to study neuronal communication in vitro. PLoS ONE 2015, 10, e0120680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Gradinaru, V.; Adamantidis, A.R.; Durand, R.; Airan, R.D.; de Lecea, L.; Deisseroth, K. Optogenetic interrogation of neural circuits: Technology for probing mammalian brain structures. Nat. Protoc. 2010, 5, 439–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, D.; Petreanu, L.; Ghitani, N.; Ranade, S.; Hromádka, T.; Mainen, Z.; Svoboda, K. Sparse optical microstimulation in barrel cortex drives learned behaviour in freely moving mice. Nature 2008, 451, 61–64. [Google Scholar] [CrossRef]
- Grosenick, L.; Marshel, J.H.; Deisseroth, K. Closed-loop and activity-guided optogenetic control. Neuron 2015, 86, 106–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagnon-Turcotte, G.; LeChasseur, Y.; Bories, C.; Messaddeq, Y.; De Koninck, Y.; Gosselin, B. A Wireless Headstage for Combined Optogenetics and Multichannel Electrophysiological Recording. IEEE Trans. Biomed. Circuits Syst 2017, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pansare, V.; Hejazi, S.; Faenza, W.; Prud’homme, R.K. Review of Long-Wavelength Optical and NIR Imaging Materials: Contrast Agents, Fluorophores and Multifunctional Nano Carriers. Chem. Mater. 2012, 24, 812–827. [Google Scholar] [CrossRef] [Green Version]
- Yue, L.; Weiland, J.D.; Roska, B.; Humayun, M.S. Retinal stimulation strategies to restore vision: Fundamentals and systems. Prog. Retin. Eye Res. 2016, 53, 21–47. [Google Scholar] [CrossRef] [Green Version]
- Bareket, L.; Barriga-Rivera, A.; Zapf, M.P.; Lovell, N.H.; Suaning, G.J. Progress in artificial vision through suprachoroidal retinal implants. J. Neural Eng. 2017, 14, 045002. [Google Scholar] [CrossRef]
- Rizzo, S.; Cinelli, L.; Finocchio, L.; Tartaro, R.; Santoro, F.; Gregori, N.Z. Assessment of Postoperative Morphologic Retinal Changes by Optical Coherence Tomography in Recipients of an Electronic Retinal Prosthesis Implant. JAMA Ophthalmol. 2019, 137, 272–278. [Google Scholar] [CrossRef]
- Hahn, P.; Fine, H.F. Practical Concepts With the Argus II Retinal Prosthesis. Ophthalmic Surg. Lasers Imaging Retina 2018, 49, 742–746. [Google Scholar] [CrossRef] [Green Version]
- Rachitskaya, A.; Lane, L.; Ehlers, J.; DeBenedictis, M.; Yuan, A. Argus II Retinal Prosthesis Implantation Using Three-Dimensional Visualization System. Retina 2019, 39 (Suppl. 1), S199–S200. [Google Scholar] [CrossRef] [PubMed]
- Stingl, K.; Bartz-Schmidt, K.U.; Besch, D.; Chee, C.K.; Cottriall, C.L.; Gekeler, F.; Groppe, M.; Jackson, T.L.; MacLaren, R.E.; Koitschev, A.; et al. Subretinal Visual Implant Alpha IMS--Clinical trial interim report. Vision Res. 2015, 111, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehlewein, L.; Troelenberg, N.; Stingl, K.; Schleehauf, S.; Kusnyerik, A.; Jackson, T.L.; MacLaren, R.E.; Chee, C.; Roider, J.; Wilhelm, B.; et al. Changes in microchip position after implantation of a subretinal vision prosthesis in humans. Acta Ophthalmol. 2019, 97, e871–e876. [Google Scholar] [CrossRef] [PubMed]
- Rachitskaya, A.; Yuan, A.; Davidson, S.; Streicher, M.; DeBenedictis, M.; Rosenfeldt, A.B.; Alberts, J. Computer-Assisted Immersive Visual Rehabilitation in Argus II Retinal Prosthesis Recipients. Ophthalmol. Retina 2020, 4, 613–619. [Google Scholar] [CrossRef]
- Winter, J.O.; Cogan, S.F.; Rizzo, J.F., 3rd. Retinal prostheses: Current challenges and future outlook. J. Biomater. Sci. Polym. Ed. 2007, 18, 1031–1055. [Google Scholar] [CrossRef]
- Dagnelie, G.; Christopher, P.; Arditi, A.; da Cruz, L.; Duncan, J.L.; Ho, A.C.; Olmos de Koo, L.C.; Sahel, J.A.; Stanga, P.E.; Thumann, G.; et al. Performance of real-world functional vision tasks by blind subjects improves after implantation with the Argus® II retinal prosthesis system. Clin. Exp. Ophthalmol. 2017, 45, 152–159. [Google Scholar] [CrossRef]
- Kitiratschky, V.B.; Stingl, K.; Wilhelm, B.; Peters, T.; Besch, D.; Sachs, H.; Gekeler, F.; Bartz-Schmidt, K.U.; Zrenner, E. Safety evaluation of “retina implant alpha IMS”—A prospective clinical trial. Graefes Arch. Clin. Exp. Ophthalmol. 2015, 253, 381–387. [Google Scholar] [CrossRef]
- Kienzler, M.A.; Isacoff, E.Y. Precise modulation of neuronal activity with synthetic photoswitchable ligands. Curr. Opin. Neurobiol. 2017, 45, 202–209. [Google Scholar] [CrossRef]
- Tochitsky, I.; Kienzler, M.A.; Isacoff, E.; Kramer, R.H. Restoring Vision to the Blind with Chemical Photoswitches. Chem. Rev. 2018, 118, 10748–10773. [Google Scholar] [CrossRef]
- Polosukhina, A.; Litt, J.; Tochitsky, I.; Nemargut, J.; Sychev, Y.; De Kouchkovsky, I.; Huang, T.; Borges, K.; Trauner, D.; Van Gelder, R.N.; et al. Photochemical restoration of visual responses in blind mice. Neuron 2012, 75, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Tochitsky, I.; Polosukhina, A.; Degtyar, V.E.; Gallerani, N.; Smith, C.M.; Friedman, A.; Van Gelder, R.N.; Trauner, D.; Kaufer, D.; Kramer, R.H. Restoring visual function to blind mice with a photoswitch that exploits electrophysiological remodeling of retinal ganglion cells. Neuron 2014, 81, 800–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerch, M.M.; Hansen, M.J.; van Dam, G.M.; Szymanski, W.; Feringa, B.L. Emerging Targets in Photopharmacology. Angew. Chem. Int. Ed. Engl. 2016, 55, 10978–10999. [Google Scholar] [CrossRef] [Green Version]
- Shim, G.; Ko, S.; Kim, D.; Le, Q.V.; Park, G.T.; Lee, J.; Kwon, T.; Choi, H.G.; Kim, Y.B.; Oh, Y.K. Light-switchable systems for remotely controlled drug delivery. J. Control. Release 2017, 267, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Bao, J.; Zhang, Y.; Li, Z.; Zhou, X.; Wan, C.; Huang, L.; Zhao, Y.; Han, G.; Xue, T. Mammalian Near-Infrared Image Vision through Injectable and Self-Powered Retinal Nanoantennae. Cell 2019, 177, 243–255.e215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hüll, K.; Benster, T.; Manookin, M.B.; Trauner, D.; Van Gelder, R.N.; Laprell, L. Photopharmacologic Vision Restoration Reduces Pathological Rhythmic Field Potentials in Blind Mouse Retina. Sci. Rep. 2019, 9, 13561. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assessment Items | Diagnosis | Reference | |
|---|---|---|---|
| History |
| Establish an initial profile.
| [131,132] |
| Clinical eye examination |
| Identifying ocular features that interfere with vision. | [133,134,135,136,137,138,139] |
| Spectral-domain optical coherence tomography (OCT) | Provides cross-sectional imaging of the fibrous layer.
| [140,141,142,143] | |
| Retinal imaging | Fundus imaging
|
| [136,144,145] |
| Most FAF cases to evaluate and monitor the progression of RP.
| [144,145,146,147] | |
| Fluorescence angiography/optical coherence tomography angiography (OCTA) | Tends to observe choroidal retinal atrophy. (Not commonly used). | [148] | |
| Adaptive optics scanning laser ophthalmoscopy (AOSLO) | High-resolution imaging modality to detect disease progression and assess the safety and efficacy of treatment.
| [149,150] | |
| Visual fields (VF) |
| Record the range of visual function from the center to the far edge. | [151,152,153] |
| Electroretin- ography |
| One of the important parameters for the diagnosis and staging of RP.
| [154,155,156,157] |
| Genetic Diagnostic Testing | Genetic counseling and targeted treatment Looking for potential new genes | [131,158] | |
| Neuroprotective Agent | Function and Progress | Reference |
|---|---|---|
| CNTF |
| [182] |
| [183,184] | |
| [185] | |
| [160,162] | |
| [186] | |
| BDNF |
| [187] |
| [162] | |
| [188] | |
| [189] | |
| FGF |
| [190] |
| [191] | |
| TUCDCA |
| [192,193,194] |
| [195,196,197] | |
| VA |
| [169,170] |
| Lutein |
| [198] |
| [199] | |
| DHA |
| [171] |
| [172] | |
| Calcium Channel Blockers |
| [200] |
| [201] | |
| Calpain Inhibitor |
| [202] |
| [203] | |
| VPA |
| [204] |
| HDACi |
| [205] |
| Status | Study Title | Interventions |
|---|---|---|
| Gene therapy | ||
| Recruiting (Phase 2) | A First-in-human, Proof of Concept Study of CPK850 in Patients With RLBP1 Retinitis Pigmentosa | Biological: CPK850 |
| Active, not recruiting (Phase 2) | Safety and Efficacy Study in Patients With Retinitis Pigmentosa Due to Mutations in PDE6B Gene | Biological: AAV2/5-hPDE6B |
| Recruiting (Phase 2) | 4D-125 in Patients With X-Linked Retinitis Pigmentosa (XLRP) | Biological: 4D-125 IVT Injection Other: Observational |
| Recruiting (Phase 3) | Gene Therapy Trial for the Treatment of X-linked Retinitis Pigmentosa Associated With Variants in the RPGR Gene | Biological: Genetic: AAV5-RPGR |
| Recruiting (Phase 3) | Follow-up Gene Therapy Trial for the Treatment of X-linked Retinitis Pigmentosa Associated With Variants in the RPGR Gene | Biological: Genetic: AAV5-RPGR 4e11 Biological: Genetic: AAV5-RPGR 2e11 |
| Recruiting (Phase 2) | Dose-escalation Study to Evaluate the Safety and Tolerability of GS030 in Subjects With Retinitis Pigmentosa | Combination Product: Gene therapy: GS030-DP AND Medical device: GS030-MD |
| Not yet recruiting (Phase 3) | A Clinical Trial Evaluating the Safety and Efficacy of a Single Subretinal Injection of AGTC-501 in Participants With X-linked Retinitis Pigmentosa Caused by RPGR Mutations | Biological: rAAV2tYF-GRK1-hRPGRco |
| Recruiting (Phase 2) | Safety and Efficacy of rAAV2tYF-GRK1-RPGR in Subjects With X-linked Retinitis Pigmentosa Caused by RPGR Mutations | Biological: rAAV2tYF-GRK1-RPGR |
| Recruiting (Phase 1/2) | Long Term Follow-Up Gene Therapy Study for XLRP RPGR | Biological: AAV-RPGR |
| Active, not recruiting (Phase 2) | Efficacy and Safety of vMCO-010 Optogenetic Therapy in Adults With Retinitis Pigmentosa [RESTORE] | Biological: Gene therapy product—vMCO-010 Procedure: Sham injection |
| Cell therapy | ||
| Recruiting (Phase 1) | Pilot Study of Intravitreal Autologous CD34+ Stem Cell Therapy for Retinitis Pigmentosa | Biological: Intravitreal autologous CD34+ cells |
| Recruiting (Phase 2) | Investigation of Therapeutic Efficacy and Safety of UMSCs for the Management of Retinitis Pigmentosa (RP) | Biological: Injection of stem cells in the sub-tenon space of eye for the management of retinitis pigmentosa Biological: Injection of stem cells in suprachoroidal space of eye for the management of retinitis pigmentosa |
| Active, not recruiting (Phase 2) | Safety of Repeat Intravitreal Injection of Human Retinal Progenitor Cells (jCell) in Adult Subjects With Retinitis Pigmentosa | Biological: human retinal progenitor cells |
| Unknown † (Phase 1) | Safety and Efficacy of Subretinal Transplantation of Clinical Human Embryonic Stem Cell Derived Retinal Pigment Epitheliums in Treatment of Retinitis Pigmentosa | Biological: Retinal pigment epitheliums transplantation |
| Unknown † (Phase 2) | Clinical Study to Evaluate Safety and Efficacy of BMMNC in Retinitis Pigmentosa | Biological: BMMNCs |
| Recruiting (Phase 1) | CNS10-NPC for the Treatment of RP | Biological: CNS10-NPC implantation |
| Unknown † (Phase 2) | Autologous Bone Marrow-Derived CD34+, CD133+, and CD271+ Stem Cell Transplantation for Retinitis Pigmentosa | Biological: Stem cell transplantation |
| Recruiting (Phase 2) | Interventional Study of Implantation of hESC-derived RPE in Patients With RP Due to Monogenic Mutation | Biological: Human embryonic stem cell-derived retinal pigment epithelium (RPE) |
| Unknown † (Early Phase 1) | Treatment of RP and LCA by Primary RPE Transplantation | Biological: Human primary retinal pigment epithelial (HuRPE) cells |
| Unknown † (Phase 1) | Stem Cells Therapy in Degenerative Diseases of the Retina | Biological: Stem/progenitor cells transplantation |
| Recruiting (Phase 1) | Safety of Cultured Allogeneic Adult Umbilical Cord Derived Mesenchymal Stem Cells for Eye Diseases | Biological: AlloRx |
| Drug treatment | ||
| Recruiting (Phase 2) | PDE6A Gene Therapy for Retinitis Pigmentosa | Drug: Subretinal injection of rAAV.hPDE6A |
| Recruiting (Phase 1/2) | The Study to Assess the Safety and Efficacy of OCU400 for Retinitis Pigmentosa | Drug: OCU400 Low Dose Drug: OCU400 Mid Dose Drug: OCU400 High Dose |
| Recruiting (Phase 1/2) | BS01 in Patients With Retinitis Pigmentosa | Drug: BS01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Liu, S.; Li, P.; Yao, K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int. J. Mol. Sci. 2022, 23, 4883. https://doi.org/10.3390/ijms23094883
Liu W, Liu S, Li P, Yao K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. International Journal of Molecular Sciences. 2022; 23(9):4883. https://doi.org/10.3390/ijms23094883
Chicago/Turabian StyleLiu, Wanqin, Shanshan Liu, Ping Li, and Kai Yao. 2022. "Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies" International Journal of Molecular Sciences 23, no. 9: 4883. https://doi.org/10.3390/ijms23094883
APA StyleLiu, W., Liu, S., Li, P., & Yao, K. (2022). Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. International Journal of Molecular Sciences, 23(9), 4883. https://doi.org/10.3390/ijms23094883