
Multi-Omics Techniques for Soybean Molecular Breeding


 ,
, 
{kind=link}
{kind=link}
Abstract
:1. Introduction
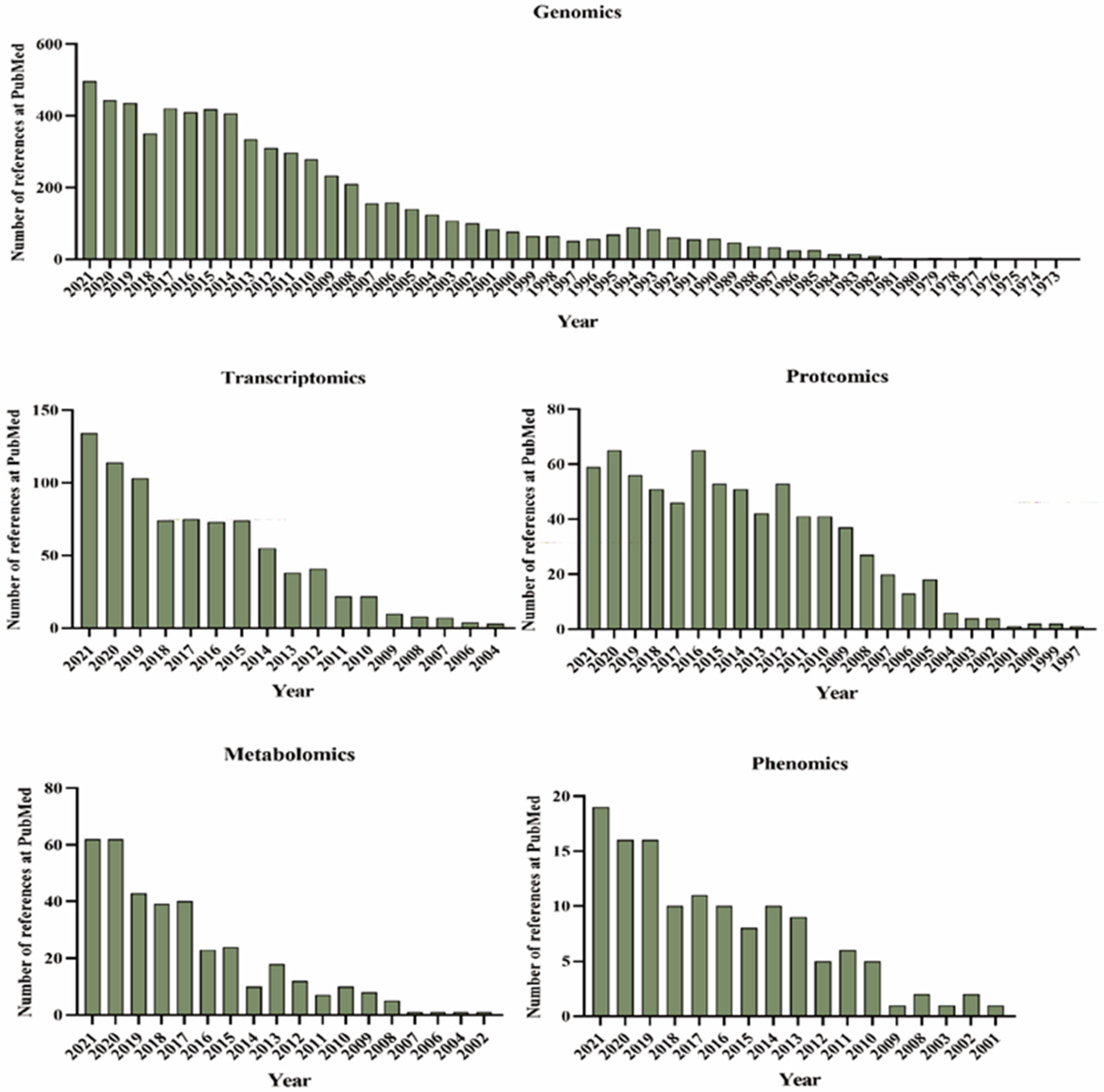
2. Multi-Omics Research Progress
2.1. Soybean Genomics Research Progress
2.2. Soybean Transcriptomics Research Progress
2.3. Soybean Proteomics Research Progress
2.4. Soybean Metabolomics Research Progress
2.5. Soybean Phenomics Research Progress
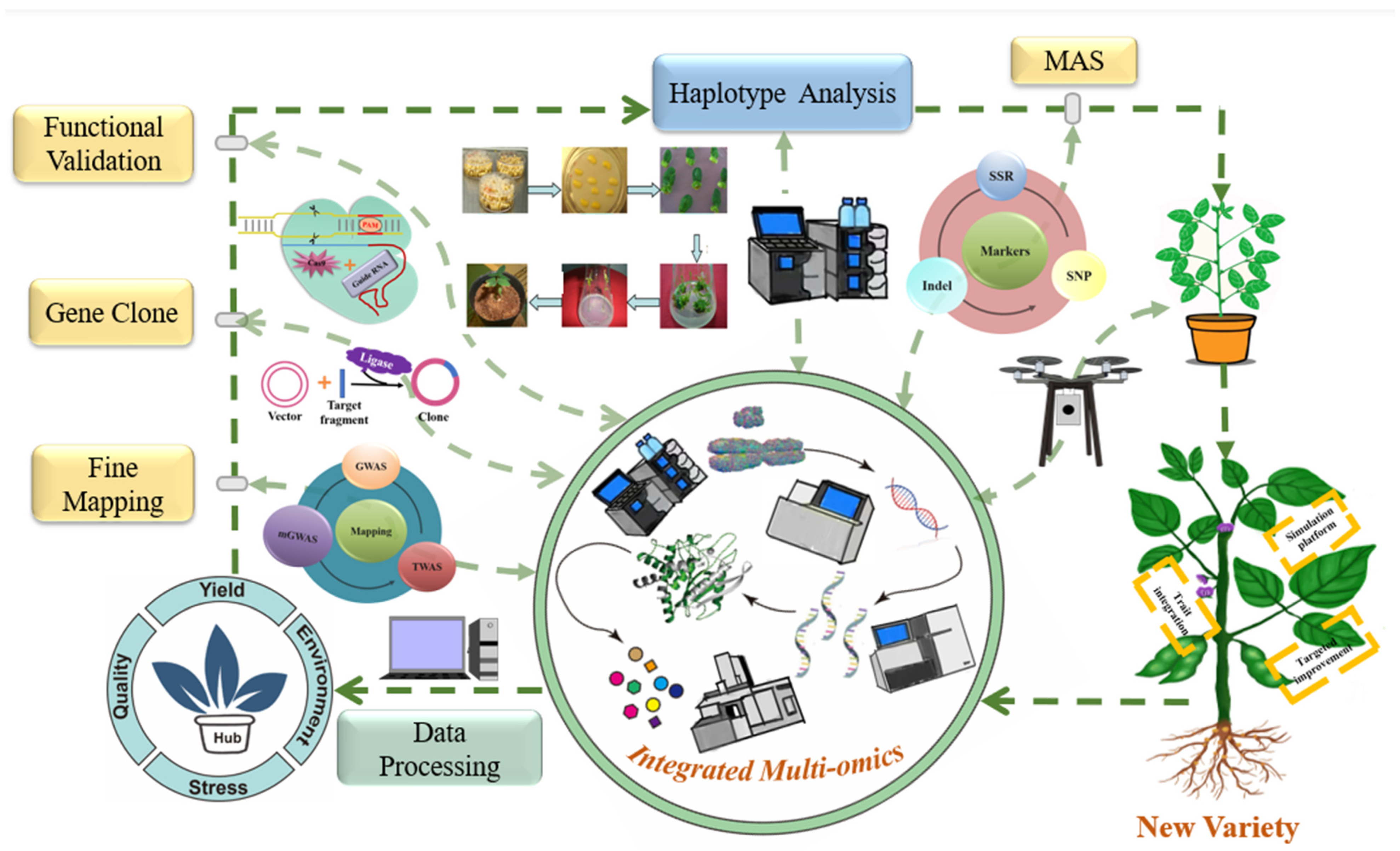
3. Molecular Breeding in Soybean
4. Further Perspectives
5. Conclusions
6. Supplementary Research Methodology
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hymowitz, T.; Shurtleff, W.R. Debunking soybean myths and legends in the historical and popular literature. Crop. Sci. 2005, 45, 473–476. [Google Scholar] [CrossRef]
- Hymowitz, T.; Harlan, J.R. Introduction of soybean to North America by Samuel Bowen in 1765. Econ. Bot. 1983, 37, 371–379. [Google Scholar] [CrossRef]
- Marra, M.C.; Piggott, N.E.; Carlson, G.A. The Net Benefits, Including convenience of roundup ready soybeans: Results from a national survey. Tech. Bull. 2004, 3. Available online: https://www.researchgate.net/publication/237717600 (accessed on 22 January 2022).
- Bradshaw, J.E. Plant breeding: Past, present and future. Euphytica 2017, 213, 60. [Google Scholar] [CrossRef]
- Orf, J.H. Breeding, Genetics, and Production of Soybeans. In Soybeans: Chemistry, Production, Processing, and Utilization; AOCS Press: St. Paul, MN, USA, 2008; pp. 33–65. [Google Scholar] [CrossRef]
- Fehr, W.R. Breeding for modified fatty acid composition in soybean. Crop. Sci. 2007, 47, S-72–S-87. [Google Scholar] [CrossRef]
- Rayaprolu, S.J.; Hettiarachchy, N.S.; Chen, P.; Kannan, A.; Mauromostakos, A. Peptides derived from high oleic acid soybean meals inhibit colon, liver and lung cancer cell growth. Food Res. Int. 2013, 50, 282–288. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, H.M.; Xu, X.; Liu, X.; Chen, W.; Yang, G.; Wong, F.L.; Li, M.W.; He, W.; Qin, N.; Wang, B.; et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 2010, 42, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Lee, S.; Van, K.; Kim, T.H.; Jeong, S.C.; Choi, I.Y.; Kim, D.S.; Lee, Y.S.; Park, D.; Ma, J.; et al. Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. and Zucc.) genome. Proc. Natl. Acad. Sci. USA 2010, 107, 22032–22037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Liu, J.; Geng, H.; Zhang, J.; Liu, Y.; Zhang, H.; Xing, S.; Du, J.; Ma, S.; Tian, Z. De novo assembly of a Chinese soybean genome. Sci. China Life Sci. 2018, 61, 871–884. [Google Scholar] [CrossRef]
- Xie, M.; Chung, C.Y.; Li, M.W.; Wong, F.L.; Wang, X.; Liu, A.; Wang, Z.; Leung, A.K.; Wong, T.H.; Tong, S.W.; et al. A reference-grade wild soybean genome. Nat. Commun. 2019, 10, 1216. [Google Scholar] [CrossRef] [Green Version]
- Valliyodan, B.; Cannon, S.B.; Bayer, P.E.; Shu, S.; Brown, A.V.; Ren, L.; Jenkins, J.; Chung, C.Y.; Chan, T.F.; Daum, C.G.; et al. Construction and comparison of three reference-quality genome assemblies for soybean. Plant J. 2019, 100, 1066–1082. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.G.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.A.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-genome of wild and cultivated soybeans. Cell 2020, 182, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tian, Z. From one linear genome to a graph-based pan-genome: A new era for genomics. Sci. China Life Sci. 2020, 63, 1938–1941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jiang, H.; Hu, Z.; Song, Q.; An, Y.C. Development of a versatile resource for post-genomic research through consolidating and characterizing 1500 diverse wild and cultivated soybean genomes. BMC Genom. 2022, 23, 250. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Jordan, D.R.; Mace, E.S. A graph-based pan-genome guides biological discovery. Mol. Plant 2020, 13, 1247–1249. [Google Scholar] [CrossRef]
- Zhao, C.; Takeshima, R.; Zhu, J.; Xu, M.; Sato, M.; Watanabe, S.; Kanazawa, A.; Liu, B.; Kong, F.; Yamada, T.; et al. A recessive allele for delayed flowering at the soybean maturity locus E9 is a leaky allele of FT2a, a FLOWERING LOCUS T ortholog. BMC Plant Biol. 2016, 16, 20. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Zhao, X.; Hu, Y.; Liu, S.; Nan, H.; Li, X.; Fang, C.; Cao, D.; Shi, X.; Kong, L.; et al. Natural variation at the soybean J locus improves adaptation to the tropics and enhances yield. Nat. Gene 2017, 49, 773–779. [Google Scholar] [CrossRef]
- Wang, S.; Liu, S.; Wang, J.; Yokosho, K.; Zhou, B.; Yu, Y.C.; Liu, Z.; Frommer, W.B.; Ma, J.F.; Chen, L.Q.; et al. Simultaneous changes in seed size, oil content and protein content driven by selection of SWEET homologues during soybean domestication. Natl. Sci. Rev. 2020, 7, 1776–1786. [Google Scholar] [CrossRef]
- Moses, L.; Pachter, L. Museum of spatial transcriptomics. Nat. Methods 2022, 87, 1–13. [Google Scholar] [CrossRef]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Li, Y.; Li, Y.; Luo, Y. Statistical and machine learning methods for spatially resolved transcriptomics data analysis. Genome Biol. 2022, 23, 83. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xie, Y.; Liu, L.; King, G.J.; White, P.; Ding, G.; Wang, S.; Cai, H.; Wang, C.; Xu, F.; et al. Genetic Control of Seed Phytate Accumulation and the Development of Low-Phytate Crops: A Review and Perspective. J. Agric. Food Chem. 2022, 70, 3375–3390. [Google Scholar] [CrossRef] [PubMed]
- Verdier, J.; Thompson, R.D. Transcriptional regulation of storage protein synthesis during dicotyledon seed filling. Plant Cell Physiol. 2008, 49, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Verdier, J.; Kakar, K.; Gallardo, K.; Le Signor, C.; Aubert, G.; Schlereth, A.; Town, C.D.; Udvardi, M.K.; Thompson, R.D. Gene expression profiling of M. truncatula transcription factors identifies putative regulators of grain legume seed filling. Plant Mol. Biol. 2008, 67, 567–580. [Google Scholar] [CrossRef]
- Hajduch, M.; Hearne, L.B.; Miernyk, J.A.; Casteel, J.E.; Joshi, T.; Agrawal, G.K.; Song, Z.; Zhou, M.; Xu, D.; Thelen, J.J. Systems analysis of seed filling in Arabidopsis: Using general linear modeling to assess concordance of transcript and protein expression. Plant Physiol. 2010, 152, 2078–2087. [Google Scholar] [CrossRef] [Green Version]
- Severin, A.J.; Woody, J.L.; Bolon, Y.T.; Joseph, B.; Diers, B.W.; Farmer, A.D.; Muehlbauer, G.J.; Nelson, R.T.; Grant, D.; Specht, J.E.; et al. RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant Biol. 2010, 10, 160. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.I.; Gonzalez, D.O.; Vodkin, L.O. Flux of transcript patterns during soybean seed development. BMC Genom. 2010, 11, 136. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.I.; Vodkin, L.O. Using RNA-Seq to profile soybean seed development from fertilization to maturity. PLoS ONE 2013, 8, e59270. [Google Scholar] [CrossRef] [Green Version]
- Crouch, M.L.; Sussex, I.M. Development and storage-protein synthesis in Brassica napus L. embryos in vivo and in vitro. Planta 1981, 153, 64–74. [Google Scholar] [CrossRef]
- Mosquna, A.; Katz, A.; Shochat, S.; Grafi, G.; Ohad, N. Interaction of FIE, a Polycomb protein, with pRb: A possible mechanism regulating endosperm development. Mol. Genet. Genom. 2004, 271, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Sinkevicius, K.W.; Selinger, D.A.; Tarczynski, M.C. The homeobox gene GLABRA2 affects seed oil content in Arabidopsis. Plant Mol. Biol. 2006, 60, 377–387. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.A.; Bolon, Y.T.; Bucciarelli, B.; Vance, C.P. Legume genomics: Understanding biology through DNA and RNA sequencing. Ann. Bot. 2014, 113, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Perry, S.E. Identification of direct targets of FUSCA3, a key regulator of Arabidopsis seed development. Plant. Physiol. 2013, 161, 1251–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, J.M.; Kwong, R.W.; Park, S.; Le, B.H.; Baden, R.; Cagliari, A.; Hashimoto, M.; Munoz, M.D.; Fischer, R.L.; Goldberg, R.B.; et al. LEC1 sequentially regulates the transcription of genes involved in diverse developmental processes during seed development. Proc. Natl. Acad. Sci. USA 2017, 114, E6710–E6719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goettel, W.; Liu, Z.; Xia, J.; Zhang, W.; Zhao, P.X.; An, Y.Q. Systems and evolutionary characterization of microRNAs and their underlying regulatory networks in soybean cotyledons. PLoS ONE 2014, 9, e86153. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.E.; Kim, M.Y.; Shim, S.; Lee, J.; Lee, S.H. Gene expression profiling for seed protein and oil synthesis during early seed development in soybean. Genes Genom. 2015, 37, 409–418. [Google Scholar] [CrossRef]
- Lu, X.; Li, Q.T.; Xiong, Q.; Li, W.; Bi, Y.D.; Lai, Y.C.; Liu, X.L.; Man, W.Q.; Zhang, W.K.; Ma, B.; et al. The transcriptomic signature of developing soybean seeds reveals the genetic basis of seed trait adaptation during domestication. Plant J. 2016, 86, 530–544. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.; Zhang, Z.; Wang, Z.; Yu, J.; Qin, H.; Mao, X.; Jiang, H.; Xin, D.; Yin, Z.; Zhu, R.; et al. Meta-analysis and transcriptome profiling reveal hub genes for soybean seed storage composition during seed development. Plant Cell Environ. 2018, 41, 2109–2127. [Google Scholar] [CrossRef]
- Wang, Y.; Ye, W.; Wang, Y. Genome-wide identification of long non-coding RNAs suggests a potential association with effector gene transcription in Phytophthora sojae. Mol. Plant Pathol. 2018, 19, 2177–2186. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Qian, L.; Wang, M.; Liu, W.; Song, X.; Cheng, H.; Yuan, F.; Zhao, M. Comparative transcriptome analysis during seeds development between two soybean cultivars. Peer J. 2021, 9, e10772. [Google Scholar] [CrossRef] [PubMed]
- Matsui, A.; Ishida, J.; Morosawa, T.; Mochizuki, Y.; Kaminuma, E.; Endo, T.A.; Okamoto, M.; Nambara, E.; Nakajima, M.; Kawashima, M.; et al. Arabidopsis transcriptome analysis under drought, cold, high-salinity and ABA treatment conditions using a tiling array. Plant Cell Physiol. 2008, 8, 1135–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Tai, H.; Li, S.; Gao, W.; Zhao, M.; Xie, C.; Li, W.X. bHLH122is important for drought and osmotic stress resistance in Arabidopsis and in the repression of ABA catabolism. New. Phytol. 2014, 201, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, S.; Bashir, K.; Matsui, A.; Tanaka, M.; Seki, M. Transcriptomic analysis of soil-grown Arabidopsis thaliana roots and shoots in response to a drought stress. Front. Plant Sci. 2016, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, S.; Bashir, K.; Nakaminami, K.; Hanada, K.; Matsui, A.; Seki, M. Drought stress differentially regulates the expression of small open reading frames (sORFs) in Arabidopsis roots and shoots. Plant Signal. Behav. 2016, 11, e1215792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Yao, Q.; Patil, G.B.; Agarwal, G.; Deshmukh, R.K.; Lin, L.; Wang, B.; Wang, Y.; Prince, S.J.; Song, L.; et al. Identification and comparative analysis of differential gene expression in soybean leaf tissue under drought and flooding stress revealed by RNA-Seq. Front. Plant Sci. 2016, 7, 1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Li, Y.; Zhu, Y.M.; Bai, X.; Lv, D.K.; Guo, D.; Ji, W.; Cai, H. Global transcriptome profiling of wild soybean (Glycine soja) roots under NaHCO 3 treatment. BMC Plant Biol. 2010, 10, 153. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, J.; Wei, J.; Liu, J.; Yang, S.; Gai, J.; Li, Y. Identification and analysis of NaHCO3 stress responsive genes in wild soybean (Glycine soja) Roots by RNA-seq. Front. Plant 2016, 7, 1842. [Google Scholar] [CrossRef] [Green Version]
- Hedlund, E.; Deng, Q. Single-cell RNA sequencing: Technical advancements and biological applications. Mol. Asp. Med. 2018, 59, 36–46. [Google Scholar] [CrossRef]
- Hurgobin, B.; Lewsey, M.G. Applications of cell- and tissue-specific ’omics to improve plant productivity. Emerg. Top. Life Sci. 2022, 6, 163–173. [Google Scholar] [CrossRef]
- Saliba, A.E.; Westermann, A.J.; Gorski, S.A.; Vogel, J. Single-cell RNA-seq: Advances and future challenges. Nucleic Acids Res. 2014, 42, 8845–8860. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Huang, Z.; Sun, J.; Cui, X.; Liu, Y. Research progress and future development trends in medicinal plant transcriptomics. Front. Plant Sci. 2021, 12, 691838. [Google Scholar] [CrossRef] [PubMed]
- Schumacher-Schuh, A.; Bieger, A.; Borelli, W.V.; Portley, M.K.; Awad, P.S.; Bandres-Ciga, S. Advances in proteomic and metabolomic profiling of neurodegenerative diseases. Front. Neurol. 2022, 12, 792227. [Google Scholar] [CrossRef] [PubMed]
- Afroz, A.; Hashiguchi, A.; Khan, M.R.; Komatsu, S. Analyses of the proteomes of the leaf, hypocotyl, and root of young soybean seedlings. Protein Pept. Lett. 2010, 17, 319–331. [Google Scholar] [CrossRef]
- Hajduch, M.; Matusova, R.; Houston, N.L.; Thelen, J.J. Comparative proteomics of seed maturation in oilseeds reveals differences in intermediary metabolism. Proteomics 2011, 11, 1619–1629. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Brechenmacher, L.; Aldrich, J.T.; Clauss, T.R.; Gritsenko, M.A.; Hixson, K.K.; Libault, M.; Tanaka, K.; Yang, F.; Yao, Q.; et al. Quantitative phosphoproteomic analysis of soybean root hairs inoculated with Bradyrhizobium japonicum. Mol. Cell Proteom. 2012, 11, 1140–1155. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Gu, F.; Liu, D.; Yin, C.; Zhao, S.; Chen, H.; Zhang, J.; Yang, C.; Zhan, X.; Zhang, M. Proteomic analysis of elite soybean Jidou17 and its parents using iTRAQ-based quantitative approaches. Proteome Sci. 2013, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.P.; Liu, H.; Tian, L.; Dong, X.B.; Shen, S.H.; Qu, L.Q. Integrated and comparative proteomics of high-oil and high-protein soybean seeds. Food Chem. 2015, 172, 105–116. [Google Scholar] [CrossRef]
- Xu, Y.; Yan, F.; Liu, Y.; Wang, Y.; Gao, H.; Zhao, S.; Zhu, Y.; Wang, Q.; Li, J. Quantitative proteomic and lipidomics analyses of high oil content GmDGAT1-2 transgenic soybean illustrate the regulatory mechanism of lipoxygenase and oleosin. Plant Cell Rep. 2021, 40, 2303–2323. [Google Scholar] [CrossRef]
- Jorrin-Novo, J.V.; Komatsu, S.; Sanchez-Lucas, R.; Rodríguez de Francisco, L.E. Gel electrophoresis-based plant proteomics: Past, present, and future. Happy 10th anniversary Journal of Proteomics! J. Proteom. 2019, 198, 1–10. [Google Scholar] [CrossRef]
- Hajduch, M.; Ganapathy, A.; Stein, J.W.; Thelen, J.J. A systematic proteomic study of seed filling in soybean. Establishment of high-resolution two-dimensional reference maps, expression profiles, and an interactive proteome database. Plant Physiol. 2005, 137, 1397–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, G.K.; Hajduch, M.; Graham, K.; Thelen, J.J. In-depth investigation of the soybean seed-filling proteome and comparison with a parallel study of rapeseed. Plant Phys. 2008, 148, 504–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Komatsu, S. Proteomic approaches to uncover the flooding and drought stress response mechanisms in soybean. J. Proteom. 2018, 17, 201–215. [Google Scholar] [CrossRef]
- Wang, X.; Khodadadi, E.; Fakheri, B.; Komatsu, S. Organ-specific proteomics of soybean seedli under flooding and drought stresses. J. Proteom. 2017, 162, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, H.; Li, F.; Yang, B.; Komatsu, S.; Zhou, S. Quantitative proteomics reveals dual effects of calcium on radicle protrusion in soybean. J. Proteom. 2021, 230, 103999. [Google Scholar] [CrossRef] [PubMed]
- Islam, N.; Bates, P.D.; Maria John, K.M.; Krishnan, H.B.; Zhang, Z.J.; Luthria, D.L.; Natarajan, S.S. Quantitative proteomic analysis of low linolenic acid transgenic soybean reveals perturbations of fatty acid metabolic pathways. Proteomics 2019, 19, 1800379. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Liu, X.; Li, L.; Zhao, H.; Liu, S.; Yu, X.; Shen, Y.; Zhou, Y.; Zhu, Y.; Shu, Y.; et al. Quantitative proteomic, physiological and biochemical analysis of cotyledon, embryo, leaf and pod reveals the effects of high temperature and humidity stress on seed vigor formation in soybean. BMC Plant Biol. 2020, 20, 127. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.M.; Elmore, J.M.; Walley, J.W. To the proteome and beyond: Advances in single-cell omics profiling for plant systems. Plant Physiol. 2022, 188, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Mergner, J.; Kuster, B. Plant Proteome Dynamics. Annu. Rev. Plant Biol. 2022, 73. [Google Scholar] [CrossRef]
- Fiehn, O.; Kopka, J.; Dörmann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000, 18, 1157–1161. [Google Scholar] [CrossRef]
- Weckwerth, W. Metabolomics in systems biology. Annu. Rev. Plant Biol. 2003, 54, 669. [Google Scholar] [CrossRef] [PubMed]
- Moco, S.; Bino, R.J.; Vos, R.; Vervoort, J. Metabolomics technologies and metabolite identification. TrAC-Trend Anal. Chem. 2007, 26, 855–866. [Google Scholar] [CrossRef]
- Allwood, J.W.; Goodacre, R. An introduction to liquid chromatography-mass spectrometry instrumentation applied in plant metabolomic analyses. Phytochem. Anal. 2010, 21, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Jogaiah, S.; Govind, S.R.; Tran, L.S. Systems biology-based approaches toward understanding drought tolerance in food crops. Crit. Rev. Biotechnol. 2013, 33, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics–the link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Hirayama, T. Practical aspects of uniform stable isotope labeling of higher plants for heteronuclear NMR-based metabolomics. Methods Mol. Biol. 2007, 358, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Choi, Y.H.; Verpoorte, R. NMR-based plant metabolomics: Where do we stand, where do we go? Trends Biotechnol. 2011, 29, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; King, R.D.; Altmann, T.; Fiehn, O. Application of metabolomics to plant genotype discrimination using statistics and machine learning. Bioinformatics 2002, 18, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Mu, X.; Liu, J.; Li, B.; Liu, H.; Zhang, B.; Xiao, P. Plant metabolomics: A new strategy and tool for quality evaluation of Chinese medicinal materials. Chin. Med. 2022, 17, 45. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Lindon, J.C. Metabonomics. Nature 2008, 455, 1054–1056. [Google Scholar] [CrossRef]
- Dolatmoradi, M.; Samarah, L.Z.; Vertes, A. Single-Cell Metabolomics by Mass Spectrometry: Opportunities and Challenges. Anal. Sens. 2022, 2, e202100032. [Google Scholar] [CrossRef]
- Lanekoff, I.; Sharma, V.V.; Marques, C. Single-cell metabolomics: Where are we and where are we going? Curr. Opin. Biotechnol. 2022, 75, 102693. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Rao, J.; Shi, J.; Hu, C.; Cheng, F.; Wilson, Z.A.; Zhang, D.; Quan, S. Seed metabolomic study reveals significant metabolite variations and correlations among different soybean cultivars. J. Integr. Plant Biol. 2014, 56, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Kim, E.H.; Park, I.; Yu, B.R.; Lim, J.D.; Lee, Y.S.; Lee, J.H.; Kim, S.H.; Chung, I.M. Isoflavones profiling of soybean [Glycine max (L.) Merrill] germplasms and their correlations with metabolic pathways. Food Chem. 2014, 153, 258–264. [Google Scholar] [CrossRef]
- Liu, J.; Hu, B.; Liu, W.; Qin, W.; Wu, H.; Zhang, J.; Yang, C.; Deng, J.; Shu, K.; Du, J.; et al. Metabolomic tool to identify soybean [Glycine max (L.) Merrill] germplasms with a high level of shade tolerance at the seedling stage. Sci. Rep. 2017, 7, 42478. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Hwang, Y.S.; Chang, W.S.; Moon, J.K.; Choung, M.G. Seed maturity differentially mediates metabolic responses in black soybean. Food Chem. 2013, 141, 2052–2059. [Google Scholar] [CrossRef]
- Wilcox, J.R.; Shibles, R.M. Interrelationships among seed quality attributes in soybean. Crop. Sci. 2001, 41, 11–14. [Google Scholar] [CrossRef]
- Feng, Z.; Ding, C.; Li, W.; Wang, D.; Cui, D. Applications of metabolomics in the research of soybean plant under abiotic stress. Food Chem. 2020, 310, 125914. [Google Scholar] [CrossRef]
- Meyer, R.C.; Steinfath, M.; Lisec, J.; Becher, M.; Witucka-Wall, H.; Törjék, O.; Fiehn, O.; Eckardt, A.; Willmitzer, L.; Selbig, J.; et al. The metabolic signature related to high plant growth rate in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2007, 104, 4759–4764. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, P.; Rabara, R.C.; Reese, R.N.; Miller, M.A.; Rohila, J.S.; Subramanian, S.; Shen, Q.J.; Morandi, D.; Bücking, H.; Shulaev, V.; et al. A toolbox of genes, proteins, metabolites and promoters for improving drought tolerance in soybean includes the metabolite coumestrol and stomatal development genes. BMC Genom. 2016, 17, 102. [Google Scholar] [CrossRef] [Green Version]
- Scandiani, M.M.; Luque, A.G.; Razori, M.V.; Ciancio Casalini, L.; Aoki, T.; O’Donnell, K.; Cervigni, G.D.; Spampinato, C.P. Metabolic profiles of soybean roots during early stages of Fusarium tucumaniae infection. J. Exp. Bot. 2015, 66, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, R.D.; D’Auria, J.C.; Silva-Ferreira, A.C.; Gibon, Y.; Kruszka, D.; Mishra, P.; Zedde, R. High-throughput plant phenotyping: A role for metabolomics? Trends Plant Sci. 2022, 1360–1385, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhou, G.; Liang, C.; Tian, Z. Omics-based interdisciplinarity is accelerating plant breeding. Curr. Opin. Plant Biol. 2022, 66, 102167. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, Y.; Du, J.; Guo, X.; Wen, W.; Gu, S.; Wang, J.; Fan, J. Crop phenomics: Current status and perspectives. Front. Plant Sci. 2019, 10, 714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lube, V.; Noyan, M.A.; Przybysz, A.; Salama, K.; Blilou, I. MultipleXLab: A high-throughput portable live-imaging root phenotyping platform using deep learning and computer vision. Plant Methods 2022, 18, 38. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Sanchez, P.; Gore, M.A.; Heun, J.T.; Thorp, K.R.; Carmo-Silva, A.E.; French, A.N.; Salvucci, M.E.; White, J.W. Development and evaluation of a field-based high-throughput phenotyping platform. Funct. Plant Biol. 2013, 41, 68–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorani, F.; Schurr, U. Future scenarios for plant phenotyping. Annu. Rev. Plant. Biol. 2013, 64, 267–291. [Google Scholar] [CrossRef] [Green Version]
- Yu, N.; Li, L.; Schmitz, N.; Tian, L.F.; Greenberg, J.A.; Diers, B.W. Development of methods to improve soybean yield estimation and predict plant maturity with an unmanned aerial vehicle based platform. Remote Sens. Environ. 2016, 187, 91–101. [Google Scholar] [CrossRef]
- Maimaitijiang, M.; Ghulam, A.; Sidike, P.; Hartling, S.; Peterson, K.; Fritschi, F. Unmanned Aerial System (UAS)-based phenotyping of soybean using multi-sensor data fusion and extreme learning machine. ISPRS J. Photogramm. 2017, 134, 43–58. [Google Scholar] [CrossRef]
- Yuan, W.; Wijewardane, N.K.; Jenkins, S.; Bai, G.; Ge, Y.; Graef, G.L. Early Prediction of Soybean Traits through Color and Texture Features of Canopy RGB Imagery. Sci. Rep. 2019, 9, 14089. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhou, J.; Ye, H.; Ali, M.L.; Nguyen, H.T.; Chen, P. Classification of soybean leaf wilting due to drought stress using UAV-based imagery. Comput. Electron. Agric. 2020, 175, 105576. [Google Scholar] [CrossRef]
- Toda, Y.; Kaga, A.; Kajiya-Kanegae, H.; Hattori, T.; Yamaoka, S.; Okamoto, M.; Tsujimoto, H.; Iwata, H. Genomic prediction modeling of soybean biomass using UAV-based remote sensing and longitudinal model parameters. Plant Genome 2021, 14, e20157. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Mou, H.; Zhou, J.; Ali, M.L.; Ye, H.; Chen, P.; Nguyen, H.T. Qualification of soybean responses to flooding stress using UAV-Based imagery and deep learning. Plant Phenom. 2021, 2021, 9892570. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Fu, X.; Zhou, S.; Zhou, J.; Ye, H.; Nguyen, H.T. Automated segmentation of soybean plants from 3D point cloud using machine learning. Comput. Electron. Agric. 2019, 162, 143–153. [Google Scholar] [CrossRef]
- Zhu, R.; Sun, K.; Yan, Z.; Yan, X.; Yu, J.; Shi, J.; Hu, Z.; Jiang, H.; Xin, D.; Zhang, Z.; et al. Analysing the phenotype development of soybean plants using low-cost 3D reconstruction. Sci. Rep. 2020, 10, 7055. [Google Scholar] [CrossRef]
- Finkel, E. Imaging. With ‘phenomics,’ Plant scientists hope to shift breeding into overdrive. Science 2009, 325, 380–381. [Google Scholar] [CrossRef]
- Naik, H.S.; Zhang, J.; Lofquist, A.; Assefa, T.; Sarkar, S.; Ackerman, D.; Singh, A.; Singh, A.K.; Ganapathysubramanian, B. A real-time phenotyping framework using machine learning for plant stress severity rating in soybean. Plant Met. 2017, 13, 23. [Google Scholar] [CrossRef] [Green Version]
- Dobbels, A.A.; Lorenz, A.J. Soybean iron deficiency chlorosis high throughput phenotyping using an unmanned aircraft system. Plant Met. 2019, 15, 97. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, H.; Zhou, J.; Fu, X.; Ye, H.; Nguyen, H.T. Development of an automated phenotyping platform for quantifying soybean dynamic responses to salinity stress in greenhouse environment. Comput. Electron. Agric. 2018, 151, 319–330. [Google Scholar] [CrossRef]
- Urbina, F.; Ekins, S. The Commoditization of AI for Molecule Design. Artif. Intell. Life Sci. 2022, 2, 100031. [Google Scholar] [CrossRef]
- Ranzato, M.A.; Hinton, G.; LeCun, Y. Guest editorial: Deep learning. Int. J. Comput. Vis. 2015, 113, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Arya, S.; Sandhu, K.S.; Singh, J. Deep learning: As the new frontier in high-throughput plant phenotyping. Euphytica 2022, 218, 47. [Google Scholar] [CrossRef]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. Imagenet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 2012, 25. [Google Scholar] [CrossRef]
- Fey, M.; Lenssen, J.E. Fast graph representation learning with PyTorch Geometric. arXiv 2019, arXiv:1903.02428. [Google Scholar]
- Jia, Y.; Shelhamer, E.; Donahue, J.; Karayev, S.; Long, J.; Girshick, R.; Darrell, T. Caffe: Convolutional architecture for fast feature embedding. In Proceedings of the 22nd ACM International Conference on Multimedia, 2014; MM ’14: Proceedings of the 22nd ACM international conference on Multimedia, Orlando, FL, USA, 3–7 November 2014; pp. 675–678. [Google Scholar] [CrossRef]
- Herrero-Huerta, M.; Rodriguez-Gonzalvez, P.; Rainey, K.M. Yield prediction by machine learning from UAS-based mulit-sensor data fusion in soybean. Plant Met. 2020, 16, 78. [Google Scholar] [CrossRef] [PubMed]
- Maimaitijiang, M.; Sagan, V.; Sidike, P.; Hartling, S.; Esposito, F.; Fritschi, F.B. Soybean yield prediction from UAV using multimodal data fusion and deep learning. Remote Sens. Environ. 2020, 237, 111599. [Google Scholar] [CrossRef]
- Riera, L.G.; Carroll, M.E.; Zhang, Z.; Shook, J.M.; Ghosal, S.; Gao, T.; Singh, A.; Bhattacharya, S.; Ganapathysubramanian, B.; Singh, A.K.; et al. Deep multiview image fusion for soybean yield estimation in breeding applications. Plant Phenom. 2021, 2021, 9846470. [Google Scholar] [CrossRef]
- Baek, J.H.; Lee, E.; Kim, N.; Kim, S.L.; Choi, I.; Ji, H.; Chung, Y.S.; Choi, M.S.; Moon, J.K.; Kim, K.H. High throughput phenotyping for various traits on soybean seeds using image analysis. Sensors 2020, 20, 248. [Google Scholar] [CrossRef] [Green Version]
- Momin, M.A.; Yamamoto, K.; Miyamoto, M.; Kondo, N.; Grift, T. Machine vision based soybean quality evaluation. Comput. Electron. Agric. 2017, 140, 452–460. [Google Scholar] [CrossRef]
- Li, Y.; Jia, J.; Zhang, L.; Khattak, A.M.; Gao, W.; Sun, S.; Wang, M. Soybean seed counting based on pod image using two-column convolution neural network. IEEE Access. 2019, 7, 64177–64185. [Google Scholar] [CrossRef]
- Yang, S.; Zheng, L.; He, P.; Wu, T.; Sun, S.; Wang, M. High-throughput soybean seeds phenotyping with convolutional neural networks and transfer learning. Plant Met. 2021, 17, 50. [Google Scholar] [CrossRef] [PubMed]
- Jubery, T.Z.; Carley, C.N.; Singh, A.; Sarkar, S.; Ganapathysubramanian, B.; Singh, A.K. Using Machine learning to develop a fully automated soybean nodule acquisition pipeline (SNAP). Plant Phenom. 2021, 2021, 9834746. [Google Scholar] [CrossRef] [PubMed]
- Santana, D.C.; de-Oliveira-Cunha, M.P.; Dos-Santos, R.G.; Cotrim, M.F.; Teodoro, L.P.R.; da-Silva-Junior, C.A.; Baio, F.H.R.; Teodoro, P.E. High-throughput phenotyping allows the selection of soybean genotypes for earliness and high grain yield. Plant Methods 2022, 18, 13. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, C.; Belin, E.; Bove, E.; Rousseau, D.; Fabre, F.; Berruyer, R.; Guillaumes, J.; Manceau, C.; Jacques, M.A.; Boureau, T. High throughput quantitative phenotyping of plant resistance using chlorophyll fluorescence image analysis. Plant Methods 2013, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feher-Juhasz, E.; Majer, P.; Sass, L.; Lantos, C.; Csiszar, J.; Turoczy, Z.; Mihaly, R.; Mai, A.; Horvath, G.V.; Vass, I.; et al. Phenotyping shows improved physiological traits and seed yield of transgenic wheat plants expressing the alfalfa aldose reductase under permanent drought stress. Acta Physiol. Plant 2014, 36, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Zhang, J.Z.; Sun, X.Y.; Zhang, X.M. Estimation of leaf chlorophyll content of rice using image color analysis. Can J. Remote Sens. 2013, 39, 185–190. [Google Scholar] [CrossRef]
- Matsuda, O.; Tanaka, A.; Fujita, T.; Iba, K. Hyperspectral imaging techniques for rapid identification of Arabidopsis mutants with altered leaf pigment status. Plant Cell Physiol. 2012, 53, 1154–1170. [Google Scholar] [CrossRef] [Green Version]
- Orel, V.; Wood, R. Early developments in artificial selection as a background to Mendel’s research. Hist. Phil. Life Sci. 1981, 3, 145–170. Available online: https://www.jstor.org/stable/23328309 (accessed on 22 January 2022).
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Hasan, N.; Choudhary, S.; Naaz, N.; Sharma, N.; Laskar, R.A. Recent advancements in molecular marker-assisted selection and applications in plant breeding programmes. J. Genet. Eng. Biotechnol. 2021, 19, 128. [Google Scholar] [CrossRef]
- Hartung, F.; Schiemann, J. Precise plant breeding using new genome editing techniques: Opportunities, safety and regulation in the EU. Plant J. 2014, 78, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.T.; Lee, J.D.; Shannon, J.G.; Bilyeu, K.D. Mutant alleles of FAD2-1A and FAD2-1B combine to produce soybeans with the high oleic acid seed oil trait. BMC Plant Biol. 2010, 10, 195. [Google Scholar] [CrossRef] [Green Version]
- Hagely, K.; Konda, A.R.; Kim, J.H.; Cahoon, E.B.; Bilyeu, K. Molecular-assisted breeding for soybean with high oleic/low linolenic acid and elevated vitamin E in the seed oil. Mol. Breed. 2021, 41, 3. [Google Scholar] [CrossRef]
- Pham, A.T.; Shannon, J.G.; Bilyeu, K.D. Combinations of mutant FAD2 and FAD3 genes to produce high oleic acid and low linolenic acid soybean oil. Theor. Appl. Genet. 2012, 125, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Hagely, K.B.; Jo, H.; Kim, J.H.; Hudson, K.A.; Bilyeu, K. Molecular-assisted breeding for improved carbohydrate profiles in soybean seed. Theor. Appl. Genet. 2020, 133, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.; Culp, C.; Škrabišová, M.; Joshi, T.; Belzile, F.; Grant, D.M.; Bilyeu, K. Molecular tools for detecting Pdh1 can improve soybean breeding efficiency by reducing yield losses due to pod shatter. Mol. Breed. 2019, 39, 27. [Google Scholar] [CrossRef]
- Kou, K.; Yang, H.; Li, H.; Fang, C.; Chen, L.; Yue, L.; Nan, H.; Kong, L.; Li, X.; Wang, F.; et al. A functionally divergent SOC1 homolog improves soybean yield and latitudinal adaptation. Curr. Biol. 2022, 32, 1728–1742.e6. [Google Scholar] [CrossRef]
- Jing, H.C.; Tian, Z.X.; Chong, K.; Li, J.Y. Progress and perspective of molecular design breeding. Sci. China Life Sci. 2021, 51, 1356. [Google Scholar] [CrossRef]
- Wang, L.; Guo, Y.; Yang, S.H. Designed breeding for adaptation of crops to environmental abiotic stresses. Sci. China Life Sci. 2021, 51, 1424–1434. [Google Scholar] [CrossRef]
- OuYang, Y.D.; Chen, L.T. Fertility Regulation and Molecular Design Hybrid Breeding in Crops. Sci. China Life Sci. 2021, 51, 1385. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, P.; Zhao, Y.; Wu, F.; Xin, D.; Liu, C.; Wu, X.; Lv, J.; Chen, Q.; Qi, Z. Multi-Omics Techniques for Soybean Molecular Breeding. Int. J. Mol. Sci. 2022, 23, 4994. https://doi.org/10.3390/ijms23094994
Cao P, Zhao Y, Wu F, Xin D, Liu C, Wu X, Lv J, Chen Q, Qi Z. Multi-Omics Techniques for Soybean Molecular Breeding. International Journal of Molecular Sciences. 2022; 23(9):4994. https://doi.org/10.3390/ijms23094994
Chicago/Turabian StyleCao, Pan, Ying Zhao, Fengjiao Wu, Dawei Xin, Chunyan Liu, Xiaoxia Wu, Jian Lv, Qingshan Chen, and Zhaoming Qi. 2022. "Multi-Omics Techniques for Soybean Molecular Breeding" International Journal of Molecular Sciences 23, no. 9: 4994. https://doi.org/10.3390/ijms23094994
APA StyleCao, P., Zhao, Y., Wu, F., Xin, D., Liu, C., Wu, X., Lv, J., Chen, Q., & Qi, Z. (2022). Multi-Omics Techniques for Soybean Molecular Breeding. International Journal of Molecular Sciences, 23(9), 4994. https://doi.org/10.3390/ijms23094994