
Cerebral Cavernous Malformation Pathogenesis: Investigating Lesion Formation and Progression with Animal Models

 and
and 
Abstract
:1. Introduction
2. CCM Clinical Presentation
3. CCM Proteins: Structure, Expression Patterns, and Molecular Functions
3.1. KRIT1/CCM1
3.2. CCM2
3.3. PDCD10/CCM3
3.4. CCM Signaling Complex (CSC)
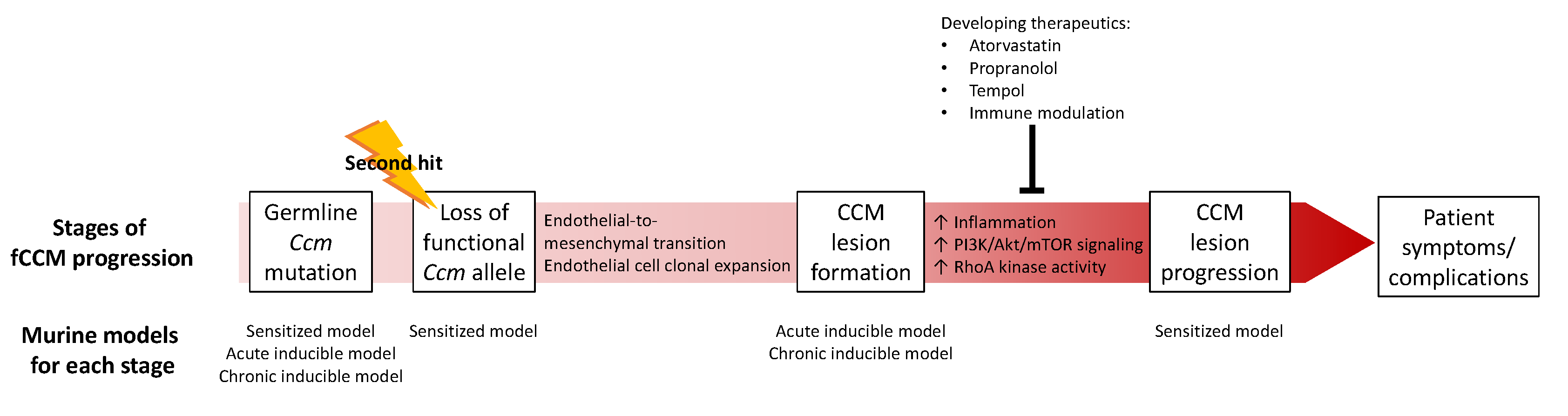
4. CCM Pathogenesis
4.1. CCM Lesion Formation
4.1.1. Familial CCM
4.1.2. Sporadic CCM
4.2. CCM Lesion Progression
5. Animal Models
5.1. Murine Models
5.1.1. Global Loss of CCM
5.1.2. Sensitized CCM Models
5.1.3. Conditional CCM Models
5.1.4. Inducible CCM Models
5.2. Zebrafish Models
5.2.1. KRIT1/CCM1
5.2.2. CCM2
5.2.3. PDCD10/CCM3
5.2.4. Other Zebrafish Models
6. Developing CCM Therapeutics
6.1. RhoA Kinase Inhibition
6.2. Propranolol
6.3. Other Therapeutics
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Batra, S.; Lin, D.; Recinos, P.F.; Zhang, J.; Rigamonti, D. Cavernous malformations: Natural history, diagnosis and treatment. Nat. Rev. Neurol. 2009, 5, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Clatterbuck, R.E.; Eberhart, C.G.; Crain, B.J.; Rigamonti, D. Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J. Neurol. Neurosurg. Psychiatry 2001, 71, 188–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noto, S.; Fujii, M.; Akimura, T.; Imoto, H.; Nomura, S.; Kajiwara, K.; Kato, S.; Fujisawa, H.; Suzuki, M. Management of patients with cavernous angiomas presenting epileptic seizures. Surg. Neurol. 2005, 64, 495–498. [Google Scholar] [CrossRef]
- Ojemann, R.G.; Ogilvy, C.S. Microsurgical treatment of supratentorial cavernous malformations. Neurosurg. Clin. N. Am. 1999, 10, 433–440. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, H.J.; Shin, H.S.; Choi, S.K.; Oh, I.H.; Lim, Y.J. Gamma Knife radiosurgery for brainstem cavernous malformations: Should a patient wait for the rebleed? Acta Neurochir. 2014, 156, 1937–1946. [Google Scholar] [CrossRef]
- Laberge-le Couteulx, S.; Jung, H.H.; Labauge, P.; Houtteville, J.P.; Lescoat, C.; Cecillon, M.; Marechal, E.; Joutel, A.; Bach, J.F.; Tournier-Lasserve, E. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat. Genet. 1999, 23, 189–193. [Google Scholar] [CrossRef]
- Sahoo, T.; Johnson, E.W.; Thomas, J.W.; Kuehl, P.M.; Jones, T.L.; Dokken, C.G.; Touchman, J.W.; Gallione, C.J.; Lee-Lin, S.Q.; Kosofsky, B.; et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum. Mol. Genet. 1999, 8, 2325–2333. [Google Scholar] [CrossRef] [Green Version]
- Denier, C.; Goutagny, S.; Labauge, P.; Krivosic, V.; Arnoult, M.; Cousin, A.; Benabid, A.L.; Comoy, J.; Frerebeau, P.; Gilbert, B.; et al. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 2004, 74, 326–337. [Google Scholar] [CrossRef] [Green Version]
- Liquori, C.; Berg, M.; Siegel, A.; Huang, E.; Zawistowski, J.S.; Stoffer, T.; Verlaan, D.; Balogun, F.; Hughes, L.; Leedom, T.P.; et al. Mutations in gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am. J. Hum. Genet. 2003, 73, 1459–1464. [Google Scholar] [CrossRef] [Green Version]
- Bergametti, F.; Denier, C.; Labauge, P.; Arnoult, M.; Boetto, S.; Clanet, M.; Coubes, P.; Echenne, B.; Ibrahim, R.; Irthum, B.; et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 2005, 76, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Gault, J.; Shenkar, R.; Recksiek, P.; Awad, I.A. Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke 2005, 36, 872–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gault, J.; Awad, I.A.; Recksiek, P.; Shenkar, R.; Breeze, R.; Handler, M.; Kleinschmidt-DeMasters, B.K. Cerebral cavernous malformations: Somatic mutations in vascular endothelial cells. Neurosurgery 2009, 65, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akers, A.L.; Johnson, E.; Steinberg, G.K.; Zabramski, J.M.; Marchuk, D.A. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): Evidence for a two-hit mechanism of CCM pathogenesis. Hum. Mol. Genet. 2009, 18, 919–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riant, F.; Bergametti, F.; Fournier, H.D.; Chapon, F.; Michalak-Provost, S.; Cecillon, M.; Lejeune, P.; Hosseini, H.; Choe, C.; Orth, M.; et al. CCM3 Mutations Are Associated with Early-Onset Cerebral Hemorrhage and Multiple Meningiomas. Mol. Syndromol. 2013, 4, 165–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labauge, P.; Fontaine, B.; Neau, J.P.; Bergametti, F.; Riant, F.; Blecon, A.; Marchelli, F.; Arnoult, M.; Lannuzel, A.; Clanet, M.; et al. Multiple dural lesions mimicking meningiomas in patients with CCM3/PDCD10 mutations. Neurology 2009, 72, 2044–2046. [Google Scholar] [CrossRef]
- Denier, C.; Labauge, P.; Bergametti, F.; Marchelli, F.; Riant, F.; Arnoult, M.; Maciazek, J.; Vicaut, E.; Brunereau, L.; Tournier-Lasserve, E.; et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann. Neurol. 2006, 60, 550–556. [Google Scholar] [CrossRef]
- Fox, C.K.; Nelson, J.; McCulloch, C.E.; Weinsheimer, S.; Pawlikowska, L.; Hart, B.; Mabray, M.; Zafar, A.; Morrison, L.; Zabramski, J.M.; et al. Seizure Incidence Rates in Children and Adults With Familial Cerebral Cavernous Malformations. Neurology 2021, 97, e1210–e1216. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, D.; Drayer, B.P.; Johnson, P.C.; Hadley, M.N.; Zabramski, J.; Spetzler, R.F. The MRI appearance of cavernous malformations (angiomas). J. Neurosurg. 1987, 67, 518–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriarity, J.L.; Clatterbuck, R.E.; Rigamonti, D. The natural history of cavernous malformations. Neurosurg. Clin. N. Am. 1999, 10, 411–417. [Google Scholar] [CrossRef]
- Rigamonti, D.; Hadley, M.N.; Drayer, B.P.; Johnson, P.C.; Hoenig-Rigamonti, K.; Knight, J.T.; Spetzler, R.F. Cerebral cavernous malformations. Incidence and familial occurrence. N. Engl. J. Med. 1988, 319, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.R.; Awad, I.A.; Little, J.R. Natural history of the cavernous angioma. J. Neurosurg. 1991, 75, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.R., Jr.; Awad, I.A.; Magdinec, M.; Paranandi, L. Factors predisposing to clinical disability in patients with cavernous malformations of the brain. Neurosurgery 1993, 32, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Porter, P.J.; Willinsky, R.A.; Harper, W.; Wallace, M.C. Cerebral cavernous malformations: Natural history and prognosis after clinical deterioration with or without hemorrhage. J. Neurosurg. 1997, 87, 190–197. [Google Scholar] [CrossRef]
- Maraire, J.N.; Awad, I.A. Intracranial cavernous malformations: Lesion behavior and management strategies. Neurosurgery 1995, 37, 591–605. [Google Scholar] [CrossRef]
- Pozzati, E.; Acciarri, N.; Tognetti, F.; Marliani, F.; Giangaspero, F. Growth, subsequent bleeding, and de novo appearance of cerebral cavernous angiomas. Neurosurgery 1996, 38, 662–669. [Google Scholar] [CrossRef]
- Serebriiskii, I.; Estojak, J.; Sonoda, G.; Testa, J.R.; Golemis, E.A. Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21-22. Oncogene 1997, 15, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Petit, N.; Blécon, A.; Denier, C.; Tournier-Lasserve, E. Patterns of expression of the three cerebral cavernous malformation (CCM) genes during embryonic and postnatal brain development. Gene Expr. Patterns 2006, 6, 495–503. [Google Scholar] [CrossRef]
- Guzeloglu-Kayisli, O.; Amankulor, N.M.; Voorhees, J.; Luleci, G.; Lifton, R.P.; Gunel, M. KRIT1/cerebral cavernous malformation 1 protein localizes to vascular endothelium, astrocytes, and pyramidal cells of the adult human cerebral cortex. Neurosurgery 2004, 54, 943–949. [Google Scholar] [CrossRef]
- Glading, A.; Han, J.; Stockton, R.A.; Ginsberg, M.H. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J. Cell Biol. 2007, 179, 247–254. [Google Scholar] [CrossRef]
- De Luca, E.; Perrelli, A.; Swamy, H.; Nitti, M.; Passalacqua, M.; Furfaro, A.L.; Salzano, A.M.; Scaloni, A.; Glading, A.J.; Retta, S.F. Protein kinase Cα regulates the nucleocytoplasmic shuttling of KRIT1. J. Cell Sci. 2021, 134, jcs250217. [Google Scholar] [CrossRef] [PubMed]
- Stockton, R.A.; Shenkar, R.; Awad, I.A.; Ginsberg, M.H. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 2010, 207, 881–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisowska, J.; Rödel, C.J.; Manet, S.; Miroshnikova, Y.A.; Boyault, C.; Planus, E.; De Mets, R.; Lee, H.H.; Destaing, O.; Mertani, H.; et al. The CCM1-CCM2 complex controls complementary functions of ROCK1 and ROCK2 that are required for endothelial integrity. J. Cell Sci. 2018, 131, jcs216093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Tang, A.T.; Wong, W.Y.; Bamezai, S.; Goddard, L.M.; Shenkar, R.; Zhou, S.; Yang, J.; Wright, A.C.; Foley, M.; et al. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature 2016, 532, 122–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehead, K.J.; Plummer, N.W.; Adams, J.A.; Marchuk, D.A.; Li, D.Y. Ccm1 is required for arterial morphogenesis: Implications for the etiology of human cavernous malformations. Development 2004, 131, 1437–1448. [Google Scholar] [CrossRef] [Green Version]
- Mably, J.D.; Chuang, L.P.; Serluca, F.C.; Mohideen, M.A.; Chen, J.N.; Fishman, M.C. santa and valentine pattern concentric growth of cardiac myocardium in the zebrafish. Development 2006, 133, 3139–3146. [Google Scholar] [CrossRef] [Green Version]
- Craig, H.D.; Günel, M.; Cepeda, O.; Johnson, E.W.; Ptacek, L.; Steinberg, G.K.; Ogilvy, C.S.; Berg, M.J.; Crawford, S.C.; Scott, R.M.; et al. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum. Mol. Genet. 1998, 7, 1851–1858. [Google Scholar] [CrossRef] [Green Version]
- Seker, A.; Pricola, K.L.; Guclu, B.; Ozturk, A.K.; Louvi, A.; Gunel, M. CCM2 expression parallels that of CCM1. Stroke 2006, 37, 518–523. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Rigamonti, D.; Dietz, H.C.; Clatterbuck, R.E. Interaction between krit1 and malcavernin: Implications for the pathogenesis of cerebral cavernous malformations. Neurosurgery 2007, 60, 353–359. [Google Scholar] [CrossRef]
- Whitehead, K.J.; Chan, A.C.; Navankasattusas, S.; Koh, W.; London, N.R.; Ling, J.; Mayo, A.H.; Drakos, S.G.; Jones, C.A.; Zhu, W.; et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat. Med. 2009, 15, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Boulday, G.; Blécon, A.; Petit, N.; Chareyre, F.; Garcia, L.A.; Niwa-Kawakita, M.; Giovannini, M.; Tournier-Lasserve, E. Tissue-specific conditional CCM2 knockout mice establish the essential role of endothelial CCM2 in angiogenesis: Implications for human cerebral cavernous malformations. Dis. Models Mech. 2009, 2, 168–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, K.; Uchida, Y.; O’Donnell, E.; Claudio, E.; Li, W.; Soneji, K.; Wang, H.; Mukouyama, Y.S.; Siebenlist, U. Conditional deletion of Ccm2 causes hemorrhage in the adult brain: A mouse model of human cerebral cavernous malformations. Hum. Mol. Genet. 2011, 20, 3198–3206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, K.; Stahl, S.; Schleider, E.; Ullrich, S.; Nickel, J.; Mueller, T.D.; Felbor, U. CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics 2007, 8, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, H.; Zhang, Y.; Ma, D. cDNA cloning and expression of an apoptosis-related gene, humanTFAR15 gene. Sci. China C Life Sci. 1999, 42, 323–329. [Google Scholar] [CrossRef]
- Li, X.; Zhang, R.; Zhang, H.; He, Y.; Ji, W.; Min, W.; Boggon, T.J. Crystal structure of CCM3, a cerebral cavernous malformation protein critical for vascular integrity. J. Biol. Chem. 2010, 285, 24099–24107. [Google Scholar] [CrossRef] [Green Version]
- Tanriover, G.; Boylan, A.J.; Diluna, M.L.; Pricola, K.L.; Louvi, A.; Gunel, M. PDCD10, the gene mutated in cerebral cavernous malformation 3, is expressed in the neurovascular unit. Neurosurgery 2008, 62, 930–938. [Google Scholar] [CrossRef]
- Fidalgo, M.; Fraile, M.; Pires, A.; Force, T.; Pombo, C.; Zalvide, J. CCM3/PDCD10 stabilizes GCKIII proteins to promote Golgi assembly and cell orientation. J. Cell Sci. 2010, 123, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Zhao, H.; Shan, J.; Long, F.; Chen, Y.; Zhang, Y.; Han, X.; Ma, D. PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol. Biol. Cell 2007, 18, 1965–1978. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Xu, C.; Di Lorenzo, A.; Kleaveland, B.; Zou, Z.; Seiler, C.; Chen, M.; Cheng, L.; Xiao, J.; He, J.; et al. CCM3 signaling through sterile 20-like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. J. Clin. Investig. 2010, 120, 2795–2804. [Google Scholar] [CrossRef]
- Yoruk, B.; Gillers, B.S.; Chi, N.C.; Scott, I.C. Ccm3 functions in a manner distinct from Ccm1 and Ccm2 in a zebrafish model of CCM vascular disease. Dev. Biol. 2012, 362, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Shenkar, R.; Shi, C.; Rebeiz, T.; Stockton, R.A.; McDonald, D.A.; Mikati, A.G.; Zhang, L.; Austin, C.; Akers, A.L.; Gallione, C.J.; et al. Exceptional aggressiveness of cerebral cavernous malformation disease associated with PDCD10 mutations. Genet. Med. 2015, 17, 188–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zhang, H.; Yu, L.; Gunel, M.; Boggon, T.J.; Chen, H.; Min, W. Stabilization of VEGFR2 signaling by cerebral cavernous malformation 3 is critical for vascular development. Sci. Signal. 2010, 3, ra26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.C.; Drakos, S.G.; Ruiz, O.E.; Smith, A.C.; Gibson, C.C.; Ling, J.; Passi, S.F.; Stratman, A.N.; Sacharidou, A.; Revelo, M.P.; et al. Mutations in 2 distinct genetic pathways result in cerebral cavernous malformations in mice. J. Clin. Investig. 2011, 121, 1871–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawistowski, J.S.; Stalheim, L.; Uhlik, M.T.; Abell, A.N.; Ancrile, B.B.; Johnson, G.L.; Marchuk, D.A. CCM1 and CCM2 protein interactions in cell signaling: Implications for cerebral cavernous malformations pathogenesis. Hum. Mol. Genet. 2005, 14, 2521–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilder, T.L.; Malone, M.H.; Bencharit, S.; Colicelli, J.; Haystead, T.A.; Johnson, G.L.; Wu, C.C. Proteomic identification of the cerebral cavernous malformation signaling complex. J. Proteome Res. 2007, 6, 4343–4355. [Google Scholar] [CrossRef]
- Stahl, S.; Gaetzner, S.; Voss, K.; Brackertz, B.; Schleider, E.; Sürücü, O.; Kunze, E.; Netzer, C.; Korenke, C.; Finckh, U.; et al. Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: In-frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex. Hum. Mutat. 2008, 29, 709–717. [Google Scholar] [CrossRef]
- Zhang, J.; Clatterbuck, R.E.; Rigamonti, D.; Chang, D.D.; Dietz, H.C. Interaction between krit1 and icap1alpha infers perturbation of integrin beta1-mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum. Mol. Genet. 2001, 10, 2953–2960. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Basu, S.; Rigamonti, D.; Dietz, H.C.; Clatterbuck, R.E. Krit1 modulates beta 1-integrin-mediated endothelial cell proliferation. Neurosurgery 2008, 63, 571–578. [Google Scholar] [CrossRef]
- Padarti, A.; Zhang, J. Recent advances in cerebral cavernous malformation research. Vessel. Plus 2018, 2, 21. [Google Scholar] [CrossRef] [Green Version]
- Goudreault, M.; D’Ambrosio, L.M.; Kean, M.J.; Mullin, M.J.; Larsen, B.G.; Sanchez, A.; Chaudhry, S.; Chen, G.I.; Sicheri, F.; Nesvizhskii, A.I.; et al. A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Mol. Cell. Proteom. 2009, 8, 157–171. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.; Pallas, D.C. STRIPAK complexes: Structure, biological function, and involvement in human diseases. Int. J. Biochem. Cell Biol. 2014, 47, 118–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagenstecher, A.; Stahl, S.; Sure, U.; Felbor, U. A two-hit mechanism causes cerebral cavernous malformations: Complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum. Mol. Genet. 2009, 18, 911–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinverno, M.; Maderna, C.; Abu Taha, A.; Corada, M.; Orsenigo, F.; Valentino, M.; Pisati, F.; Fusco, C.; Graziano, P.; Giannotta, M.; et al. Endothelial cell clonal expansion in the development of cerebral cavernous malformations. Nat. Commun. 2019, 10, 2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detter, M.R.; Snellings, D.A.; Marchuk, D.A. Cerebral Cavernous Malformations Develop Through Clonal Expansion of Mutant Endothelial Cells. Circ. Res. 2018, 123, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Rath, M.; Pagenstecher, A.; Hoischen, A.; Felbor, U. Postzygotic mosaicism in cerebral cavernous malformation. J. Med. Genet. 2020, 57, 212–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, D.A.; Shi, C.; Shenkar, R.; Gallione, C.J.; Akers, A.L.; Li, S.; De Castro, N.; Berg, M.J.; Corcoran, D.L.; Awad, I.A.; et al. Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: Evidence for a common biochemical pathway for CCM pathogenesis. Hum. Mol. Genet. 2014, 23, 4357–4370. [Google Scholar] [CrossRef]
- D’Angelo, R.; Alafaci, C.; Scimone, C.; Ruggeri, A.; Salpietro, F.M.; Bramanti, P.; Tomasello, F.; Sidoti, A. Sporadic cerebral cavernous malformations: Report of further mutations of CCM genes in 40 Italian patients. Biomed. Res. Int. 2013, 2013, 459253. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, R.; Marini, V.; Rinaldi, C.; Origone, P.; Dorcaratto, A.; Avolio, M.; Goitre, L.; Forni, M.; Capra, V.; Alafaci, C.; et al. Mutation analysis of CCM1, CCM2 and CCM3 genes in a cohort of Italian patients with cerebral cavernous malformation. Brain Pathol. 2011, 21, 215–224. [Google Scholar] [CrossRef]
- Peyre, M.; Miyagishima, D.; Bielle, F.; Chapon, F.; Sierant, M.; Venot, Q.; Lerond, J.; Marijon, P.; Abi-Jaoude, S.; Le Van, T.; et al. Somatic PIK3CA Mutations in Sporadic Cerebral Cavernous Malformations. N. Engl. J. Med. 2021, 385, 996. [Google Scholar] [CrossRef]
- Eren Gozel, H.; Kök, K.; Ozlen, F.; Isler, C.; Pence, S. A novel insight into differential expression profiles of sporadic cerebral cavernous malformation patients with different symptoms. Sci. Rep. 2021, 11, 19351. [Google Scholar] [CrossRef]
- Scimone, C.; Donato, L.; Alibrandi, S.; Esposito, T.; Alafaci, C.; D’Angelo, R.; Sidoti, A. Transcriptome analysis provides new molecular signatures in sporadic Cerebral Cavernous Malformation endothelial cells. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165956. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Xiao, X.; Ren, J.; Cui, B.; Zong, Y.; Zou, J.; Kou, Z.; Jiang, N.; Meng, G.; Zeng, G.; et al. Somatic MAP3K3 and PIK3CA mutations in sporadic cerebral and spinal cord cavernous malformations. Brain 2021, 144, 2648–2658. [Google Scholar] [CrossRef] [PubMed]
- Zabramski, J.M.; Wascher, T.M.; Spetzler, R.F.; Johnson, B.; Golfinos, J.; Drayer, B.P.; Brown, B.; Rigamonti, D.; Brown, G. The natural history of familial cavernous malformations: Results of an ongoing study. J. Neurosurg. 1994, 80, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Girard, R.; Fam, M.D.; Zeineddine, H.A.; Tan, H.; Mikati, A.G.; Shi, C.; Jesselson, M.; Shenkar, R.; Wu, M.; Cao, Y.; et al. Vascular permeability and iron deposition biomarkers in longitudinal follow-up of cerebral cavernous malformations. J. Neurosurg. 2017, 127, 102–110. [Google Scholar] [CrossRef]
- Mikati, A.G.; Khanna, O.; Zhang, L.; Girard, R.; Shenkar, R.; Guo, X.; Shah, A.; Larsson, H.B.; Tan, H.; Li, L.; et al. Vascular permeability in cerebral cavernous malformations. J. Cereb. Blood Flow Metab. 2015, 35, 1632–1639. [Google Scholar] [CrossRef]
- Girard, R.; Zeineddine, H.A.; Fam, M.D.; Mayampurath, A.; Cao, Y.; Shi, C.; Shenkar, R.; Polster, S.P.; Jesselson, M.; Duggan, R.; et al. Plasma Biomarkers of Inflammation Reflect Seizures and Hemorrhagic Activity of Cerebral Cavernous Malformations. Transl. Stroke Res. 2018, 9, 34–43. [Google Scholar] [CrossRef]
- Koskimäki, J.; Zhang, D.; Li, Y.; Saadat, L.; Moore, T.; Lightle, R.; Polster, S.P.; Carrión-Penagos, J.; Lyne, S.B.; Zeineddine, H.A.; et al. Transcriptome clarifies mechanisms of lesion genesis versus progression in models of Ccm3 cerebral cavernous malformations. Acta Neuropathol. Commun. 2019, 7, 132. [Google Scholar] [CrossRef]
- Shi, C.; Shenkar, R.; Zeineddine, H.A.; Girard, R.; Fam, M.D.; Austin, C.; Moore, T.; Lightle, R.; Zhang, L.; Wu, M.; et al. B-Cell Depletion Reduces the Maturation of Cerebral Cavernous Malformations in Murine Models. J. Neuroimmune Pharmacol. 2016, 11, 369–377. [Google Scholar] [CrossRef]
- Yau, A.C.Y.; Globisch, M.A.; Onyeogaziri, F.C.; Conze, L.L.; Smith, R.; Jauhiainen, S.; Corada, M.; Orsenigo, F.; Huang, H.; Herre, M.; et al. Inflammation and neutrophil extracellular traps in cerebral cavernous malformation. Cell. Mol. Life Sci. 2022, 79, 206. [Google Scholar] [CrossRef]
- Castro, C.; Oyamada, H.A.A.; Cafasso, M.; Lopes, L.M.; Monteiro, C.; Sacramento, P.M.; Alves-Leon, S.V.; da Fontoura Galvão, G.; Hygino, J.; de Souza, J.; et al. Elevated proportion of TLR2- and TLR4-expressing Th17-like cells and activated memory B cells was associated with clinical activity of cerebral cavernous malformations. J. Neuroinflammation 2022, 19, 28. [Google Scholar] [CrossRef]
- McDonald, D.A.; Shi, C.; Shenkar, R.; Stockton, R.A.; Liu, F.; Ginsberg, M.H.; Marchuk, D.A.; Awad, I.A. Fasudil decreases lesion burden in a murine model of cerebral cavernous malformation disease. Stroke 2012, 43, 571–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenkar, R.; Shi, C.; Austin, C.; Moore, T.; Lightle, R.; Cao, Y.; Zhang, L.; Wu, M.; Zeineddine, H.A.; Girard, R.; et al. RhoA Kinase Inhibition With Fasudil Versus Simvastatin in Murine Models of Cerebral Cavernous Malformations. Stroke 2017, 48, 187–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, A.A.; Snellings, D.A.; Su, Y.S.; Hong, C.C.; Castro, M.; Tang, A.T.; Detter, M.R.; Hobson, N.; Girard, R.; Romanos, S.; et al. PIK3CA and CCM mutations fuel cavernomas through a cancer-like mechanism. Nature 2021, 594, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Kleaveland, B.; Zheng, X.; Liu, J.J.; Blum, Y.; Tung, J.J.; Zou, Z.; Sweeney, S.M.; Chen, M.; Guo, L.; Lu, M.M.; et al. Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat. Med. 2009, 15, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Plummer, N.W.; Squire, T.L.; Srinivasan, S.; Huang, E.; Zawistowski, J.S.; Matsunami, H.; Hale, L.P.; Marchuk, D.A. Neuronal expression of the Ccm2 gene in a new mouse model of cerebral cavernous malformations. Mamm. Genome 2006, 17, 119–128. [Google Scholar] [CrossRef]
- McDonald, D.A.; Shenkar, R.; Shi, C.; Stockton, R.A.; Akers, A.L.; Kucherlapati, M.H.; Kucherlapati, R.; Brainer, J.; Ginsberg, M.H.; Awad, I.A.; et al. A novel mouse model of cerebral cavernous malformations based on the two-hit mutation hypothesis recapitulates the human disease. Hum. Mol. Genet. 2011, 20, 211–222. [Google Scholar] [CrossRef]
- Plummer, N.W.; Gallione, C.J.; Srinivasan, S.; Zawistowski, J.S.; Louis, D.N.; Marchuk, D.A. Loss of p53 sensitizes mice with a mutation in Ccm1 (KRIT1) to development of cerebral vascular malformations. Am. J. Pathol. 2004, 165, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Shenkar, R.; Venkatasubramanian, P.N.; Wyrwicz, A.M.; Zhao, J.C.; Shi, C.; Akers, A.; Marchuk, D.A.; Awad, I.A. Advanced magnetic resonance imaging of cerebral cavernous malformations: Part II. Imaging of lesions in murine models. Neurosurgery 2008, 63, 790–797. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.M.; Roach, J.P.; Hu, A.; Stamatovic, S.M.; Zochowski, M.R.; Keep, R.F.; Andjelkovic, A.V. Connexin 43 gap junctions contribute to brain endothelial barrier hyperpermeability in familial cerebral cavernous malformations type III by modulating tight junction structure. FASEB J. 2018, 32, 2615–2629. [Google Scholar] [CrossRef] [Green Version]
- de Wind, N.; Dekker, M.; Berns, A.; Radman, M.; te Riele, H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 1995, 82, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Reitmair, A.H.; Schmits, R.; Ewel, A.; Bapat, B.; Redston, M.; Mitri, A.; Waterhouse, P.; Mittrücker, H.W.; Wakeham, A.; Liu, B.; et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat. Genet. 1995, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Zeineddine, H.A.; Girard, R.; Saadat, L.; Shen, L.; Lightle, R.; Moore, T.; Cao, Y.; Hobson, N.; Shenkar, R.; Avner, K.; et al. Phenotypic characterization of murine models of cerebral cavernous malformations. Lab. Investig. 2019, 99, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Louvi, A.; Chen, L.; Two, A.M.; Zhang, H.; Min, W.; Günel, M. Loss of cerebral cavernous malformation 3 (Ccm3) in neuroglia leads to CCM and vascular pathology. Proc. Natl. Acad. Sci. USA 2011, 108, 3737–3742. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhang, H.; He, Y.; Jiang, Q.; Tanaka, Y.; Park, I.H.; Pober, J.S.; Min, W.; Zhou, H.J. Mural Cell-Specific Deletion of Cerebral Cavernous Malformation 3 in the Brain Induces Cerebral Cavernous Malformations. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2171–2186. [Google Scholar] [CrossRef]
- Boulday, G.; Rudini, N.; Maddaluno, L.; Blécon, A.; Arnould, M.; Gaudric, A.; Chapon, F.; Adams, R.H.; Dejana, E.; Tournier-Lasserve, E. Developmental timing of CCM2 loss influences cerebral cavernous malformations in mice. J. Exp. Med. 2011, 208, 1835–1847. [Google Scholar] [CrossRef]
- Zhou, H.J.; Qin, L.; Jiang, Q.; Murray, K.N.; Zhang, H.; Li, B.; Lin, Q.; Graham, M.; Liu, X.; Grutzendler, J.; et al. Caveolae-mediated Tie2 signaling contributes to CCM pathogenesis in a brain endothelial cell-specific Pdcd10-deficient mouse model. Nat. Commun. 2021, 12, 504. [Google Scholar] [CrossRef]
- Tang, A.T.; Choi, J.P.; Kotzin, J.J.; Yang, Y.; Hong, C.C.; Hobson, N.; Girard, R.; Zeineddine, H.A.; Lightle, R.; Moore, T.; et al. Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature 2017, 545, 305–310. [Google Scholar] [CrossRef]
- Maderna, C.; Pisati, F.; Tripodo, C.; Dejana, E.; Malinverno, M. A murine model of cerebral cavernous malformations with acute hemorrhage. iScience 2022, 25, 103943. [Google Scholar] [CrossRef]
- Detter, M.R.; Shenkar, R.; Benavides, C.R.; Neilson, C.A.; Moore, T.; Lightle, R.; Hobson, N.; Shen, L.; Cao, Y.; Girard, R.; et al. Novel Murine Models of Cerebral Cavernous Malformations. Angiogenesis 2020, 23, 651–666. [Google Scholar] [CrossRef]
- Cardoso, C.; Arnould, M.; De Luca, C.; Otten, C.; Abdelilah-Seyfried, S.; Heredia, A.; Leutenegger, A.L.; Schwaninger, M.; Tournier-Lasserve, E.; Boulday, G. Novel Chronic Mouse Model of Cerebral Cavernous Malformations. Stroke 2020, 51, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; van der Flier, L.G.; Sato, T.; van Es, J.H.; van den Born, M.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; van Rheenen, J.; Simons, B.D.; et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livet, J.; Weissman, T.A.; Kang, H.; Draft, R.W.; Lu, J.; Bennis, R.A.; Sanes, J.R.; Lichtman, J.W. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 2007, 450, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Shenkar, R.; Peiper, A.; Pardo, H.; Moore, T.; Lightle, R.; Girard, R.; Hobson, N.; Polster, S.P.; Koskimäki, J.; Zhang, D.; et al. Rho Kinase Inhibition Blunts Lesion Development and Hemorrhage in Murine Models of Aggressive Pdcd10/Ccm3 Disease. Stroke 2019, 50, 738–744. [Google Scholar] [CrossRef]
- Hogan, B.M.; Bussmann, J.; Wolburg, H.; Schulte-Merker, S. Ccm1 cell autonomously regulates endothelial cellular morphogenesis and vascular tubulogenesis in zebrafish. Hum. Mol. Genet. 2008, 17, 2424–2432. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.N.; Haffter, P.; Odenthal, J.; Vogelsang, E.; Brand, M.; van Eeden, F.J.; Furutani-Seiki, M.; Granato, M.; Hammerschmidt, M.; Heisenberg, C.P.; et al. Mutations affecting the cardiovascular system and other internal organs in zebrafish. Development 1996, 123, 293–302. [Google Scholar] [CrossRef]
- Stainier, D.Y.; Fouquet, B.; Chen, J.N.; Warren, K.S.; Weinstein, B.M.; Meiler, S.E.; Mohideen, M.A.; Neuhauss, S.C.; Solnica-Krezel, L.; Schier, A.F.; et al. Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development 1996, 123, 285–292. [Google Scholar] [CrossRef]
- Rödel, C.J.; Otten, C.; Donat, S.; Lourenço, M.; Fischer, D.; Kuropka, B.; Paolini, A.; Freund, C.; Abdelilah-Seyfried, S. Blood Flow Suppresses Vascular Anomalies in a Zebrafish Model of Cerebral Cavernous Malformations. Circ. Res. 2019, 125, e43–e54. [Google Scholar] [CrossRef]
- Li, W.; Tran, V.; Shaked, I.; Xue, B.; Moore, T.; Lightle, R.; Kleinfeld, D.; Awad, I.A.; Ginsberg, M.H. Abortive intussusceptive angiogenesis causes multi-cavernous vascular malformations. Elife 2021, 10, e62155. [Google Scholar] [CrossRef]
- Voss, K.; Stahl, S.; Hogan, B.M.; Reinders, J.; Schleider, E.; Schulte-Merker, S.; Felbor, U. Functional analyses of human and zebrafish 18-amino acid in-frame deletion pave the way for domain mapping of the cerebral cavernous malformation 3 protein. Hum. Mutat. 2009, 30, 1003–1011. [Google Scholar] [CrossRef]
- Donat, S.; Abdelilah-Seyfried, S. Generation of Transgenic Lines of Zebrafish Expressing Fluorescently Tagged CCM Proteins to Study Their Function and Subcellular Localization Within the Vasculature. Methods Mol. Biol. 2020, 2152, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Karaaslan, B.; Gülsuna, B.; Erol, G.; Dağli, Ö.; Emmez, H.; Kurt, G.; Çeltikçi, E.; Börcek, A. Stereotactic radiosurgery for cerebral cavernous malformation: Comparison of hemorrhage rates before and after stereotactic radiosurgery. J. Neurosurg. 2022, 136, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Bertalanffy, H.; Kar, S.; Tsuji, Y. Microsurgical management of midbrain cavernous malformations: Does lesion depth influence the outcome? Acta Neurochir. 2021, 163, 2739–2754. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, V.; Sumi, S. Molecular Biomarkers and Drug Targets in Brain Arteriovenous and Cavernous Malformations: Where Are We? Stroke 2022, 53, 279–289. [Google Scholar] [CrossRef]
- McKerracher, L.; Shenkar, R.; Abbinanti, M.; Cao, Y.; Peiper, A.; Liao, J.K.; Lightle, R.; Moore, T.; Hobson, N.; Gallione, C.; et al. A Brain-Targeted Orally Available ROCK2 Inhibitor Benefits Mild and Aggressive Cavernous Angioma Disease. Transl. Stroke Res. 2020, 11, 365–376. [Google Scholar] [CrossRef]
- Moghadasian, M.H. Clinical pharmacology of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. Life Sci. 1999, 65, 1329–1337. [Google Scholar] [CrossRef]
- Polster, S.P.; Stadnik, A.; Akers, A.L.; Cao, Y.; Christoforidis, G.A.; Fam, M.D.; Flemming, K.D.; Girard, R.; Hobson, N.; Koenig, J.I.; et al. Atorvastatin Treatment of Cavernous Angiomas with Symptomatic Hemorrhage Exploratory Proof of Concept (AT CASH EPOC) Trial. Neurosurgery 2019, 85, 843–853. [Google Scholar] [CrossRef]
- Reinhard, M.; Schuchardt, F.; Meckel, S.; Heinz, J.; Felbor, U.; Sure, U.; Geisen, U. Propranolol stops progressive multiple cerebral cavernoma in an adult patient. J. Neurol. Sci. 2016, 367, 15–17. [Google Scholar] [CrossRef]
- Zabramski, J.M.; Kalani, M.Y.S.; Filippidis, A.S.; Spetzler, R.F. Propranolol Treatment of Cavernous Malformations with Symptomatic Hemorrhage. World Neurosurg. 2016, 88, 631–639. [Google Scholar] [CrossRef]
- Moschovi, M.; Alexiou, G.A.; Stefanaki, K.; Tourkantoni, N.; Prodromou, N. Propranolol treatment for a giant infantile brain cavernoma. J. Child Neurol. 2010, 25, 653–655. [Google Scholar] [CrossRef]
- Berti, I.; Marchetti, F.; Skabar, A.; Zennaro, F.; Zanon, D.; Ventura, A. Propranolol for cerebral cavernous angiomatosis: A magic bullet. Clin. Pediatrics 2014, 53, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Miquel, J.; Bruneau, B.; Dupuy, A. Successful treatment of multifocal intracerebral and spinal hemangiomas with propranolol. J. Am. Acad. Dermatol. 2014, 70, e83–e84. [Google Scholar] [CrossRef] [PubMed]
- Léauté-Labrèze, C.; Dumas de la Roque, E.; Hubiche, T.; Boralevi, F.; Thambo, J.B.; Taïeb, A. Propranolol for severe hemangiomas of infancy. N. Engl. J. Med. 2008, 358, 2649–2651. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.E.; Ryan, M.; Wittenberg, B.; Armstrong, J.; Greenan, K.; Wilkinson, C. Successful treatment of hemorrhagic brainstem cavernous malformation with hematoma evacuation and postoperative propranolol. Childs Nerv. Syst. 2020, 36, 2109–2112. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shenkar, R.; Detter, M.R.; Moore, T.; Benavides, C.; Lightle, R.; Girard, R.; Hobson, N.; Cao, Y.; Li, Y.; et al. Propranolol inhibits cavernous vascular malformations by β1 adrenergic receptor antagonism in animal models. J. Clin. Investig. 2021, 131, e144893. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; Malinverno, M.; Globisch, M.A.; Maderna, C.; Corada, M.; Orsenigo, F.; Conze, L.L.; Rorsman, C.; Sundell, V.; Arce, M.; et al. Propranolol Reduces the Development of Lesions and Rescues Barrier Function in Cerebral Cavernous Malformations: A Preclinical Study. Stroke 2021, 52, 1418–1427. [Google Scholar] [CrossRef]
- Goldberg, J.; Jaeggi, C.; Schoeni, D.; Mordasini, P.; Raabe, A.; Bervini, D. Bleeding risk of cerebral cavernous malformations in patients on β-blocker medication: A cohort study. J. Neurosurg. 2018, 130, 1931–1936. [Google Scholar] [CrossRef] [Green Version]
- Zuurbier, S.M.; Hickman, C.R.; Rinkel, L.A.; Berg, R.; Sure, U.; Al-Shahi Salman, R. Association Between Beta-Blocker or Statin Drug Use and the Risk of Hemorrhage From Cerebral Cavernous Malformations. Stroke 2022. [Google Scholar] [CrossRef]
- Lanfranconi, S.; Scola, E.; Bertani, G.A.; Zarino, B.; Pallini, R.; d’Alessandris, G.; Mazzon, E.; Marino, S.; Carriero, M.R.; Scelzo, E.; et al. Propranolol for familial cerebral cavernous malformation (Treat_CCM): Study protocol for a randomized controlled pilot trial. Trials 2020, 21, 401. [Google Scholar] [CrossRef]
- Shi, C.; Shenkar, R.; Du, H.; Duckworth, E.; Raja, H.; Batjer, H.H.; Awad, I.A. Immune response in human cerebral cavernous malformations. Stroke 2009, 40, 1659–1665. [Google Scholar] [CrossRef] [Green Version]
- Gibson, C.C.; Zhu, W.; Davis, C.T.; Bowman-Kirigin, J.A.; Chan, A.C.; Ling, J.; Walker, A.E.; Goitre, L.; Delle Monache, S.; Retta, S.F.; et al. Strategy for identifying repurposed drugs for the treatment of cerebral cavernous malformation. Circulation 2015, 131, 289–299. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Species | Model Type | Modeling Strategy | Strength(s) | Weakness(es) |
|---|---|---|---|---|
| Mus musculus (Mouse) | Sensitized model | Crossing mice heterozygous for a Ccm allele with a genetically unstable mouse line (e.g., Trp53−/− or Msh2−/− mice) |
|
|
| Acute inducible model | Crossing Ccm-flox mice with CreERT2 lines; high TMX dose immediately post-birth |
|
| |
| Chronic inducible model | Crossing Ccm-flox mice with CreERT2 lines; low TMX dose post-birth |
|
| |
| Danio rerio (Zebrafish) | ccm mutants | ccm mutations identified via genetic screening |
|
|
| ccm morphants | Morpholino targeting to either deplete expression or mimic mutation |
|
| |
| ccm mosaic mutants | CRISPR-Cas9-mediated ccm deletion |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phillips, C.M.; Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Cerebral Cavernous Malformation Pathogenesis: Investigating Lesion Formation and Progression with Animal Models. Int. J. Mol. Sci. 2022, 23, 5000. https://doi.org/10.3390/ijms23095000
Phillips CM, Stamatovic SM, Keep RF, Andjelkovic AV. Cerebral Cavernous Malformation Pathogenesis: Investigating Lesion Formation and Progression with Animal Models. International Journal of Molecular Sciences. 2022; 23(9):5000. https://doi.org/10.3390/ijms23095000
Chicago/Turabian StylePhillips, Chelsea M., Svetlana M. Stamatovic, Richard F. Keep, and Anuska V. Andjelkovic. 2022. "Cerebral Cavernous Malformation Pathogenesis: Investigating Lesion Formation and Progression with Animal Models" International Journal of Molecular Sciences 23, no. 9: 5000. https://doi.org/10.3390/ijms23095000
APA StylePhillips, C. M., Stamatovic, S. M., Keep, R. F., & Andjelkovic, A. V. (2022). Cerebral Cavernous Malformation Pathogenesis: Investigating Lesion Formation and Progression with Animal Models. International Journal of Molecular Sciences, 23(9), 5000. https://doi.org/10.3390/ijms23095000