
Transcriptomic Profiling of Peripheral Edge of Lesions to Elucidate the Pathogenesis of Psoriasis Vulgaris

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
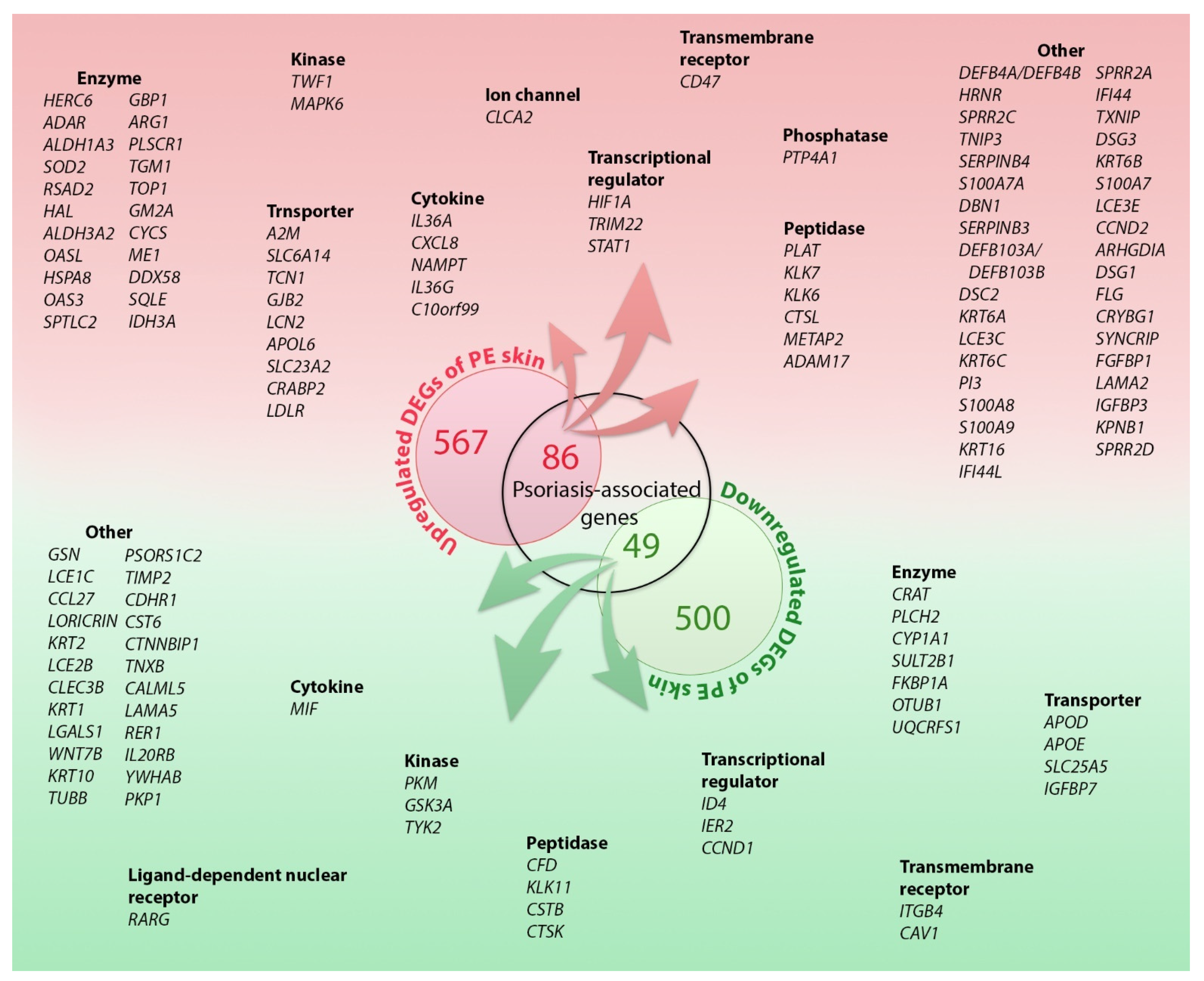
2.1. DEGs in the PE Skin
2.2. DEG Analysis
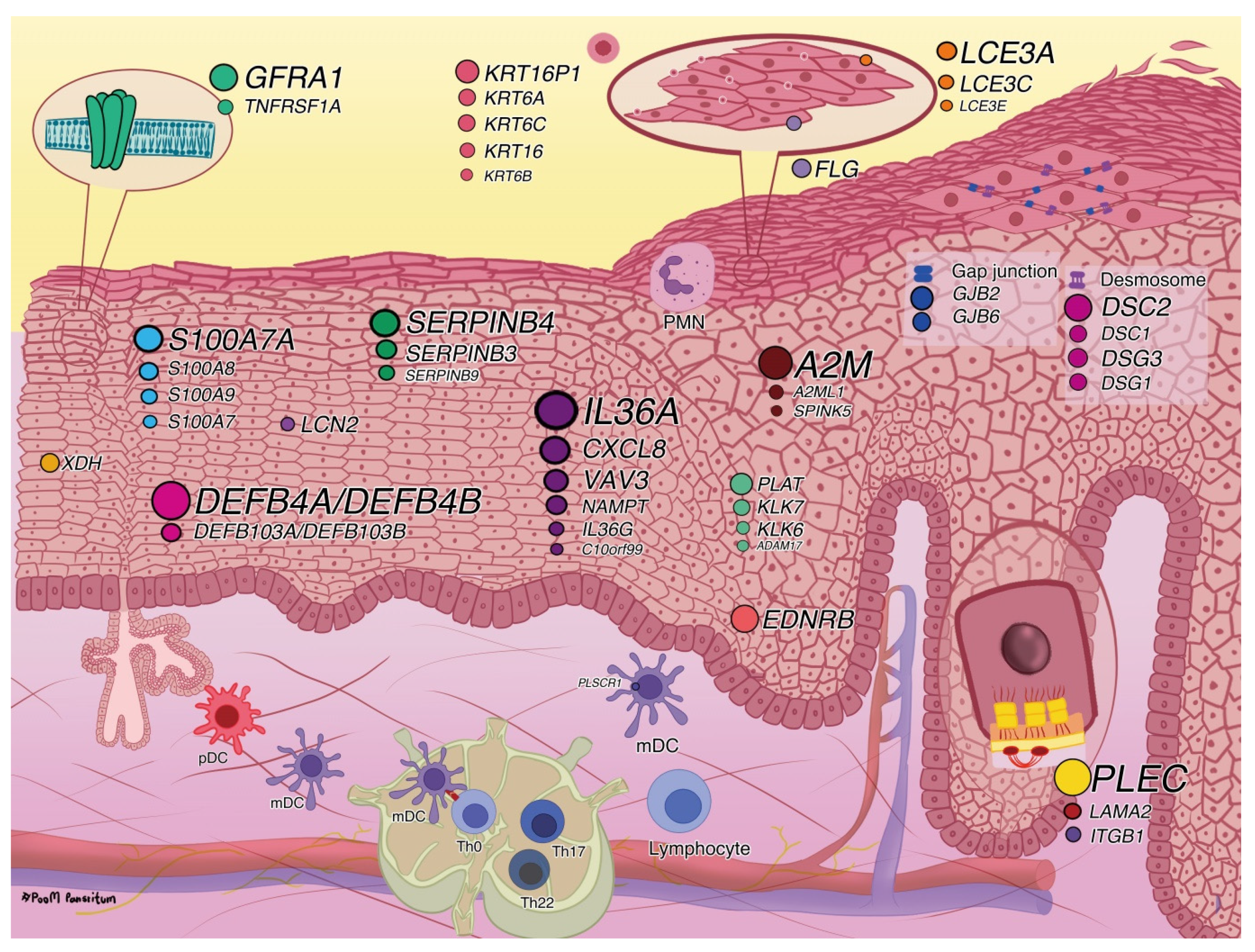
2.2.1. Disease Associations and Biological Functions
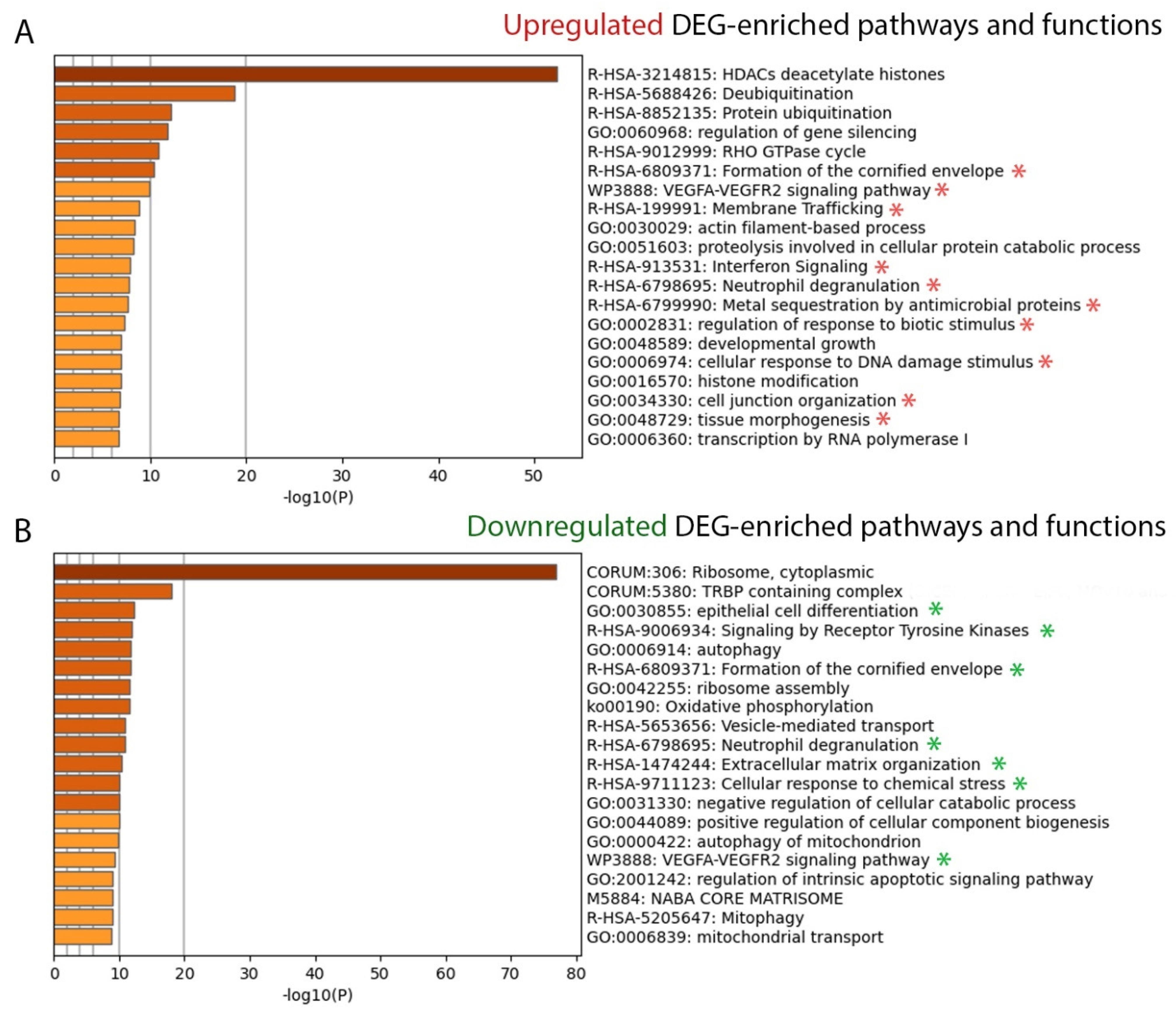
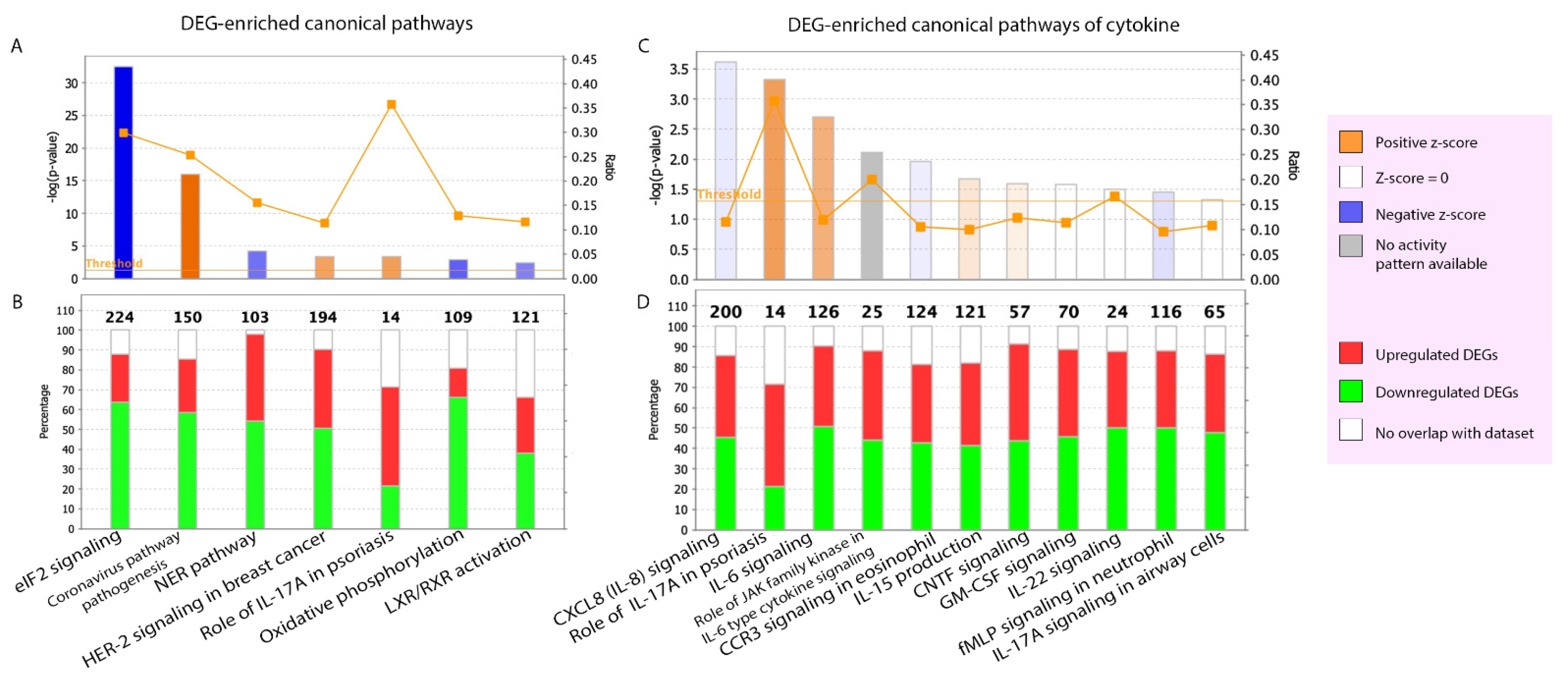
2.2.2. Canonical Pathways
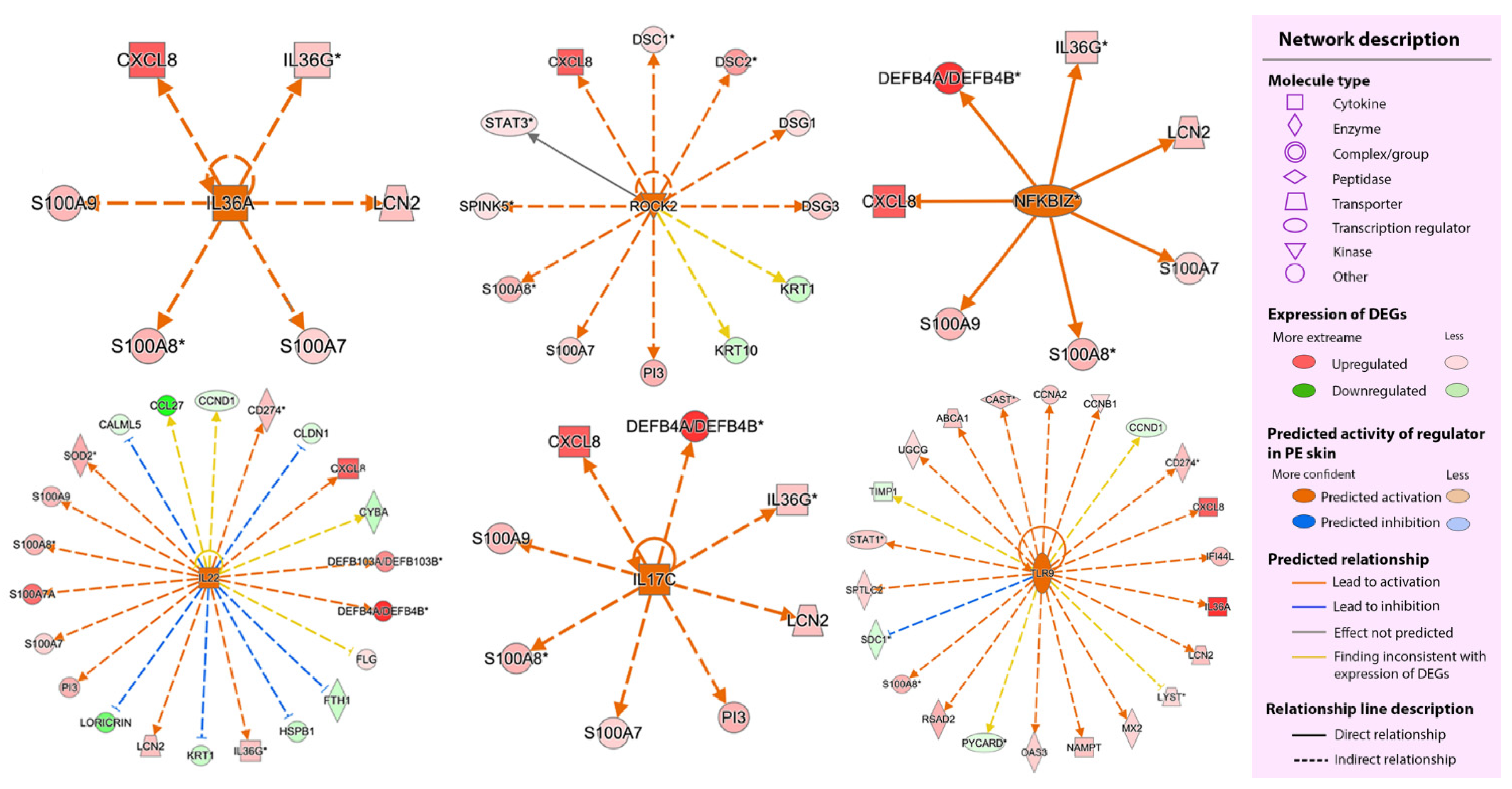
2.2.3. Upstream Regulators
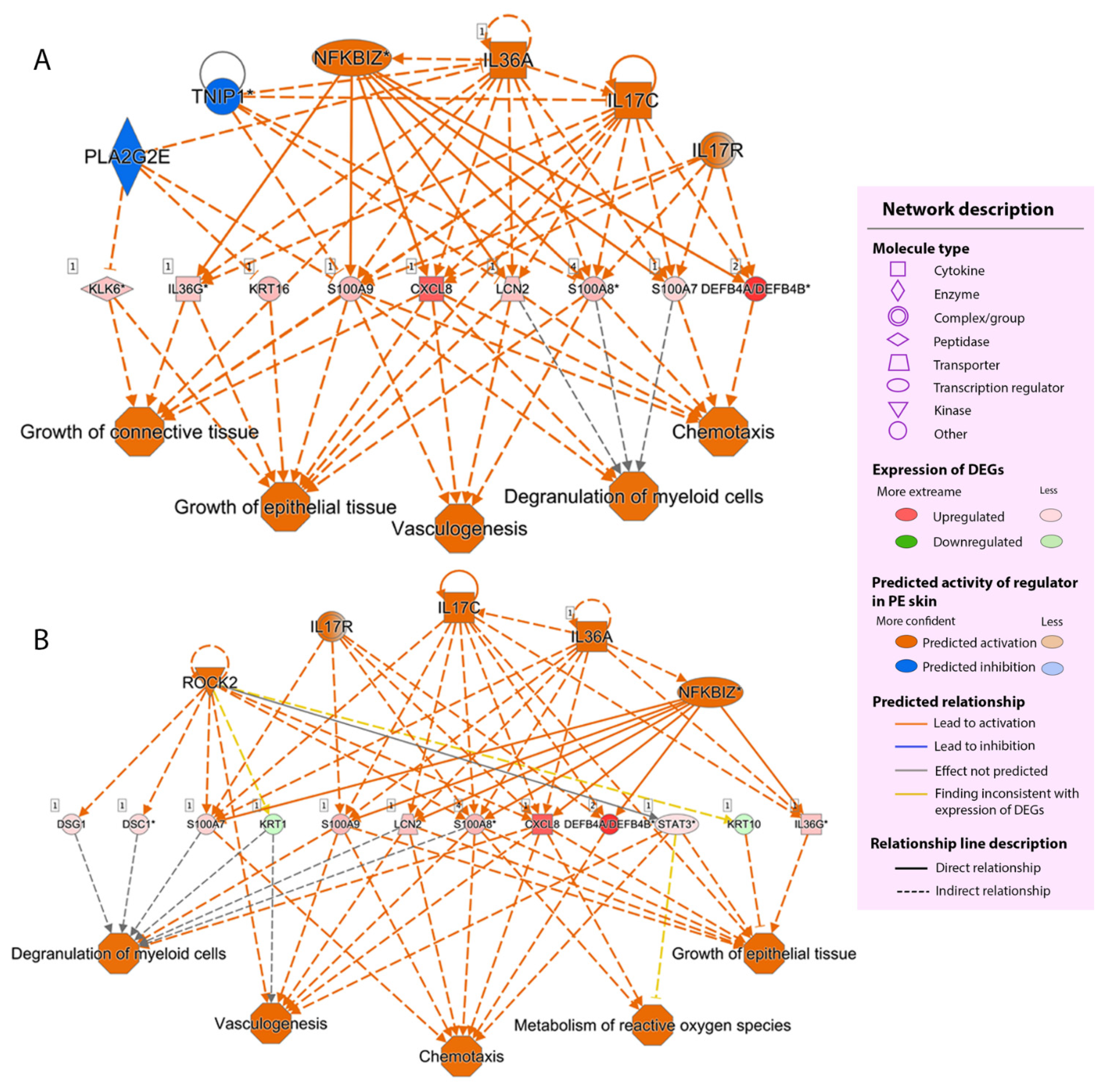
2.2.4. Regulator Effect
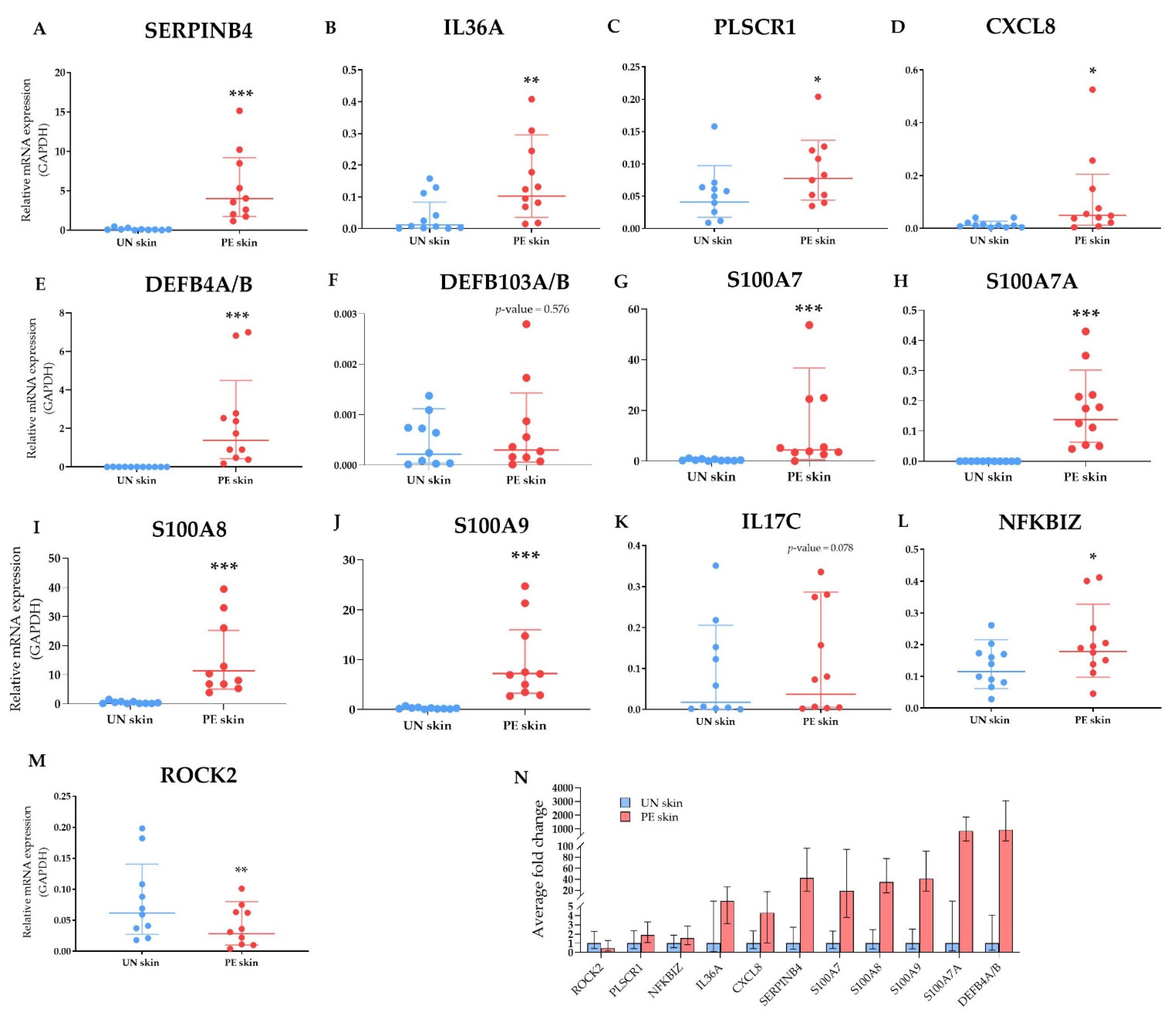
2.2.5. Validation of Selected DEGs by Multiplex Real-Time PCR
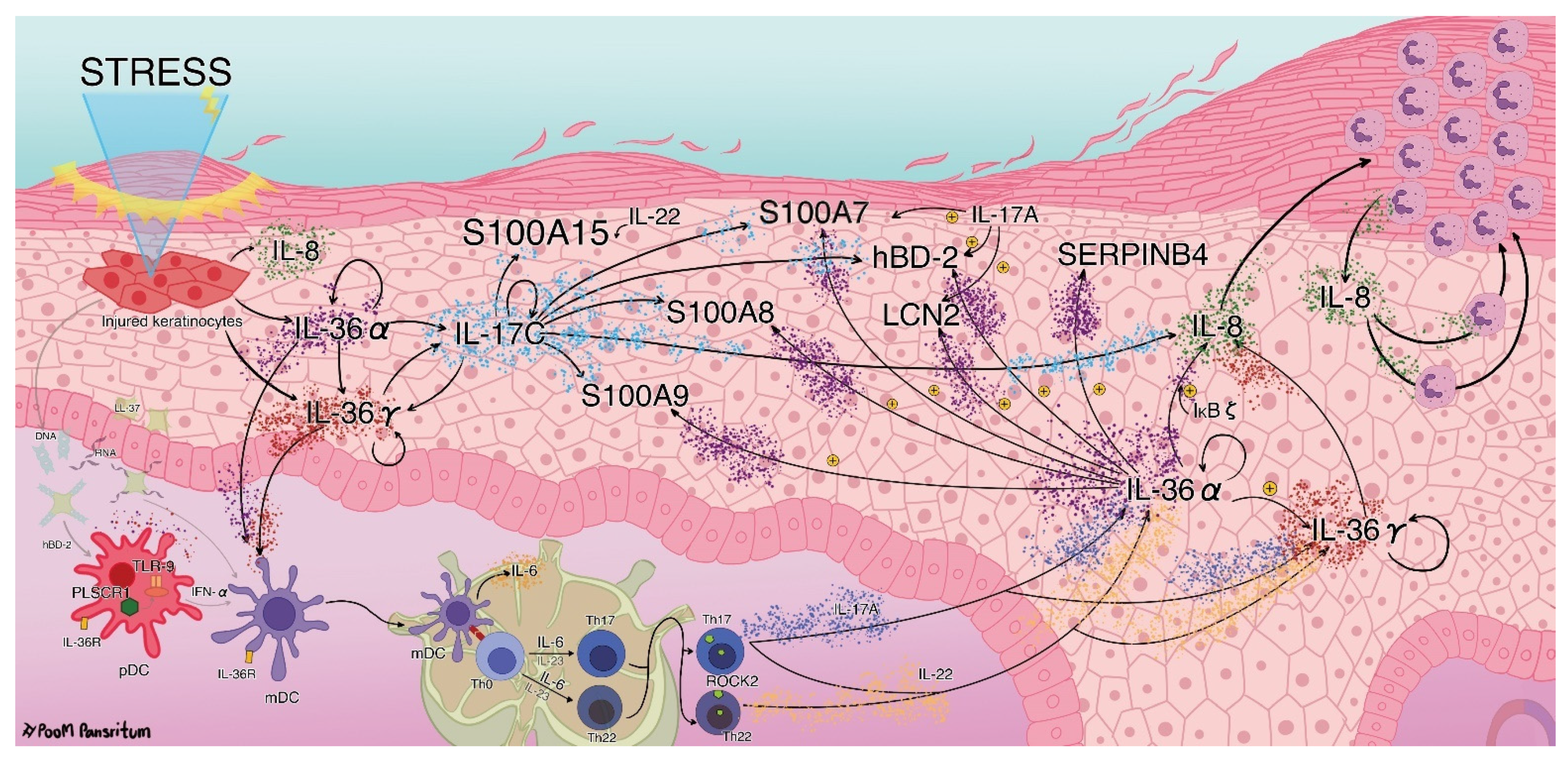
3. Discussion
4. Materials and Methods
4.1. Patients
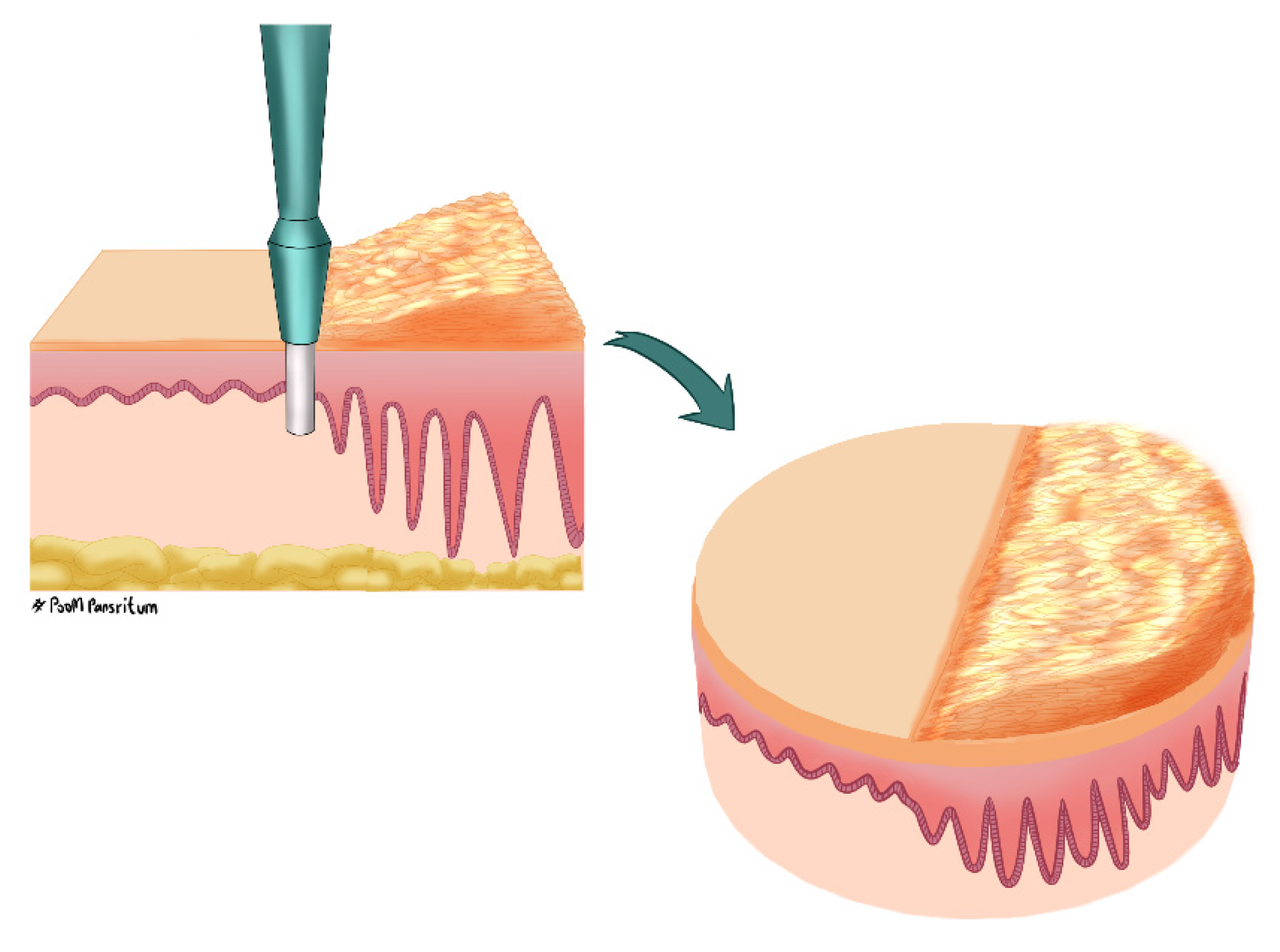
4.2. Tissue Sampling
4.3. High-Throughput Next-Generation Sequencing (NGS)
4.4. Sequencing Data Analysis
4.5. DEGs Analysis
4.6. Quantitative Multiplex Real-Time PCR (Multiplex qPCR)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griffiths, C.E.M.; Armstrong, A.W.; Gudjonsson, J.E.; Barker, J.N.W.N. Psoriasis. Lancet 2021, 397, 1301–1315. [Google Scholar] [CrossRef]
- Dastoli, S.; Nisticò, S.P.; Morrone, P.; Patruno, C.; Leo, A.; Citraro, R.; Gallelli, L.; Russo, E.; De Sarro, G.; Bennardo, L. Colchicine in Managing Skin Conditions: A Systematic Review. Pharmaceutics 2022, 14, 294. [Google Scholar] [CrossRef] [PubMed]
- Amoruso, G.; Nisticò, S.; Iannone, L.; Russo, E.; Rago, G.; Patruno, C.; Bennardo, L. Ixekizumab May Improve Renal Function in Psoriasis. Healthcare 2021, 9, 543. [Google Scholar] [CrossRef] [PubMed]
- Harden, J.L.; Krueger, J.G.; Bowcock, A.M. The immunogenetics of Psoriasis: A comprehensive review. J. Autoimmun. 2015, 64, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Di Meglio, P.; Villanova, F.; Nestle, F.O. Psoriasis. Cold Spring Harb. Perspect. Med. 2014, 4, a015354. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Luo, S.; Huang, Y.; Lu, Q. Critical role of environmental factors in the pathogenesis of psoriasis. J. Dermatol. 2017, 44, 863–872. [Google Scholar] [CrossRef] [Green Version]
- Perera, G.K.; Di Meglio, P.; Nestle, F.O. Psoriasis. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 385–422. [Google Scholar] [CrossRef]
- Kim, J.; Krueger, J.G. The Immunopathogenesis of Psoriasis. Dermatol. Clin. 2015, 33, 13–23. [Google Scholar] [CrossRef]
- Hawkes, J.E.; Chan, T.C.; Krueger, J.G. Psoriasis pathogenesis and the development of novel targeted immune therapies. J. Allergy Clin. Immunol. 2017, 140, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Iannone, L.F.; Bennardo, L.; Palleria, C.; Roberti, R.; De Sarro, C.; Naturale, M.D.; Dastoli, S.; Donato, L.; Manti, A.; Valenti, G.; et al. Safety profile of biologic drugs for psoriasis in clinical practice: An Italian prospective pharmacovigilance study. PLoS ONE 2020, 15, e0241575. [Google Scholar] [CrossRef]
- Jabbari, A.; Suárez-Fariñas, M.; Dewell, S.; Krueger, J.G. Transcriptional Profiling of Psoriasis Using RNA-seq Reveals Previously Unidentified Differentially Expressed Genes. J. Investig. Dermatol. 2012, 132, 246–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Tsoi, L.C.; Swindell, W.R.; Gudjonsson, J.E.; Tejasvi, T.; Johnston, A.; Ding, J.; Stuart, P.E.; Xing, X.; Kochkodan, J.J.; et al. Transcriptome Analysis of Psoriasis in a Large Case–Control Sample: RNA-Seq Provides Insights into Disease Mechanisms. J. Investig. Dermatol. 2014, 134, 1828–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swindell, W.R.; Xing, X.; Voorhees, J.J.; Elder, J.T.; Johnston, A.; Gudjonsson, J.E. Integrative RNA-seq and microarray data analysis reveals GC content and gene length biases in the psoriasis transcriptome. Physiol. Genom. 2014, 46, 533–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, R.; Yan, D.; Chang, H.-W.; Lee, K.; Bhattarai, S.; Huang, Z.-M.; Nakamura, M.; Singh, R.; Afifi, L.; Taravati, K.; et al. RNA-seq and flow-cytometry of conventional, scalp, and palmoplantar psoriasis reveal shared and distinct molecular pathways. Sci. Rep. 2018, 8, 11368. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Gong, Y.; Cui, L.; Hu, Y.; Zhou, Q.; Chen, Z.; Yu, Y.; Chen, Y.; Xu, P.; Zhang, X.; et al. High-throughput transcriptome and pathogenesis analysis of clinical psoriasis. J. Dermatol. Sci. 2020, 98, 109–118. [Google Scholar] [CrossRef]
- Kõks, S.; Keermann, M.; Reimann, E.; Prans, E.; Abram, K.; Silm, H.; Kõks, G.; Kingo, K. Psoriasis-Specific RNA Isoforms Identified by RNA-Seq Analysis of 173,446 Transcripts. Front. Med. 2016, 3, 46. [Google Scholar] [CrossRef] [Green Version]
- Zolotarenko, A.; Chekalin, E.; Mesentsev, A.; Kiseleva, L.; Gribanova, E.; Mehta, R.; Baranova, A.; Tatarinova, T.; Piruzian, E.S.; Bruskin, S. Integrated computational approach to the analysis of RNA-seq data reveals new transcriptional regulators of psoriasis. Exp. Mol. Med. 2016, 48, e268. [Google Scholar] [CrossRef] [Green Version]
- Ahn, R.; Gupta, R.; Lai, K.; Chopra, N.; Arron, S.T.; Liao, W. Network analysis of psoriasis reveals biological pathways and roles for coding and long non-coding RNAs. BMC Genom. 2016, 17, 841. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Fariñas, M.; Li, K.; Fuentes-Duculan, J.; Hayden, K.; Brodmerkel, C.; Krueger, J.G. Expanding the Psoriasis Disease Profile: Interrogation of the Skin and Serum of Patients with Moderate-to-Severe Psoriasis. J. Investig. Dermatol. 2012, 132, 2552–2564. [Google Scholar] [CrossRef] [Green Version]
- Zolotarenko, A.; Chekalin, E.; Mehta, R.; Baranova, A.; Tatarinova, T.V.; Bruskin, S. Identification of Transcriptional Regulators of Psoriasis from RNA-Seq Experiments. Methods Mol. Biol. 2017, 1613, 355–370. [Google Scholar] [CrossRef]
- De Oliveira, P.S.S.; Pereira, M.C.; De Paula, S.K.S.; Lima, E.V.A.; Lima, M.M.D.A.; De Arruda, R.G.; De Oliveira, W.L.M.; Duarte, L.B.P.; Pitta, I.D.R.; Rêgo, M.J.M.B.; et al. Increased IL17A, IFNG, and FOXP3 Transcripts in Moderate-Severe Psoriasis: A Major Influence Exerted by IL17A in Disease Severity. Mediat. Inflamm. 2016, 2016, 4395276. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, L.; Srivastava, A.; Meisgen, F.; Mahapatra, K.; Xia, P.; Landén, N.; Pivarcsi, A.; Sonkoly, E. The Keratinocyte Transcriptome in Psoriasis: Pathways Related to Immune Responses, Cell Cycle and Keratinization. Acta Derm. Venereol. 2019, 99, 196–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swindell, W.R.; Sarkar, M.; Liang, Y.; Xing, X.; Baliwag, J.; Elder, J.T.; Johnston, A.; Ward, N.L.; Gudjonsson, J.E. RNA-seq identifies a diminished differentiation gene signature in primary monolayer keratinocytes grown from lesional and uninvolved psoriatic skin. Sci. Rep. 2017, 7, 18045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lee, J.; Kim, H.J.; Kameyama, N.; Nazarian, R.; Der, E.; Cohen, S.; Guttman-Yassky, E.; Putterman, C.; Krueger, J.G. Single-cell transcriptomics applied to emigrating cells from psoriasis elucidate pathogenic versus regulatory immune cell subsets. J. Allergy Clin. Immunol. 2021, 148, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Goodfield, M.; Hull, S.M.; Holland, D.; Roberts, G.; Wood, E.; Reid, S.; Cunliffe, W. Investigations of the ‘active’ edge of plaque psoriasis: Vascular proliferation precedes changes in epidermal keratin. Br. J. Dermatol. 1994, 131, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Hull, S.M.; Goodfield, M.; Wood, E.J.; Cunliffe, W.J. Active and Inactive Edges of Psoriatic Plaques: Identification by Tracing and Investigation by Laser-Doppler Flowmetry and Immunocytochemical Techniques. J. Investig. Dermatol. 1989, 92, 782–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komine, M.; Karakawa, M.; Takekoshi, T.; Sakurai, N.; Minatani, Y.; Mitsui, H.; Tada, Y.; Saeki, H.; Asahina, A.; Tamaki, K. Early Inflammatory Changes in the “Perilesional Skin” of Psoriatic Plaques: Is there Interaction between Dendritic Cells and Keratinocytes? J. Investig. Dermatol. 2007, 127, 1915–1922. [Google Scholar] [CrossRef] [Green Version]
- Alshenawy, H.A.; Hasby, E.A. Immunophenotyping of dendritic cells in lesional, perilesional and distant skin of chronic plaque psoriasis. Cell. Immunol. 2011, 269, 115–119. [Google Scholar] [CrossRef]
- Nattkemper, L.A.; Tey, H.L.; Valdes-Rodriguez, R.; Lee, H.; Mollanazar, N.K.; Albornoz, C.; Sanders, K.M.; Yosipovitch, G. The Genetics of Chronic Itch: Gene Expression in the Skin of Patients with Atopic Dermatitis and Psoriasis with Severe Itch. J. Investig. Dermatol. 2018, 138, 1311–1317. [Google Scholar] [CrossRef] [Green Version]
- Swindell, W.R.; Sarkar, M.; Liang, Y.; Xing, X.; Gudjonsson, J.E. Cross-Disease Transcriptomics: Unique IL-17A Signaling in Psoriasis Lesions and an Autoimmune PBMC Signature. J. Investig. Dermatol. 2016, 136, 1820–1830. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Lan, Z.; Ye, J.; Pang, L.; Liu, Y.; Wu, W.; Qin, X.; Guo, Y.; Zhang, P. Cytokine Storm: The Primary Determinant for the Pathophysiological Evolution of COVID-19 Deterioration. Front. Immunol. 2021, 12, 589095. [Google Scholar] [CrossRef] [PubMed]
- Büchau, A.S.; Gallo, R.L. Innate immunity and antimicrobial defense systems in psoriasis. Clin. Dermatol. 2007, 25, 616–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Yamasaki, K. Psoriasis and Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 6791. [Google Scholar] [CrossRef] [PubMed]
- de Jongh, G.J.; Zeeuwen, P.L.; Kucharekova, M.; Pfundt, R.; van der Valk, P.G.; Blokx, W.; Dogan, A.; Hiemstra, P.S.; van de Kerkhof, P.C.; Schalkwijk, J. High Expression Levels of Keratinocyte Antimicrobial Proteins in Psoriasis Compared with Atopic Dermatitis. J. Investig. Dermatol. 2005, 125, 1163–1173. [Google Scholar] [CrossRef]
- Steinz, K.; Schubert, S.; Harder, J.; Gerdes, S.; Mrowietz, U.; Gläser, R. Bacterial soft tissue infection in psoriasis despite induction of epidermal antimicrobial peptides. Exp. Dermatol. 2014, 23, 862–864. [Google Scholar] [CrossRef]
- Bierkarre, H.; Harder, J.; Cuthbert, R.; Emery, P.; Leuschner, I.; Mrowietz, U.; Hedderich, J.; McGonagle, D.; Gläser, R. Differential expression of antimicrobial peptides in psoriasis and psoriatic arthritis as a novel contributory mechanism for skin and joint disease heterogeneity. Scand. J. Rheumatol. 2015, 45, 188–196. [Google Scholar] [CrossRef]
- Tewary, P.; De La Rosa, G.; Sharma, N.; Rodriguez, L.G.; Tarasov, S.G.; Howard, O.M.Z.; Shirota, H.; Steinhagen, F.; Klinman, D.M.; Yang, D.; et al. β-Defensin 2 and 3 Promote the Uptake of Self or CpG DNA, Enhance IFN-α Production by Human Plasmacytoid Dendritic Cells, and Promote Inflammation. J. Immunol. 2013, 191, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Wolf, R.; Mirmohammadsadegh, A.; Walz, M.; Lysa, B.; Tartler, U.; Remus, R.; Hengge, U.; Michel, G.; Ruzicka, T. Molecular cloning and characterization of alternatively spliced mRNA isoforms from psoriatic skin encoding a novel member of the S100 family. FASEB J. 2003, 17, 1969–1971. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Jang, S.; Min, J.-K.; Lee, K.; Sohn, K.-C.; Lim, J.-S.; Im, M.; Lee, H.-E.; Seo, Y.-J.; Kim, C.-D.; et al. S100A8 and S100A9 are messengers in the crosstalk between epidermis and dermis modulating a psoriatic milieu in human skin. Biochem. Biophys. Res. Commun. 2012, 423, 647–653. [Google Scholar] [CrossRef]
- Hegyi, Z.; Zwicker, S.; Bureik, D.; Peric, M.; Koglin, S.; Batycka-Baran, A.; Prinz, J.C.; Ruzicka, T.; Schauber, J.; Wolf, R. Vitamin D Analog Calcipotriol Suppresses the Th17 Cytokine–Induced Proinflammatory S100 “Alarmins” Psoriasin (S100A7) and Koebnerisin (S100A15) in Psoriasis. J. Investig. Dermatol. 2012, 132, 1416–1424. [Google Scholar] [CrossRef] [Green Version]
- Wolf, R.; Mascia, F.; Dharamsi, A.; Howard, O.M.Z.; Cataisson, C.; Bliskovski, V.; Winston, J.; Feigenbaum, L.; Lichti, U.; Ruzicka, T.; et al. Gene from a Psoriasis Susceptibility Locus Primes the Skin for Inflammation. Sci. Transl. Med. 2010, 2, 61ra90. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Yang, G.; Xiao, F.; Xie, J.; Wang, S.; Lu, L.; Cui, D. Role of Th22 Cells in the Pathogenesis of Autoimmune Diseases. Front. Immunol. 2021, 12, 688066. [Google Scholar] [CrossRef] [PubMed]
- Rosen, T.; Nolan, E.M. Metal Sequestration and Antimicrobial Activity of Human Calprotectin Are pH-Dependent. Biochemistry 2020, 59, 2468–2478. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Higuchi, D.; Takahashi, T.; Ogo, M.; Baciu, P.; Goetinck, P.F.; Hibino, T. Overexpression of Serpin Squamous Cell Carcinoma Antigens in Psoriatic Skin. J. Investig. Dermatol. 2002, 118, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Izuhara, K.; Yamaguchi, Y.; Ohta, S.; Nunomura, S.; Nanri, Y.; Azuma, Y.; Nomura, N.; Noguchi, Y.; Aihara, M. Squamous Cell Carcinoma Antigen 2 (SCCA2, SERPINB4): An Emerging Biomarker for Skin Inflammatory Diseases. Int. J. Mol. Sci. 2018, 19, 1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iversen, O.-J.; Lysvand, H.; Hagen, L. The autoantigen Pso p27: A post-translational modification of SCCA molecules. Autoimmunity 2010, 44, 229–234. [Google Scholar] [CrossRef]
- Lysvand, H.; Hagen, L.; Klubicka, L.; Slupphaug, G.; Iversen, O.-J. Psoriasis pathogenesis—Pso p27 is generated from SCCA1 with chymase. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 734–738. [Google Scholar] [CrossRef] [Green Version]
- Dalaker, T.J.M. Expression of the Psoriasis-associated Antigen, Pso p27, is Inhibited by Cyclosporin A. Acta Derm. Venereol. 1999, 79, 281–284. [Google Scholar] [CrossRef]
- Åsbakk, K.; Bergh, K.; Iversen, O. The psoriasis-associated antigen, pso p27, participates in the formation of complement activating immune-complexes in psoriatic scale. APMIS 1990, 98, 143–149. [Google Scholar] [CrossRef]
- Johnston, A.; Xing, X.; Guzman, A.M.; Riblett, M.; Loyd, C.M.; Ward, N.L.; Wohn, C.; Prens, E.P.; Wang, F.; Maier, L.E.; et al. IL-1F5, -F6, -F8, and -F9: A Novel IL-1 Family Signaling System That Is Active in Psoriasis and Promotes Keratinocyte Antimicrobial Peptide Expression. J. Immunol. 2011, 186, 2613–2622. [Google Scholar] [CrossRef] [Green Version]
- Keermann, M.; Koks, S.; Reimann, E.; Abram, K.; Erm, T.; Silm, H.; Kingo, K. Expression of IL-36 family cytokines and IL-37 but not IL-38 is altered in psoriatic skin. J. Dermatol. Sci. 2015, 80, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Boutet, M.; Bart, G.; Penhoat, M.; Amiaud, J.; Brulin, B.; Charrier, C.; Morel, F.; Lecron, J.; Rolli-Derkinderen, M.; Bourreille, A.; et al. Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin. Exp. Immunol. 2016, 184, 159–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Liu, Y.; Li, C.; Chang, L.; Wang, W.; Wang, Z.; Gao, X.; Ryffel, B.; Wu, Y.; Lai, Y. IL-36γ Induced by the TLR3-SLUG-VDR Axis Promotes Wound Healing via REG3A. J. Investig. Dermatol. 2017, 137, 2620–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furue, K.; Ito, T.; Tanaka, Y.; Yumine, A.; Hashimoto-Hachiya, A.; Takemura, M.; Murata, M.; Yamamura, K.; Tsuji, G.; Furue, M. Cyto/chemokine profile of in vitro scratched keratinocyte model: Implications of significant upregulation of CCL20, CXCL8 and IL36G in Koebner phenomenon. J. Dermatol. Sci. 2019, 94, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, S.; Garcet, S.; Salud-Gnilo, C.; Gonzalez, J.; Li, X.; Murai-Yamamura, M.; Yamamura, K.; Rambhia, D.; Kunjravia, N.; Guttman-Yassky, E.; et al. IL-36 and IL-17A Cooperatively Induce a Psoriasis-Like Gene Expression Response in Human Keratinocytes. J. Investig. Dermatol. 2021, 141, 2086–2090. [Google Scholar] [CrossRef]
- Catapano, M.; Vergnano, M.; Romano, M.; Mahil, S.K.; Choon, S.-E.; Burden, A.D.; Young, H.S.; Carr, I.M.; Lachmann, H.J.; Lombardi, G.; et al. IL-36 Promotes Systemic IFN-I Responses in Severe Forms of Psoriasis. J. Investig. Dermatol. 2020, 140, 816–826.e3. [Google Scholar] [CrossRef] [Green Version]
- Foster, A.M.; Baliwag, J.; Chen, C.S.; Guzman, A.M.; Stoll, S.W.; Gudjonsson, J.E.; Ward, N.L.; Johnston, A. IL-36 Promotes Myeloid Cell Infiltration, Activation, and Inflammatory Activity in Skin. J. Immunol. 2014, 192, 6053–6061. [Google Scholar] [CrossRef] [Green Version]
- Akdis, M.; Palomares, O.; Van De Veen, W.; Van Splunter, M.; Akdis, C.A. TH17 and TH22 cells: A confusion of antimicrobial response with tissue inflammation versus protection. J. Allergy Clin. Immunol. 2012, 129, 1438–1449. [Google Scholar] [CrossRef]
- Srivastava, A.; Luo, L.; Lohcharoenkal, W.; Meisgen, F.; Pasquali, L.; Pivarcsi, A.; Sonkoly, E. Cross-talk between IFN-γ and TWEAK through miR-149 amplifies skin inflammation in psoriasis. J. Allergy Clin. Immunol. 2021, 147, 2225–2235. [Google Scholar] [CrossRef]
- Fritz, Y.; Klenotic, P.A.; Swindell, W.R.; Yin, Z.Q.; Groft, S.G.; Zhang, L.; Baliwag, J.; Camhi, M.I.; Diaconu, D.; Young, A.B.; et al. Induction of Alternative Proinflammatory Cytokines Accounts for Sustained Psoriasiform Skin Inflammation in IL-17C+IL-6KO Mice. J. Investig. Dermatol. 2017, 137, 696–705. [Google Scholar] [CrossRef] [Green Version]
- Carrier, Y.; Ma, H.-L.; Ramon, H.E.; Napierata, L.; Small, C.; O’Toole, M.; Young, D.A.; Fouser, L.A.; Nickerson-Nutter, C.; Collins, M.; et al. Inter-Regulation of Th17 Cytokines and the IL-36 Cytokines In Vitro and In Vivo: Implications in Psoriasis Pathogenesis. J. Investig. Dermatol. 2011, 131, 2428–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wawrzycki, B.; Pietrzak, A.; Grywalska, E.; Krasowska, D.; Chodorowska, G.; Roliński, J. Interleukin-22 and Its Correlation with Disease Activity in Plaque Psoriasis. Arch. Immunol. Ther. Exp. 2019, 67, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.; Hennig, A.; Lorscheid, S.; Grondona, P.; Schulze-Osthoff, K.; Hailfinger, S.; Kramer, D. IκBζ is a key transcriptional regulator of IL-36–driven psoriasis-related gene expression in keratinocytes. Proc. Natl. Acad. Sci. USA 2018, 115, 10088–10093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashiguchi, Y.; Yabe, R.; Chung, S.-H.; Murayama, M.A.; Yoshida, K.; Matsuo, K.; Kubo, S.; Saijo, S.; Nakamura, Y.; Matsue, H.; et al. IL-36α from Skin-Resident Cells Plays an Important Role in the Pathogenesis of Imiquimod-Induced Psoriasiform Dermatitis by Forming a Local Autoamplification Loop. J. Immunol. 2018, 201, 167–182. [Google Scholar] [CrossRef]
- Johnston, A.; Fritz, Y.; Dawes, S.M.; Diaconu, D.; Al-Attar, P.M.; Guzman, A.M.; Chen, C.S.; Fu, W.; Gudjonsson, J.E.; McCormick, T.S.; et al. Keratinocyte Overexpression of IL-17C Promotes Psoriasiform Skin Inflammation. J. Immunol. 2013, 190, 2252–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Carrozzi, V.; Sambandam, A.; Luis, E.; Lin, Z.; Jeet, S.; Lesch, J.; Hackney, J.; Kim, J.; Zhou, M.; Lai, J.; et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat. Immunol. 2011, 12, 1159–1166. [Google Scholar] [CrossRef]
- Lauffer, F.; Jargosch, M.; Baghin, V.; Krause, L.; Kempf, W.; Absmaier-Kijak, M.; Morelli, M.; Madonna, S.; Marsais, F.; Lepescheux, L.; et al. IL-17C amplifies epithelial inflammation in human psoriasis and atopic eczema. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 800–809. [Google Scholar] [CrossRef]
- Li, H.; Chen, J.; Huang, A.; Stinson, J.; Heldens, S.; Foster, J.; Dowd, P.; Gurney, A.L.; Wood, W.I. Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proc. Natl. Acad. Sci. USA 2000, 97, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Johansen, C.; Mose, M.; Ommen, P.; Bertelsen, T.; Vinter, H.; Hailfinger, S.; Lorscheid, S.; Schulze-Osthoff, K.; Iversen, L. IκBζ is a key driver in the development of psoriasis. Proc. Natl. Acad. Sci. USA 2015, 112, E5825–E5833. [Google Scholar] [CrossRef] [Green Version]
- Muromoto, R.; Hirao, T.; Tawa, K.; Hirashima, K.; Kon, S.; Kitai, Y.; Matsuda, T. IL-17A plays a central role in the expression of psoriasis signature genes through the induction of IκB-ζ in keratinocytes. Int. Immunol. 2016, 28, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Ovesen, S.; Schulze-Osthoff, K.; Iversen, L.; Johansen, C. IkBζ is a Key Regulator of Tumour Necrosis Factor-a and Interleukin-17A-mediated Induction of Interleukin-36g in Human Keratinocytes. Acta Derm. Venereol. 2021, 101, adv00386. [Google Scholar] [CrossRef]
- Johansen, C.; Bertelsen, T.; Ljungberg, C.; Mose, M.; Iversen, L. Characterization of TNF-α– and IL-17A–Mediated Synergistic Induction of DEFB4 Gene Expression in Human Keratinocytes through IκBζ. J. Investig. Dermatol. 2016, 136, 1608–1616. [Google Scholar] [CrossRef] [Green Version]
- Barker, J.N.; Jones, M.L.; Mitra, R.S.; Crockett-Torabe, E.; Fantone, J.C.; Kunkel, S.L.; Warren, J.S.; Dixit, V.M.; Nickoloff, B.J. Modulation of keratinocyte-derived interleukin-8 which is chemotactic for neutrophils and T lymphocytes. Am. J. Pathol. 1991, 139, 869–876. [Google Scholar] [PubMed]
- Koch, A.E.; Polverini, P.J.; Kunkel, S.L.; Harlow, L.A.; DiPietro, L.A.; Elner, V.M.; Elner, S.G.; Strieter, R.M. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 1992, 258, 1798–1801. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, B.J.; Mitra, R.S.; Varani, J.; Dixit, V.M.; Polverini, P.J. Aberrant production of interleukin-8 and thrombospondin-1 by psoriatic keratinocytes mediates angiogenesis. Am. J. Pathol. 1994, 144, 820–828. [Google Scholar] [PubMed]
- Tuschil, A.; Lam, C.; Haslberger, A.; Lindley, I. Interleukin-8 Stimulates Calcium Transients and Promotes Epidermal Cell Proliferation. J. Investig. Dermatol. 1992, 99, 294–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, H.; Koga, T.; Kohda, F.; Hara, H.; Urabe, K.; Furue, M. Interleukin-8-positive neutrophils in psoriasis. J. Dermatol. Sci. 2001, 26, 119–124. [Google Scholar] [CrossRef]
- Guilloteau, K.; Paris, I.; Pedretti, N.; Boniface, K.; Juchaux, F.; Huguier, V.; Guillet, G.; Bernard, F.-X.; Lecron, J.-C.; Morel, F. Skin Inflammation Induced by the Synergistic Action of IL-17A, IL-22, Oncostatin M, IL-1α, and TNF-α Recapitulates Some Features of Psoriasis. J. Immunol. 2010, 184, 5263–5270. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Nikamo, P.; Lohcharoenkal, W.; Li, D.; Meisgen, F.; Landén, N.X.; Ståhle, M.; Pivarcsi, A.; Sonkoly, E. MicroRNA-146a suppresses IL-17–mediated skin inflammation and is genetically associated with psoriasis. J. Allergy Clin. Immunol. 2017, 139, 550–561. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Nyuydzefe, M.S.; Weiss, J.M.; Zhang, J.; Waksal, S.D.; Zanin-Zhorov, A. ROCK2, but not ROCK1 interacts with phosphorylated STAT3 and co-occupies TH17/TFH gene promoters in TH17-activated human T cells. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Lock, F.E.; Hotchin, N.A. Distinct Roles for ROCK1 and ROCK2 in the Regulation of Keratinocyte Differentiation. PLoS ONE 2009, 4, e8190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, B.A.; Dennstedt, E.; Mitchell, D.C.; Walshe, T.E.; Noma, K.; Loureiro, R.; Saint-Geniez, M.; Campaigniac, J.; Liao, J.K.; Patricia, D.A. RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB J. 2010, 24, 3186–3195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanin-Zhorov, A.; Weiss, J.M.; Nyuydzefe, M.S.; Chen, W.; Scher, J.U.; Mo, R.; Depoil, D.; Rao, N.; Liu, B.; Wei, J.; et al. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, 16814–16819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanin-Zhorov, A.; Weiss, J.M.; Trzeciak, A.; Chen, W.; Zhang, J.; Nyuydzefe, M.S.; Arencibia, C.; Polimera, S.; Schueller, O.; Fuentes-Duculan, J.; et al. Cutting Edge: Selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients with Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10. J. Immunol. 2017, 198, 3809–3814. [Google Scholar] [CrossRef] [Green Version]
- Chuang, H.-H.; Yang, C.-H.; Tsay, Y.-G.; Hsu, C.-Y.; Tseng, L.-M.; Chang, Z.-F.; Lee, H.-H. ROCKII Ser1366 phosphorylation reflects the activation status. Biochem. J. 2012, 443, 145–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowery, D.M.; Clauser, K.; Hjerrild, M.; Lim, D.; Alexander, J.; Kishi, K.; Ong, S.-E.; Gammeltoft, S.; A Carr, S.; Yaffe, M.B. Proteomic screen defines the Polo-box domain interactome and identifies Rock2 as a Plk1 substrate. EMBO J. 2007, 26, 2262–2273. [Google Scholar] [CrossRef] [PubMed]
- Couzens, A.L.; Saridakis, V.; Scheid, M.P. The hydrophobic motif of ROCK2 requires association with the N-terminal extension for kinase activity. Biochem. J. 2009, 419, 141–148. [Google Scholar] [CrossRef]
- Vacharanukrauh, P.; Meephansan, J.; Tangtanatakul, P.; Soonthornchai, W.; Wongpiyabovorn, J.; Serirat, O.; Komine, M. High-Throughput RNA Sequencing Reveals the Effect of NB-UVB Phototherapy on Major Inflammatory Molecules of Lesional Psoriasis. Psoriasis Targets Ther. 2021, 11, 133–149. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Loraine, A.E.; Blakley, I.C.; Jagadeesan, S.; Harper, J.; Miller, G.; Firon, N. Analysis and Visualization of RNA-Seq Expression Data Using RStudio, Bioconductor, and Integrated Genome Browser. In Plant Functional Genomics: Methods and Protocols; Alonso, J.M., Stepanova, A.N., Eds.; Springer: New York, NY, USA, 2015; pp. 481–501. [Google Scholar]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Molecule Type * | Log 2FC | p-Value | Symbol | Molecule Type * | Log 2FC | p-Value | |
|---|---|---|---|---|---|---|---|---|
| Upregulated DEGs | Downregulated DEGs | |||||||
| 1 | A2M | transporter | 15.982 | 8.600 × 10−6 | ATP5F1A | transporter | −27.343 | 7.380 × 10−6 |
| 2 | H3C2 | other | 15.367 | 9.340 × 10−5 | SEPTIN5 | enzyme | −25.527 | 1.410 × 10−3 |
| 3 | PLEC | other | 15.295 | 1.360 × 10−5 | KHNYN | other | −24.254 | 3.700 × 10−3 |
| 4 | ITSN2 | other | 14.968 | 3.200 × 10−4 | PLD3 | enzyme | −23.010 | 5.630 × 10−3 |
| 5 | UGGT1 | enzyme | 14.906 | 1.080 × 10−4 | FHL1 | other | −21.983 | 9.840 × 10−3 |
| 6 | STT3A | enzyme | 14.745 | 3.460 × 10−3 | HEXA | enzyme | −20.826 | 8.320 × 10−3 |
| 7 | HERC6 | enzyme | 14.724 | 1.120 × 10−3 | DNM2 | enzyme | −19.584 | 7.500 × 10−3 |
| 8 | DEFB4A/DEFB4B | other | 14.690 | 1.400 × 10−4 | TLE5 | transcription regulator | −18.613 | 3.990 × 10−3 |
| 9 | CAND1 | transcription regulator | 14.654 | 8.540 × 10−3 | MYH14 | enzyme | −16.846 | 1.460 × 10−3 |
| 10 | HRNR | other | 14.597 | 1.340 × 10−5 | GSN | other | −16.812 | 1.490 × 10−3 |
| 11 | IL36A | cytokine | 14.572 | 1.340 × 10−4 | SPSB3 | other | −16.593 | 1.100 × 10−3 |
| 12 | SMARCA4 | transcription regulator | 14.517 | 2.630 × 10−5 | LCE1C | other | −16.153 | 1.620 × 10−3 |
| 13 | CYFIP1 | translation regulator | 14.500 | 2.820 × 10−3 | LAMTOR4 | other | −16.058 | 9.610 × 10−3 |
| 14 | HIPK3 | kinase | 14.365 | 6.200 × 10−3 | SYT8 | transporter | −16.014 | 9.260 × 10−3 |
| 15 | PLA2G4E-AS1 | other | 14.274 | 8.590 × 10−5 | LTBP4 | growth factor | −15.985 | 2.600 × 10−3 |
| 16 | TRIP12 | enzyme | 14.146 | 4.660 × 10−3 | ITGB4 | transmembrane receptor | −15.717 | 3.270 × 10−3 |
| 17 | CLIC1 | ion channel | 14.091 | 4.110 × 10−5 | DCTN1 | other | −15.591 | 3.700 × 10−3 |
| 18 | CD164 | other | 14.069 | 1.280 × 10−3 | RTN4 | other | −15.532 | 3.930 × 10−3 |
| 19 | BZW1 | translation regulator | 14.025 | 6.550 × 10−3 | CAMSAP3 | other | −15.517 | 3.990 × 10−3 |
| 20 | NRBP1 | kinase | 13.939 | 1.280 × 10−4 | NFIX | transcription regulator | −15.266 | 5.070 × 10−3 |
| 21 | PSMD12 | other | 13.697 | 8.810 × 10−5 | CRAT | enzyme | −15.226 | 4.210 × 10−3 |
| 22 | EPB41L3 | other | 13.662 | 2.000 × 10−3 | RAB40C | enzyme | −15.156 | 5.860 × 10−3 |
| 23 | SCYL2 | other | 13.652 | 1.490 × 10−4 | CCL27 | other | −15.021 | 3.820 × 10−3 |
| 24 | H2BC10 | other | 13.610 | 1.870 × 10−4 | SIK1/SIK1B | kinase | −14.994 | 6.560 × 10−3 |
| 25 | WDR75 | other | 13.554 | 1.180 × 10−3 | CAPG | other | −14.970 | 2.080 × 10−3 |
| 26 | H4C13 | other | 13.550 | 1.080 × 10−3 | EPN2 | other | −14.879 | 7.780 × 10−3 |
| 27 | EXT2 | enzyme | 13.432 | 4.740 × 10−4 | ARAP1 | other | −14.871 | 7.950 × 10−3 |
| 28 | H3C7 | other | 13.405 | 1.480 × 10−3 | PDCD4 | other | −14.858 | 7.870 × 10−3 |
| 29 | PLEKHG5 | other | 13.319 | 2.030 × 10−4 | XPO6 | other | −14.735 | 9.330 × 10−3 |
| 30 | DMXL1 | other | 13.302 | 1.930 × 10−3 | PODN | other | −14.705 | 9.560 × 10−3 |
| 31 | GBP6 | enzyme | 13.278 | 2.320 × 10−3 | TUBGCP2 | peptidase | −14.694 | 9.730 × 10−3 |
| 32 | H3C11 | other | 13.099 | 4.590 × 10−4 | CIRBP | translation regulator | −14.658 | 9.860 × 10−3 |
| 33 | H2AC14 | other | 13.092 | 4.200 × 10−6 | TSPAN14 | other | −14.640 | 9.940 × 10−3 |
| 34 | SMC6 | other | 12.917 | 2.030 × 10−4 | ATP2B4 | transporter | −14.506 | 1.540 × 10−3 |
| 35 | H2AC21 | other | 12.847 | 7.450 × 10−4 | CNOT1 | other | −14.414 | 8.590 × 10−3 |
| 36 | H2AC4 | other | 12.793 | 5.310 × 10−4 | WBP2 | transcription regulator | −14.403 | 3.680 × 10−3 |
| 37 | CD59 | other | 12.792 | 9.370 × 10−4 | MAN2C1 | enzyme | −14.324 | 2.770 × 10−3 |
| 38 | TMEM14EP | other | 12.753 | 6.240 × 10−4 | POLD2 | enzyme | −14.293 | 6.080 × 10−3 |
| 39 | GLB1 | enzyme | 12.717 | 1.270 × 10−3 | MPST | enzyme | −14.237 | 5.410 × 10−3 |
| 40 | CTNNBL1 | other | 12.703 | 2.090 × 10−3 | ACSS2 | enzyme | −14.029 | 4.050 × 10−3 |
| 41 | GFPT1 | enzyme | 12.637 | 3.910 × 10−3 | AP2A1 | transporter | −13.944 | 4.740 × 10−3 |
| 42 | H3C8 | other | 12.599 | 1.300 × 10−4 | SCRIB | other | −13.900 | 6.080 × 10−3 |
| 43 | ZDHHC6 | enzyme | 12.565 | 4.940 × 10−3 | TAP2 | transporter | −13.808 | 8.590 × 10−3 |
| 44 | IKBKB | kinase | 12.437 | 2.280 × 10−3 | PHYHIP | other | −13.783 | 6.750 × 10−3 |
| 45 | GANAB | enzyme | 12.412 | 2.170 × 10−4 | PLCH2 | enzyme | −13.78 | 6.560 × 10−3 |
| 46 | TMEM184B | other | 12.410 | 1.410 × 10−3 | TMEM63B | ion channel | −13.617 | 6.260 × 10−3 |
| 47 | SEPTIN11 | other | 12.377 | 9.900 × 10−4 | TAX1BP1 | other | −13.600 | 3.020 × 10−3 |
| 48 | RMDN3 | other | 12.322 | 3.100 × 10−3 | PKM | kinase | −13.594 | 5.310 × 10−3 |
| 49 | LPP | other | 12.292 | 5.600 × 10−3 | EPHX3 | enzyme | −13.472 | 8.680 × 10−3 |
| 50 | H2BC13 | other | 12.292 | 2.600 × 10−3 | PTBP1 | enzyme | −13.167 | 8.660 × 10−3 |
| Gene Symbol | Log2FC | p-Value | Gene Symbol | Log2FC | p-Value |
|---|---|---|---|---|---|
| Upregulation | Downregulation | ||||
| Anti-microbial peptides (AMPs) | S100 proteins | ||||
| DEFB4A/DEFB4B | 14.690 | 1.400 × 10−4 | S100A16 | −5.334 | 9.920 × 10−3 |
| DEFB103A/DEFB103B | 7.624 | 2.290 × 10−4 | S100A10 | −4.100 | 2.780 × 10−5 |
| S100A7A (S100A15) | 9.559 | 1.180 × 10−5 | S100A4 | −3.819 | 1.450 × 10−4 |
| S100A8 | 4.944 | 2.590 × 10−4 | Cytokines | ||
| S100A9 | 4.722 | 3.440 × 10−5 | FBRS | −3.204 | 2.140 × 10−3 |
| S100A7 | 3.002 | 3.930 × 10−5 | SLURP1 | −1.975 | 3.420 × 10−3 |
| LCN2 | 4.182 | 8.240 × 10−4 | TIMP1 | −1.857 | 4.290 × 10−3 |
| Cytokines | MIF | −1.842 | 4.860 × 10−3 | ||
| IL36A | 14.572 | 1.340 × 10−4 | CXCL14 | −1.752 | 1.650 × 10−3 |
| CXCL8 | 10.681 | 6.580 × 10−4 | Transmembrane receptor | ||
| VAV3 | 9.178 | 1.290 × 10−4 | ITGB4 | −15.717 | 3.270 × 10−3 |
| NAMPT | 3.919 | 8.090 × 10−4 | Cell adhesion molecules | ||
| IL36G | 3.897 | 2.090 × 10−3 | GJB3 | −5.454 | 9.220 × 10−3 |
| C10orf99 | 2.567 | 2.490 × 10−3 | GJB5 | −2.317 | 9.110 × 10−3 |
| Transmembrane receptors | LAMA5 | −2.125 | 7.100 × 10−3 | ||
| GFRA1 | 9.023 | 5.660 × 10−3 | Keratin and late cornified envelope | ||
| TNFRSF1A | 3.317 | 1.180 × 10−3 | KRT2 | −5.484 | 9.530 × 10−4 |
| ITGB1 | 2.030 | 8.210 × 10−3 | KRT1 | −3.700 | 3.250 × 10−4 |
| Cell adhesion molecules | KRT10 | −3.521 | 1.510 × 10−4 | ||
| DSC2 | 6.595 | 1.260 × 10−4 | KRT77 | −3.416 | 4.850 × 10−4 |
| DSC1 | 2.478 | 6.390 × 10−3 | LCE1C | −16.153 | 1.620 × 10−3 |
| DSG3 | 3.599 | 1.590 × 10−3 | LCE2C/LCE2D | −5.366 | 9.450 × 10−4 |
| DSG1 | 2.456 | 7.280 × 10−3 | LCE2B | −4.689 | 6.280 × 10−5 |
| GJB2 | 5.468 | 3.430 × 10−6 | LCE1B | −3.871 | 8.320 × 10−5 |
| GJB6 | 5.135 | 4.940 × 10−3 | LCE6A | −3.563 | 5.910 × 10−4 |
| PLEC | 15.295 | 1.360 × 10−5 | LCE2A | −3.467 | 1.230 × 10−4 |
| LAMA2 | 1.626 | 8.630 × 10−3 | LCE1D | −3.371 | 9.840 × 10−4 |
| Keratin and late cornified envelope | LCE1A | −3.132 | 1.310 × 10−3 | ||
| KRT16P1 | 8.120 | 8.130 × 10−3 | LCE1F | −2.956 | 1.890 × 10−3 |
| KRT6A | 5.740 | 3.170 × 10−4 | Peptidase | ||
| KRT6C | 5.310 | 1.170 × 10−4 | KLK11 | −5.992 | 2.640 × 10−5 |
| KRT16 | 4.675 | 8.870 × 10−5 | Other interesting DEGs | ||
| KRT6B | 3.384 | 2.000 × 10−3 | APOE | −3.300 | 6.710 × 10−3 |
| LCE3A | 7.383 | 2.430 × 10−5 | CALML5 | −2.153 | 6.690 × 10−4 |
| LCE3C | 5.451 | 1.000 × 10−3 | CAV1 | −2.121 | 5.430 × 10−3 |
| LCE3E | 2.652 | 2.210 × 10−3 | CCL27 | −15.021 | 3.820 × 10−3 |
| Peptidase | CCND1 | −1.827 | 4.370 × 10−3 | ||
| PLAT | 4.388 | 5.750 × 10−4 | CD81 | −3.603 | 1.190 × 10−4 |
| KLK7 | 4.224 | 7.300 × 10−4 | COL16A1 | −2.658 | 7.960 × 10−4 |
| KLK6 | 3.927 | 2.030 × 10−3 | COL1A1 | −2.636 | 1.870 × 10−4 |
| ADAM17 | 2.146 | 3.400 × 10−3 | COL6A1 | −3.547 | 8.270 × 10−5 |
| Protease inhibitor | COL6A2 | −3.357 | 3.740 × 10−3 | ||
| A2M | 15.982 | 8.600 × 10−6 | COL7A1 | −1.766 | 1.970 × 10−3 |
| A2ML1 | 1.703 | 7.220 × 10−3 | FBL | −3.122 | 4.290 × 10−3 |
| SPINK5 | 1.574 | 4.340 × 10−3 | GSN | −16.812 | 1.490 × 10−3 |
| Other interesting DEGs | GPX4 | −3.137 | 4.430 × 10−3 | ||
| CD59 | 12.792 | 9.370 × 10−3 | SERPINF1 | −2.115 | 3.820 × 10−3 |
| IKBKB | 12.437 | 2.280 × 10−3 | IL20RB | −1.788 | 2.680 × 10−3 |
| EDNRB | 11.815 | 5.820 × 10−3 | IGFBP6 | −5.399 | 1.260 × 10−3 |
| TNIP3 | 10.794 | 6.580 × 10−3 | IGFBP4 | −3.248 | 1.140 × 10−4 |
| SERPINB4 | 10.691 | 7.430 × 10−3 | IGFBP7 | −1.574 | 6.940 × 10−3 |
| SERPINB3 | 7.630 | 3.290 × 10−5 | ITGB5 | −2.077 | 7.180 × 10−3 |
| SERPINB9 | 3.960 | 6.270 × 10−3 | LGALS7/LGALS7B | −4.880 | 8.880 × 10−5 |
| XDH | 5.565 | 6.140 × 10−3 | LGALS1 | −3.681 | 1.030 × 10−3 |
| PI3 | 5.139 | 5.430 × 10−5 | LORICRIN | −9.244 | 8.540 × 10−4 |
| STAT1 | 3.384 | 3.940 × 10−3 | MMP28 | −11.574 | 7.540 × 10−3 |
| STAT3 | 1.617 | 8.030 × 10−3 | NUPR1 | −2.183 | 3.910 × 10−3 |
| DDX21 | 2.520 | 2.580 × 10−3 | PECAM1 | −2.517 | 2.980 × 10−3 |
| PLSCR1 | 2.352 | 3.520 × 10−3 | PSORS1C2 | −3.373 | 8.360 × 10−3 |
| RICTOR | 2.338 | 8.330 × 10−3 | TIMP2 | −3.092 | 3.820 × 10−4 |
| FLG | 2.135 | 6.550 × 10−4 | TNXB | −2.238 | 2.460 × 10−3 |
| KDM5A | 1.905 | 2.380 × 10−3 | TYK2 | −1.915 | 5.780 × 10−3 |
| HLA-A | 1.825 | 1.170 × 10−3 | VEGFB | −3.382 | 6.940 × 10−3 |
| IGFBP3 | 1.600 | 1.200 × 10−3 | WNT7B | −3.574 | 4.310 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boonpethkaew, S.; Meephansan, J.; Jumlongpim, O.; Tangtanatakul, P.; Soonthornchai, W.; Wongpiyabovorn, J.; Vipanurat, R.; Komine, M. Transcriptomic Profiling of Peripheral Edge of Lesions to Elucidate the Pathogenesis of Psoriasis Vulgaris. Int. J. Mol. Sci. 2022, 23, 4983. https://doi.org/10.3390/ijms23094983
Boonpethkaew S, Meephansan J, Jumlongpim O, Tangtanatakul P, Soonthornchai W, Wongpiyabovorn J, Vipanurat R, Komine M. Transcriptomic Profiling of Peripheral Edge of Lesions to Elucidate the Pathogenesis of Psoriasis Vulgaris. International Journal of Molecular Sciences. 2022; 23(9):4983. https://doi.org/10.3390/ijms23094983
Chicago/Turabian StyleBoonpethkaew, Suphagan, Jitlada Meephansan, Onjira Jumlongpim, Pattarin Tangtanatakul, Wipasiri Soonthornchai, Jongkonnee Wongpiyabovorn, Ratchanee Vipanurat, and Mayumi Komine. 2022. "Transcriptomic Profiling of Peripheral Edge of Lesions to Elucidate the Pathogenesis of Psoriasis Vulgaris" International Journal of Molecular Sciences 23, no. 9: 4983. https://doi.org/10.3390/ijms23094983
APA StyleBoonpethkaew, S., Meephansan, J., Jumlongpim, O., Tangtanatakul, P., Soonthornchai, W., Wongpiyabovorn, J., Vipanurat, R., & Komine, M. (2022). Transcriptomic Profiling of Peripheral Edge of Lesions to Elucidate the Pathogenesis of Psoriasis Vulgaris. International Journal of Molecular Sciences, 23(9), 4983. https://doi.org/10.3390/ijms23094983