
Like Brothers in Arms: How Hormonal Stimuli and Changes in the Metabolism Signaling Cooperate, Leading HPV Infection to Drive the Onset of Cervical Cancer




{kind=link}
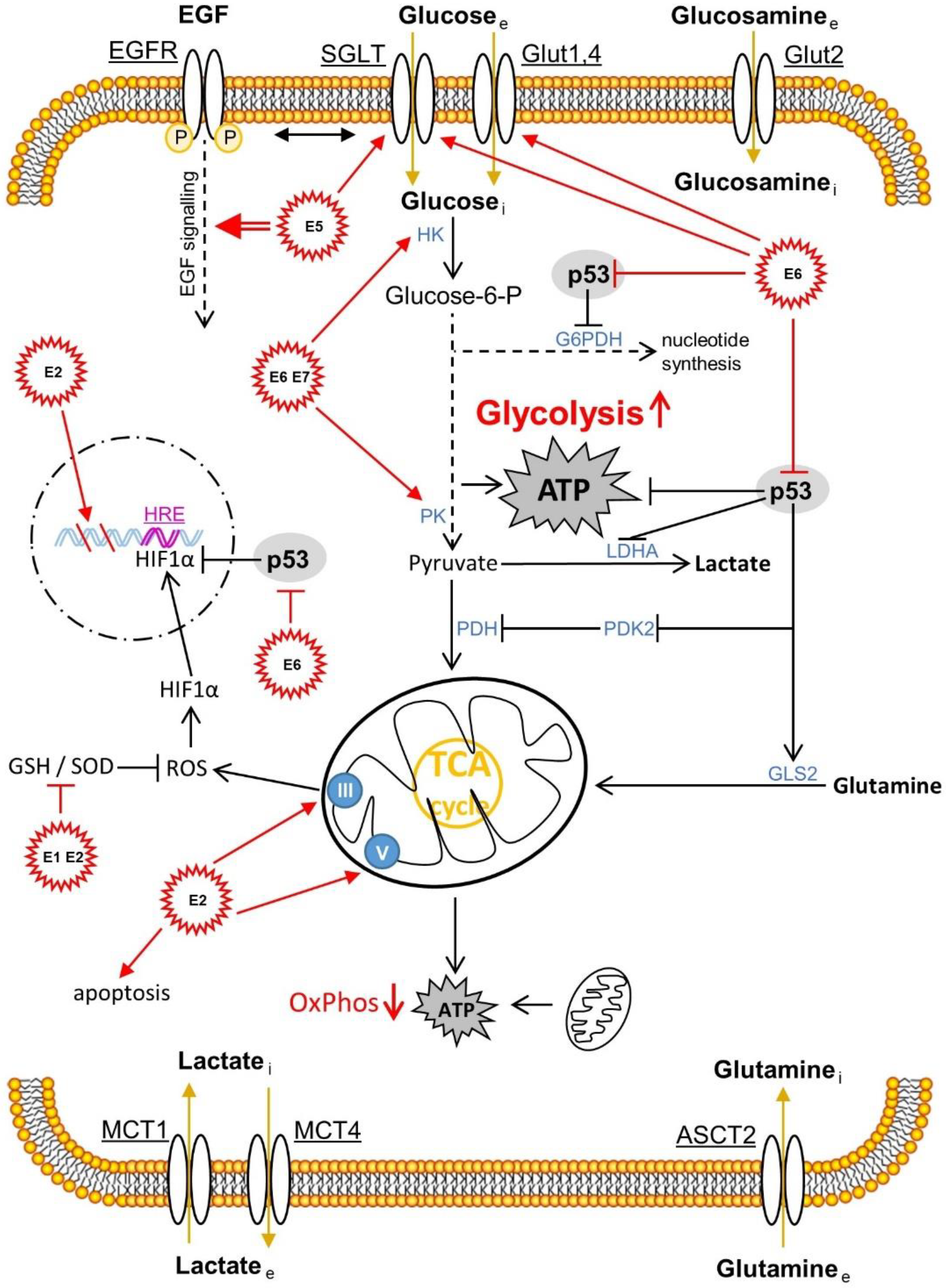{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Biological Background of HPV Infection, Structure of Viral DNA and HPV Replication Cycle in the Stratified Epithelium of the Cervix
2.1. Viral Proteins Determining Host Cell Fate in Terms of Immune Evasion and Duration of HPV Infection and Transformation
2.2. The Demand for Oxygen and Nutrients within HPV-Infected Lesions Promotes Angiogenesis
2.3. The Effects of HPV Oncoproteins on Cellular Metabolism within the Cervical Intraepithelial Lesions
3. The Effects of the Hormone Microenvironment on Cervical Carcinogenesis
3.1. General Information about Sex Steroid Hormones, Their Biosynthesis and Their Localization in the Female Body
3.2. Estrogen Signaling
3.3. Influence of Estrogen Signaling on the Microenvironment of Cervical Intraepithelial Lesion, Cervical Pre-Cancer and in Tumor
3.3.1. Estrogen Distribution in the Tumor and the Surrounding Microenvironment
3.3.2. Different Estrogen Signaling Pathways Causing Both, Pro- and Anti-Tumorigenic Effects in HPV-Positive Lesions of the Cervical Epithelium
3.3.3. The Importance of the ERα for the Tumor Stroma and the Tumor Microenvironment in Relation to the Development of Precancerous Lesions up to the Invasive Form of Cervical Carcinoma
3.3.4. Estrogen Signaling in the Infiltrating Cells of the Immune Response in Cervical Carcinogenesis
3.4. The Effect of ER Antagonists in Modulating the Immune Microenvironment of the Premalignant Cervical Lesion and CxCa
4. Conclusions and Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef] [PubMed]
- De Vuyst, H.; Clifford, G.M.; Nascimento, M.C.; Madeleine, M.M.; Franceschi, S. Prevalence and type distribution of human papillomavirus in carcinoma and intraepithelial neoplasia of the vulva, vagina and anus: A meta-analysis. Int. J. Cancer 2009, 124, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Wittekindt, C.; Wagner, S.; Sharma, S.J.; Wurdemann, N.; Knuth, J.; Reder, H.; Klussmann, J.P. HPV—A different view on Head and Neck Cancer. Laryngorhinootologie 2018, 97, S48–S113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brianti, P.; De Flammineis, E.; Mercuri, S.R. Review of HPV-related diseases and cancers. New Microbiol. 2017, 40, 80–85. [Google Scholar] [PubMed]
- Chesson, H.W.; Dunne, E.F.; Hariri, S.; Markowitz, L.E. The estimated lifetime probability of acquiring human papillomavirus in the United States. Sex Transm. Dis. 2014, 41, 660–664. [Google Scholar] [CrossRef] [PubMed]
- de Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.D.; Chatterjee, S.; Alam, S.; Salzberg, A.C.; Milici, J.; van der Burg, S.H.; Meyers, C. Effect of Productive Human Papillomavirus 16 Infection on Global Gene Expression in Cervical Epithelium. J. Virol. 2018, 92, e01261-18. [Google Scholar] [CrossRef] [Green Version]
- McBride, A.A.; Munger, K. Expert Views on HPV Infection. Viruses 2018, 10, 94. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin-Drubin, M.E.; Meyers, C. Evidence for the coexistence of two genital HPV types within the same host cell in vitro. Virology 2004, 321, 173–180. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin-Drubin, M.E.; Meyers, J.; Munger, K. Cancer associated human papillomaviruses. Curr. Opin. Virol. 2012, 2, 459–466. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin-Drubin, M.E.; Munger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef] [Green Version]
- James, C.D.; Morgan, I.M.; Bristol, M.L. The Relationship between Estrogen-Related Signaling and Human Papillomavirus Positive Cancers. Pathogens 2020, 9, 403. [Google Scholar] [CrossRef]
- Marks, M.A.; Gravitt, P.E.; Burk, R.D.; Studentsov, Y.; Farzadegan, H.; Klein, S.L. Progesterone and 17beta-estradiol enhance regulatory responses to human papillomavirus type 16 virus-like particles in peripheral blood mononuclear cells from healthy women. Clin. Vaccine Immunol. 2010, 17, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Jayshree, R.S. The Immune Microenvironment in Human Papilloma Virus-Induced Cervical Lesions-Evidence for Estrogen as an Immunomodulator. Front. Cell. Infect. Microbiol. 2021, 11, 649815. [Google Scholar] [CrossRef]
- Siegel, R.L.; Fedewa, S.A.; Miller, K.D.; Goding-Sauer, A.; Pinheiro, P.S.; Martinez-Tyson, D.; Jemal, A. Cancer statistics for Hispanics/Latinos, 2015. CA Cancer J. Clin. 2015, 65, 457–480. [Google Scholar] [CrossRef] [Green Version]
- Viens, L.J.; Henley, S.J.; Watson, M.; Markowitz, L.E.; Thomas, C.C.; Thompson, T.D.; Razzaghi, H.; Saraiya, M. Human Papillomavirus-Associated Cancers—United States, 2008–2012. Morb. Mortal. Wkly. Rep. 2016, 65, 661–666. [Google Scholar] [CrossRef]
- Hellberg, D. Sex steroids and cervical cancer. Anticancer Res. 2012, 32, 3045–3054. [Google Scholar]
- Li, B.; Zhang, L.; Zhao, J.; Tan, G.; Zhang, W.; Zhang, N.; Tian, J.; Qu, P. The value of cytokine levels in triage and risk prediction for women with persistent high-risk human papilloma virus infection of the cervix. Infect. Agents Cancer 2019, 14, 16. [Google Scholar] [CrossRef]
- Nguyen, H.H.; Broker, T.R.; Chow, L.T.; Alvarez, R.D.; Vu, H.L.; Andrasi, J.; Brewer, L.R.; Jin, G.; Mestecky, J. Immune responses to human papillomavirus in genital tract of women with cervical cancer. Gynecol. Oncol. 2005, 96, 452–461. [Google Scholar] [CrossRef]
- Samir, R.; Asplund, A.; Tot, T.; Pekar, G.; Hellberg, D. High-risk HPV infection and CIN grade correlates to the expression of c-myc, CD4+, FHIT, E-cadherin, Ki-67, and p16INK4a. J. Low. Genit. Tract Dis. 2011, 15, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Silins, I.; Kallings, I.; Dillner, J. Correlates of the spread of human papillomavirus infection. Cancer Epidemiol. Biomarkers Prev. 2000, 9, 953–959. [Google Scholar] [PubMed]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen-gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Ratten, L.K.; Plummer, E.L.; Bradshaw, C.S.; Fairley, C.K.; Murray, G.L.; Garland, S.M.; Bateson, D.; Tachedjian, G.; Masson, L.; Vodstrcil, L.A. The Effect of Exogenous Sex Steroids on the Vaginal Microbiota: A Systematic Review. Front. Cell. Infect. Microbiol. 2021, 11, 732423. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.W.; Long, H.Z.; Cheng, Y.; Luo, H.Y.; Wen, D.D.; Gao, L.C. From Microbiome to Inflammation: The Key Drivers of Cervical Cancer. Front. Microbiol. 2021, 12, 767931. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Chen, L.R.; Chen, K.H. In Vitro and Vivo Identification, Metabolism and Action of Xenoestrogens: An Overview. Int. J. Mol. Sci. 2021, 22, 4013. [Google Scholar] [CrossRef]
- Läsche, M.; Urban, H.; Gallwas, J.; Gründker, C. HPV and Other Microbiota; Who’s Good and Who’s Bad: Effects of the Microbial Environment on the Development of Cervical Cancer-A Non-Systematic Review. Cells 2021, 10, 714. [Google Scholar] [CrossRef]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568. [Google Scholar] [CrossRef]
- Taneja, V. Sex Hormones Determine Immune Response. Front. Immunol. 2018, 9, 1931. [Google Scholar] [CrossRef]
- Amador-Molina, A.; Hernández-Valencia, J.F.; Lamoyi, E.; Contreras-Paredes, A.; Lizano, M. Role of innate immunity against human papillomavirus (HPV) infections and effect of adjuvants in promoting specific immune response. Viruses 2013, 5, 2624–2642. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.R.; James, C.D.; Bristol, M.L.; Nulton, T.J.; Wang, X.; Kaur, N.; White, E.A.; Windle, B.; Morgan, I.M. Human Papillomavirus 16 E2 Regulates Keratinocyte Gene Expression Relevant to Cancer and the Viral Life Cycle. J. Virol. 2019, 93, e01941-18. [Google Scholar] [CrossRef] [Green Version]
- Woodworth, C.D. HPV innate immunity. Front. Biosci. 2002, 7, d2058–d2071. [Google Scholar] [CrossRef]
- Timmons, B.; Akins, M.; Mahendroo, M. Cervical remodeling during pregnancy and parturition. Trends Endocrinol. Metab. 2010, 21, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, P.; Gottschalk, E.Y.; Meneses, P.I. HPV entry into cells. Mutat. Res. Rev. Mutat. Res. 2017, 772, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Letian, T.; Tianyu, Z. Cellular receptor binding and entry of human papillomavirus. Virol. J. 2010, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Cardenas-Mora, J.; Spindler, J.E.; Jang, M.K.; McBride, A.A. Dimerization of the papillomavirus E2 protein is required for efficient mitotic chromosome association and Brd4 binding. J. Virol. 2008, 82, 7298–7305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clower, R.V.; Hu, Y.; Melendy, T. Papillomavirus E2 protein interacts with and stimulates human topoisomerase I. Virology 2006, 348, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Laimins, L.A. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol. 2013, 8, 1547–1557. [Google Scholar] [CrossRef] [Green Version]
- Kadaja, M.; Sumerina, A.; Verst, T.; Ojarand, M.; Ustav, E.; Ustav, M. Genomic instability of the host cell induced by the human papillomavirus replication machinery. EMBO J. 2007, 26, 2180–2191. [Google Scholar] [CrossRef] [Green Version]
- Lehoux, M.; Fradet-Turcotte, A.; Archambault, J. Methods to assess the nucleocytoplasmic shuttling of the HPV E1 helicase and its effects on cellular proliferation and induction of a DNA damage response. Methods Mol. Biol. 2015, 1249, 67–80. [Google Scholar] [CrossRef]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPhillips, M.G.; Oliveira, J.G.; Spindler, J.E.; Mitra, R.; McBride, A.A. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol. 2006, 80, 9530–9543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melendy, T.; Sedman, J.; Stenlund, A. Cellular factors required for papillomavirus DNA replication. J. Virol. 1995, 69, 7857–7867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [Green Version]
- Narahari, J.; Fisk, J.C.; Melendy, T.; Roman, A. Interactions of the cellular CCAAT displacement protein and human papillomavirus E2 protein with the viral origin of replication can regulate DNA replication. Virology 2006, 350, 302–311. [Google Scholar] [CrossRef] [Green Version]
- Weitzman, M.D.; Lilley, C.E.; Chaurushiya, M.S. Genomes in conflict: Maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 2010, 64, 61–81. [Google Scholar] [CrossRef]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009, 5, e1000397. [Google Scholar] [CrossRef]
- Graham, S.V. Human papillomavirus: Gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol. 2010, 5, 1493–1506. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.V. The human papillomavirus replication cycle, and its links to cancer progression: A comprehensive review. Clin. Sci. 2017, 131, 2201–2221. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.M.; Baker, C.C. Papillomavirus genome structure, expression, and post-transcriptional regulation. Front. Biosci. 2006, 11, 2286–2302. [Google Scholar] [CrossRef] [Green Version]
- Frattini, M.G.; Lim, H.B.; Laimins, L.A. In vitro synthesis of oncogenic human papillomaviruses requires episomal genomes for differentiation-dependent late expression. Proc. Natl. Acad. Sci. USA 1996, 93, 3062–3067. [Google Scholar] [CrossRef] [Green Version]
- Park, R.B.; Androphy, E.J. Genetic analysis of high-risk e6 in episomal maintenance of human papillomavirus genomes in primary human keratinocytes. J. Virol. 2002, 76, 11359–11364. [Google Scholar] [CrossRef] [Green Version]
- Stubenrauch, F.; Lim, H.B.; Laimins, L.A. Differential requirements for conserved E2 binding sites in the life cycle of oncogenic human papillomavirus type 31. J. Virol. 1998, 72, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.T.; Hubert, W.G.; Ruesch, M.N.; Laimins, L.A. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 8449–8454. [Google Scholar] [CrossRef] [Green Version]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef] [Green Version]
- Suprynowicz, F.A.; Krawczyk, E.; Hebert, J.D.; Sudarshan, S.R.; Simic, V.; Kamonjoh, C.M.; Schlegel, R. The human papillomavirus type 16 E5 oncoprotein inhibits epidermal growth factor trafficking independently of endosome acidification. J. Virol. 2010, 84, 10619–10629. [Google Scholar] [CrossRef] [Green Version]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The smallest oncoprotein with many functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef] [Green Version]
- Wetherill, L.F.; Holmes, K.K.; Verow, M.; Müller, M.; Howell, G.; Harris, M.; Fishwick, C.; Stonehouse, N.; Foster, R.; Blair, G.E.; et al. High-risk human papillomavirus E5 oncoprotein displays channel-forming activity sensitive to small-molecule inhibitors. J. Virol. 2012, 86, 5341–5351. [Google Scholar] [CrossRef] [Green Version]
- Wasson, C.W.; Morgan, E.L.; Müller, M.; Ross, R.L.; Hartley, M.; Roberts, S.; Macdonald, A. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget 2017, 8, 103581–103600. [Google Scholar] [CrossRef] [Green Version]
- Morgan, E.L.; Wasson, C.W.; Hanson, L.; Kealy, D.; Pentland, I.; McGuire, V.; Scarpini, C.; Coleman, N.; Arthur, J.S.C.; Parish, J.L.; et al. STAT3 activation by E6 is essential for the differentiation-dependent HPV18 life cycle. PLoS Pathog. 2018, 14, e1006975. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 activation in HPV positive cervical cancer through a virus-driven Rac1-NFκB-IL-6 signalling axis. PLoS Pathog. 2019, 15, e1007835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppe-Seyler, K.; Honegger, A.; Bossler, F.; Sponagel, J.; Bulkescher, J.; Lohrey, C.; Hoppe-Seyler, F. Viral E6/E7 oncogene and cellular hexokinase 2 expression in HPV-positive cancer cell lines. Oncotarget 2017, 8, 106342–106351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, S.; Banks, L. Molecular mechanisms underlying human papillomavirus E6 and E7 oncoprotein-induced cell transformation. Mutat. Res. Rev. Mutat. Res. 2017, 772, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [Green Version]
- Songock, W.K.; Kim, S.M.; Bodily, J.M. The human papillomavirus E7 oncoprotein as a regulator of transcription. Virus Res. 2017, 231, 56–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.A. Manipulation of Epithelial Differentiation by HPV Oncoproteins. Viruses 2019, 11, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarth, J.A.; Patterson, M.R.; Morgan, E.L.; Macdonald, A. The human papillomavirus oncoproteins: A review of the host pathways targeted on the road to transformation. J. Gen. Virol. 2021, 102, 001540. [Google Scholar] [CrossRef]
- Schwartz, S. HPV-16 RNA processing. Front. Biosci. 2008, 13, 5880–5891. [Google Scholar] [CrossRef] [Green Version]
- Middleton, K.; Peh, W.; Southern, S.; Griffin, H.; Sotlar, K.; Nakahara, T.; El-Sherif, A.; Morris, L.; Seth, R.; Hibma, M.; et al. Organization of human papillomavirus productive cycle during neoplastic progression provides a basis for selection of diagnostic markers. J. Virol. 2003, 77, 10186–10201. [Google Scholar] [CrossRef] [Green Version]
- Peh, W.L.; Middleton, K.; Christensen, N.; Nicholls, P.; Egawa, K.; Sotlar, K.; Brandsma, J.; Percival, A.; Lewis, J.; Liu, W.J.; et al. Life cycle heterogeneity in animal models of human papillomavirus-associated disease. J. Virol. 2002, 76, 10401–10416. [Google Scholar] [CrossRef] [Green Version]
- Ruesch, M.N.; Laimins, L.A. Human papillomavirus oncoproteins alter differentiation-dependent cell cycle exit on suspension in semisolid medium. Virology 1998, 250, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Stanley, M. Immunobiology of HPV and HPV vaccines. Gynecol. Oncol. 2008, 109, S15–S21. [Google Scholar] [CrossRef]
- Heck, D.V.; Yee, C.L.; Howley, P.M.; Münger, K. Efficiency of binding the retinoblastoma protein correlates with the transforming capacity of the E7 oncoproteins of the human papillomaviruses. Proc. Natl. Acad. Sci. USA 1992, 89, 4442–4446. [Google Scholar] [CrossRef] [Green Version]
- Hummel, M.; Hudson, J.B.; Laimins, L.A. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J. Virol. 1992, 66, 6070–6080. [Google Scholar] [CrossRef] [Green Version]
- Ozbun, M.A.; Meyers, C. Characterization of late gene transcripts expressed during vegetative replication of human papillomavirus type 31b. J. Virol. 1997, 71, 5161–5172. [Google Scholar] [CrossRef] [Green Version]
- Ozbun, M.A.; Meyers, C. Temporal usage of multiple promoters during the life cycle of human papillomavirus type 31b. J. Virol. 1998, 72, 2715–2722. [Google Scholar] [CrossRef] [Green Version]
- Bedell, M.A.; Hudson, J.B.; Golub, T.R.; Turyk, M.E.; Hosken, M.; Wilbanks, G.D.; Laimins, L.A. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J. Virol. 1991, 65, 2254–2260. [Google Scholar] [CrossRef] [Green Version]
- Fehrmann, F.; Klumpp, D.J.; Laimins, L.A. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J. Virol. 2003, 77, 2819–2831. [Google Scholar] [CrossRef] [Green Version]
- Peh, W.L.; Brandsma, J.L.; Christensen, N.D.; Cladel, N.M.; Wu, X.; Doorbar, J. The viral E4 protein is required for the completion of the cottontail rabbit papillomavirus productive cycle in vivo. J. Virol. 2004, 78, 2142–2151. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.; Fehrmann, F.; Laimins, L.A. Role of the E1–E4 protein in the differentiation-dependent life cycle of human papillomavirus type 31. J. Virol. 2005, 79, 6732–6740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, C.A.; Fradet-Turcotte, A.; Archambault, J.; Laimins, L.A. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification. Proc. Natl. Acad. Sci. USA 2007, 104, 19541–19546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, M.A. Immune responses to human papilloma viruses. Indian J. Med. Res. 2009, 130, 266–276. [Google Scholar] [PubMed]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.; Nakagawa, M.; Moscicki, A.B. Cell-mediated immune response to human papillomavirus infection. Clin. Diagn. Lab. Immunol. 2001, 8, 209–220. [Google Scholar] [CrossRef] [Green Version]
- zur Hausen, H. Papillomavirus infections—A major cause of human cancers. Biochim. Biophys. Acta 1996, 1288, F55–F78. [Google Scholar] [CrossRef]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.E.; Laimins, L.A. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J. Virol. 2000, 74, 4174–4182. [Google Scholar] [CrossRef] [Green Version]
- Nees, M.; Geoghegan, J.M.; Hyman, T.; Frank, S.; Miller, L.; Woodworth, C.D. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J. Virol. 2001, 75, 4283–4296. [Google Scholar] [CrossRef] [Green Version]
- Hebner, C.M.; Wilson, R.; Rader, J.; Bidder, M.; Laimins, L.A. Human papillomaviruses target the double-stranded RNA protein kinase pathway. J. Gen. Virol. 2006, 87, 3183–3193. [Google Scholar] [CrossRef]
- Kubo, M.; Hanada, T.; Yoshimura, A. Suppressors of cytokine signaling and immunity. Nat. Immunol. 2003, 4, 1169–1176. [Google Scholar] [CrossRef]
- Ghittoni, R.; Accardi, R.; Hasan, U.; Gheit, T.; Sylla, B.; Tommasino, M. The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes 2010, 40, 1–13. [Google Scholar] [CrossRef]
- Rincon-Orozco, B.; Halec, G.; Rosenberger, S.; Muschik, D.; Nindl, I.; Bachmann, A.; Ritter, T.M.; Dondog, B.; Ly, R.; Bosch, F.X.; et al. Epigenetic silencing of interferon-kappa in human papillomavirus type 16-positive cells. Cancer Res. 2009, 69, 8718–8725. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Amerio, P.; Sauder, D.N. Role of cytokines in epidermal Langerhans cell migration. J. Leukoc. Biol. 1999, 66, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Alcocer-González, J.M.; Berumen, J.; Taméz-Guerra, R.; Bermúdez-Morales, V.; Peralta-Zaragoza, O.; Hernández-Pando, R.; Moreno, J.; Gariglio, P.; Madrid-Marina, V. In vivo expression of immunosuppressive cytokines in human papillomavirus-transformed cervical cancer cells. Viral Immunol. 2006, 19, 481–491. [Google Scholar] [CrossRef]
- Arany, I.; Tyring, S.K. Status of local cellular immunity in interferon-responsive and -nonresponsive human papillomavirus-associated lesions. Sex Transm. Dis. 1996, 23, 475–480. [Google Scholar] [CrossRef]
- Fichorova, R.N.; Anderson, D.J. Differential expression of immunobiological mediators by immortalized human cervical and vaginal epithelial cells. Biol. Reprod. 1999, 60, 508–514. [Google Scholar] [CrossRef]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef]
- Hubert, P.; van den Brüle, F.; Giannini, S.L.; Franzen-Detrooz, E.; Boniver, J.; Delvenne, P. Colonization of in vitro-formed cervical human papillomavirus- associated (pre)neoplastic lesions with dendritic cells: Role of granulocyte/macrophage colony-stimulating factor. Am. J. Pathol. 1999, 154, 775–784. [Google Scholar] [CrossRef]
- Fahey, L.M.; Raff, A.B.; Da Silva, D.M.; Kast, W.M. A major role for the minor capsid protein of human papillomavirus type 16 in immune escape. J. Immunol. 2009, 183, 6151–6156. [Google Scholar] [CrossRef]
- O’Brien, P.M.; Saveria Campo, M. Evasion of host immunity directed by papillomavirus-encoded proteins. Virus Res. 2002, 88, 103–117. [Google Scholar] [CrossRef]
- Van der Burg, S.H.; Palefsky, J.M. Human Immunodeficiency Virus and Human Papilloma Virus—Why HPV-induced lesions do not spontaneously resolve and why therapeutic vaccination can be successful. J. Transl. Med. 2009, 7, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Smith-McCune, K.; Zhu, Y.H.; Hanahan, D.; Arbeit, J. Cross-species comparison of angiogenesis during the premalignant stages of squamous carcinogenesis in the human cervix and K14-HPV16 transgenic mice. Cancer Res. 1997, 57, 1294–1300. [Google Scholar]
- Smith-McCune, K.K.; Weidner, N. Demonstration and characterization of the angiogenic properties of cervical dysplasia. Cancer Res. 1994, 54, 800–804. [Google Scholar] [PubMed]
- Bárdos, J.I.; Ashcroft, M. Negative and positive regulation of HIF-1: A complex network. Biochim. Biophys. Acta 2005, 1755, 107–120. [Google Scholar] [CrossRef]
- Bodily, J.M.; Mehta, K.P.; Laimins, L.A. Human papillomavirus E7 enhances hypoxia-inducible factor 1-mediated transcription by inhibiting binding of histone deacetylases. Cancer Res. 2011, 71, 1187–1195. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Li, F.; Mead, L.; White, H.; Walker, J.; Ingram, D.A.; Roman, A. Human papillomavirus causes an angiogenic switch in keratinocytes which is sufficient to alter endothelial cell behavior. Virology 2007, 367, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Clere, N.; Bermont, L.; Fauconnet, S.; Lascombe, I.; Saunier, M.; Vettoretti, L.; Plissonnier, M.L.; Mougin, C. The human papillomavirus type 18 E6 oncoprotein induces Vascular Endothelial Growth Factor 121 (VEGF121) transcription from the promoter through a p53-independent mechanism. Exp. Cell Res. 2007, 313, 3239–3250. [Google Scholar] [CrossRef]
- Nakamura, M.; Bodily, J.M.; Beglin, M.; Kyo, S.; Inoue, M.; Laimins, L.A. Hypoxia-specific stabilization of HIF-1alpha by human papillomaviruses. Virology 2009, 387, 442–448. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Zhang, Q.; Nishitani, J.; Brown, J.; Shi, S.; Le, A.D. Overexpression of human papillomavirus type 16 oncoproteins enhances hypoxia-inducible factor 1 alpha protein accumulation and vascular endothelial growth factor expression in human cervical carcinoma cells. Clin. Cancer Res. 2007, 13, 2568–2576. [Google Scholar] [CrossRef] [Green Version]
- Toussaint-Smith, E.; Donner, D.B.; Roman, A. Expression of human papillomavirus type 16 E6 and E7 oncoproteins in primary foreskin keratinocytes is sufficient to alter the expression of angiogenic factors. Oncogene 2004, 23, 2988–2995. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, A.C.; Schiffman, M.; Herrero, R.; Wacholder, S.; Hildesheim, A.; Castle, P.E.; Solomon, D.; Burk, R. Rapid clearance of human papillomavirus and implications for clinical focus on persistent infections. J. Natl. Cancer Inst. 2008, 100, 513–517. [Google Scholar] [CrossRef] [Green Version]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef] [Green Version]
- Vinokurova, S.; Wentzensen, N.; Kraus, I.; Klaes, R.; Driesch, C.; Melsheimer, P.; Kisseljov, F.; Dürst, M.; Schneider, A.; von Knebel Doeberitz, M. Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res. 2008, 68, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Chaiwongkot, A.; Vinokurova, S.; Pientong, C.; Ekalaksananan, T.; Kongyingyoes, B.; Kleebkaow, P.; Chumworathayi, B.; Patarapadungkit, N.; Reuschenbach, M.; von Knebel Doeberitz, M. Differential methylation of E2 binding sites in episomal and integrated HPV 16 genomes in preinvasive and invasive cervical lesions. Int. J. Cancer 2013, 132, 2087–2094. [Google Scholar] [CrossRef]
- Dooley, K.E.; Warburton, A.; McBride, A.A. Tandemly Integrated HPV16 Can Form a Brd4-Dependent Super-Enhancer-Like Element That Drives Transcription of Viral Oncogenes. mBio 2016, 7, e01446-16. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Mahata, S.; Shishodia, G.; Pande, S.; Verma, G.; Hedau, S.; Bhambhani, S.; Kumari, A.; Batra, S.; Basir, S.F.; et al. Physical state & copy number of high risk human papillomavirus type 16 DNA in progression of cervical cancer. Indian J. Med. Res. 2014, 139, 531–543. [Google Scholar]
- Itahana, Y.; Itahana, K. Emerging Roles of p53 Family Members in Glucose Metabolism. Int. J. Mol. Sci. 2018, 19, 776. [Google Scholar] [CrossRef] [Green Version]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Läsche, M.; Emons, G.; Gründker, C. Shedding New Light on Cancer Metabolism: A Metabolic Tightrope Between Life and Death. Front. Oncol. 2020, 10, 409. [Google Scholar] [CrossRef] [PubMed]
- Veldman, T.; Liu, X.; Yuan, H.; Schlegel, R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 8211–8216. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. Über den Stoffwechsel der Carcinomzelle. Naturwissenschaften 1924, 12, 1131–1137. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors In The Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Ferber, M.J.; Thorland, E.C.; Brink, A.A.; Rapp, A.K.; Phillips, L.A.; McGovern, R.; Gostout, B.S.; Cheung, T.H.; Chung, T.K.; Fu, W.Y.; et al. Preferential integration of human papillomavirus type 18 near the c-myc locus in cervical carcinoma. Oncogene 2003, 22, 7233–7242. [Google Scholar] [CrossRef] [Green Version]
- Peter, M.; Rosty, C.; Couturier, J.; Radvanyi, F.; Teshima, H.; Sastre-Garau, X. MYC activation associated with the integration of HPV DNA at the MYC locus in genital tumors. Oncogene 2006, 25, 5985–5993. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.P.; Friedman, C.L.; Bryant, E.M.; McDougall, J.K. Viral integration and fragile sites in human papillomavirus-immortalized human keratinocyte cell lines. Genes Chromosomes Cancer 1992, 5, 150–157. [Google Scholar] [CrossRef]
- Spangle, J.M.; Münger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Semenza, G.L. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget 2011, 2, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Rodolico, V.; Arancio, W.; Amato, M.C.; Aragona, F.; Cappello, F.; Di Fede, O.; Pannone, G.; Campisi, G. Hypoxia inducible factor-1 alpha expression is increased in infected positive HPV16 DNA oral squamous cell carcinoma and positively associated with HPV16 E7 oncoprotein. Infect. Agents Cancer 2011, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Crusius, K.; Auvinen, E.; Alonso, A. Enhancement of EGF- and PMA-mediated MAP kinase activation in cells expressing the human papillomavirus type 16 E5 protein. Oncogene 1997, 15, 1437–1444. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Zhang, Q.; Ishida, Y.; Hajjar, S.; Tang, X.; Shi, H.; Dang, C.V.; Le, A.D. EGF induces epithelial-mesenchymal transition and cancer stem-like cell properties in human oral cancer cells via promoting Warburg effect. Oncotarget 2017, 8, 9557–9571. [Google Scholar] [CrossRef] [Green Version]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef]
- Gonzalez-Menendez, P.; Hevia, D.; Mayo, J.C.; Sainz, R.M. The dark side of glucose transporters in prostate cancer: Are they a new feature to characterize carcinomas? Int. J. Cancer 2018, 142, 2414–2424. [Google Scholar] [CrossRef] [Green Version]
- Medina, R.A.; Owen, G.I. Glucose transporters: Expression, regulation and cancer. Biol. Res. 2002, 35, 9–26. [Google Scholar] [CrossRef]
- Nualart, F.; Los Angeles García, M.; Medina, R.A.; Owen, G.I. Glucose transporters in sex steroid hormone related cancer. Curr. Vasc. Pharmacol. 2009, 7, 534–548. [Google Scholar] [CrossRef]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.M.; Turk, E. The sodium/glucose cotransport family SLC5. Pflugers Arch. 2004, 447, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Leiprecht, N.; Munoz, C.; Alesutan, I.; Siraskar, G.; Sopjani, M.; Föller, M.; Stubenrauch, F.; Iftner, T.; Lang, F. Regulation of Na(+)-coupled glucose carrier SGLT1 by human papillomavirus 18 E6 protein. Biochem. Biophys. Res. Commun. 2011, 404, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Weihua, Z.; Tsan, R.; Huang, W.C.; Wu, Q.; Chiu, C.H.; Fidler, I.J.; Hung, M.C. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 2008, 13, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, R.; Hou, W.J.; Zhao, Y.J.; Liu, S.L.; Qiu, X.S.; Wang, E.H.; Wu, G.P. Overexpression of HPV16 E6/E7 mediated HIF-1α upregulation of GLUT1 expression in lung cancer cells. Tumor Biol. 2016, 37, 4655–4663. [Google Scholar] [CrossRef]
- Guo, Y.; Meng, X.; Ma, J.; Zheng, Y.; Wang, Q.; Wang, Y.; Shang, H. Human papillomavirus 16 E6 contributes HIF-1α induced Warburg effect by attenuating the VHL-HIF-1α interaction. Int. J. Mol. Sci. 2014, 15, 7974–7986. [Google Scholar] [CrossRef] [Green Version]
- Gründker, C.; Läsche, M.; Hellinger, J.W.; Emons, G. Mechanisms of Metastasis and Cell Mobility—The Role of Metabolism. Geburtshilfe Frauenheilkd 2019, 79, 184–188. [Google Scholar] [CrossRef]
- Li, X.; Coffino, P. High-risk human papillomavirus E6 protein has two distinct binding sites within p53, of which only one determines degradation. J. Virol. 1996, 70, 4509–4516. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef]
- Mazurek, S.; Zwerschke, W.; Jansen-Dürr, P.; Eigenbrodt, E. Effects of the human papilloma virus HPV-16 E7 oncoprotein on glycolysis and glutaminolysis: Role of pyruvate kinase type M2 and the glycolytic-enzyme complex. Biochem. J. 2001, 356, 247–256. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Singh, P.K.; Mehla, K.; Hollingsworth, M.A.; Johnson, K.R. Regulation of Aerobic Glycolysis by microRNAs in Cancer. Mol. Cell Pharmacol. 2011, 3, 125–134. [Google Scholar]
- Slabáková, E.; Culig, Z.; Remšík, J.; Souček, K. Alternative mechanisms of miR-34a regulation in cancer. Cell Death Dis. 2017, 8, e3100. [Google Scholar] [CrossRef]
- Zhang, R.; Su, J.; Xue, S.L.; Yang, H.; Ju, L.L.; Ji, Y.; Wu, K.H.; Zhang, Y.W.; Zhang, Y.X.; Hu, J.F.; et al. HPV E6/p53 mediated down-regulation of miR-34a inhibits Warburg effect through targeting LDHA in cervical cancer. Am. J. Cancer Res. 2016, 6, 312–320. [Google Scholar]
- Stambolsky, P.; Weisz, L.; Shats, I.; Klein, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Regulation of AIF expression by p53. Cell Death Differ. 2006, 13, 2140–2149. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Chen, X. The ferredoxin reductase gene is regulated by the p53 family and sensitizes cells to oxidative stress-induced apoptosis. Oncogene 2002, 21, 7195–7204. [Google Scholar] [CrossRef] [Green Version]
- McDonald, A.E.; Pichaud, N.; Darveau, C.A. “Alternative” fuels contributing to mitochondrial electron transport: Importance of non-classical pathways in the diversity of animal metabolism. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2018, 224, 185–194. [Google Scholar] [CrossRef]
- Blachon, S.; Bellanger, S.; Demeret, C.; Thierry, F. Nucleo-cytoplasmic shuttling of high risk human Papillomavirus E2 proteins induces apoptosis. J. Biol. Chem. 2005, 280, 36088–36098. [Google Scholar] [CrossRef] [Green Version]
- Bellanger, S.; Tan, C.L.; Nei, W.; He, P.P.; Thierry, F. The human papillomavirus type 18 E2 protein is a cell cycle-dependent target of the SCFSkp2 ubiquitin ligase. J. Virol. 2010, 84, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Lai, D.; Tan, C.L.; Gunaratne, J.; Quek, L.S.; Nei, W.; Thierry, F.; Bellanger, S. Localization of HPV-18 E2 at mitochondrial membranes induces ROS release and modulates host cell metabolism. PLoS ONE 2013, 8, e75625. [Google Scholar] [CrossRef] [Green Version]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brèthes, D.; di Rago, J.P.; Velours, J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef]
- Nohl, H.; Gille, L.; Staniek, K. Intracellular generation of reactive oxygen species by mitochondria. Biochem. Pharmacol. 2005, 69, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Cruz-Gregorio, A.; Manzo-Merino, J.; Gonzaléz-García, M.C.; Pedraza-Chaverri, J.; Medina-Campos, O.N.; Valverde, M.; Rojas, E.; Rodríguez-Sastre, M.A.; García-Cuellar, C.M.; Lizano, M. Human Papillomavirus Types 16 and 18 Early-expressed Proteins Differentially Modulate the Cellular Redox State and DNA Damage. Int. J. Biol. Sci. 2018, 14, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riera Leal, A.; Ortiz-Lazareno, P.C.; Jave-Suarez, L.F.; Ramirez De Arellano, A.; Aguilar-Lemarroy, A.; Ortiz-Garcia, Y.M.; Barron-Gallardo, C.A.; Solis-Martinez, R.; Luquin De Anda, S.; Munoz-Valle, J.F.; et al. 17betaestradiolinduced mitochondrial dysfunction and Warburg effect in cervical cancer cells allow cell survival under metabolic stress. Int. J. Oncol. 2020, 56, 33–46. [Google Scholar] [CrossRef]
- Shin, S.; Kwon, Y.J.; Ye, D.J.; Baek, H.S.; Kwon, T.U.; Kim, D.; Chun, Y.J. Human steroid sulfatase enhances aerobic glycolysis through induction of HIF1alpha and glycolytic enzymes. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2464–2474. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Homaei, A.; Raju, A.B.; Meher, B.R. Estrogen: The necessary evil for human health, and ways to tame it. Biomed. Pharmacother. 2018, 102, 403–411. [Google Scholar] [CrossRef]
- Arias-Pulido, H.; Peyton, C.L.; Joste, N.E.; Vargas, H.; Wheeler, C.M. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J. Clin. Microbiol. 2006, 44, 1755–1762. [Google Scholar] [CrossRef] [Green Version]
- Cannarella, R.; Condorelli, R.A.; Mongioì, L.M.; Barbagallo, F.; Calogero, A.E.; La Vignera, S. Effects of the selective estrogen receptor modulators for the treatment of male infertility: A systematic review and meta-analysis. Expert Opin. Pharmacother. 2019, 20, 1517–1525. [Google Scholar] [CrossRef]
- Cauley, J.A. Estrogen and bone health in men and women. Steroids 2015, 99, 11–15. [Google Scholar] [CrossRef]
- Dobbs, R.W.; Malhotra, N.R.; Greenwald, D.T.; Wang, A.Y.; Prins, G.S.; Abern, M.R. Estrogens and prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 185–194. [Google Scholar] [CrossRef]
- Harrison, R.F.; Bonnar, J. Clinical uses of estrogens. Pharmacol. Ther. 1980, 11, 451–467. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Itonaga, T.; Ikegawa, K.; Nishigaki, S.; Kawai, M.; Koga, E.; Sakakibara, H.; Ross, J.L. Ultra-low-dose estrogen therapy for female hypogonadism. Clin. Pediatr. Endocrinol. 2020, 29, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Knowlton, A.A.; Lee, A.R. Estrogen and the cardiovascular system. Pharmacol. Ther. 2012, 135, 54–70. [Google Scholar] [CrossRef] [Green Version]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.L.; Herndon, C. New roles for neuronal estrogen receptors. Neurogastroenterol. Motil. 2017, 29, e13121. [Google Scholar] [CrossRef]
- Marquardt, R.M.; Kim, T.H.; Shin, J.H.; Jeong, J.W. Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis? Int. J. Mol. Sci. 2019, 20, 3822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef] [Green Version]
- Nakano, T.; Kadono, Y.; Iwamoto, H.; Yaegashi, H.; Iijima, M.; Kawaguchi, S.; Nohara, T.; Shigehara, K.; Izumi, K.; Mizokami, A. Therapeutic Effect of Ethinylestradiol in Castration-resistant Prostate Cancer. Anticancer Res. 2020, 40, 2291–2296. [Google Scholar] [CrossRef]
- Pett, M.; Coleman, N. Integration of high-risk human papillomavirus: A key event in cervical carcinogenesis? J. Pathol. 2007, 212, 356–367. [Google Scholar] [CrossRef]
- Regidor, P.A. Clinical relevance in present day hormonal contraception. Horm. Mol. Biol. Clin. Investig. 2018, 37. [Google Scholar] [CrossRef]
- Simpson, E.R. Sources of estrogen and their importance. J. Steroid Biochem. Mol. Biol. 2003, 86, 225–230. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef] [PubMed]
- Hariri, L.; Rehman, A. Estradiol. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Caroccia, B.; Seccia, T.M.; Barton, M.; Rossi, G.P. Estrogen Signaling in the Adrenal Cortex: Implications for Blood Pressure Sex Differences. Hypertension 2016, 68, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Nelson, L.R.; Bulun, S.E. Estrogen production and action. J. Am. Acad. Dermatol. 2001, 45, S116–S124. [Google Scholar] [CrossRef]
- Wasada, T.; Akamine, Y.; Kato, K.; Ibayashi, H.; Nomura, Y. Adrenal contribution to circulating estrogens in woman. Endocrinol. Jpn 1978, 25, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Barakat, R.; Oakley, O.; Kim, H.; Jin, J.; Ko, C.J. Extra-gonadal sites of estrogen biosynthesis and function. BMB Rep. 2016, 49, 488–496. [Google Scholar] [CrossRef]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plottel, C.S.; Blaser, M.J. Microbiome and malignancy. Cell Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, C.J.; Tschugguel, W.; Schneeberger, C.; Huber, J.C. Production and actions of estrogens. N. Engl. J. Med. 2002, 346, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.T.; Han, G.Z.; Shim, J.Y.; Wen, Y.; Jiang, X.R. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology 2006, 147, 4132–4150. [Google Scholar] [CrossRef]
- Raftogianis, R.; Creveling, C.; Weinshilboum, R.; Weisz, J. Estrogen metabolism by conjugation. J. Natl. Cancer Inst. Monogr. 2000, 2000, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Dabek, M.; McCrae, S.I.; Stevens, V.J.; Duncan, S.H.; Louis, P. Distribution of beta-glucosidase and beta-glucuronidase activity and of beta-glucuronidase gene gus in human colonic bacteria. FEMS Microbiol. Ecol. 2008, 66, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.L.; Madak-Erdogan, Z. Estrogen and Microbiota Crosstalk: Should We Pay Attention? Trends Endocrinol. Metab. 2016, 27, 752–755. [Google Scholar] [CrossRef]
- Ervin, S.M.; Li, H.; Lim, L.; Roberts, L.R.; Liang, X.; Mani, S.; Redinbo, M.R. Gut microbial β-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens. J. Biol. Chem. 2019, 294, 18586–18599. [Google Scholar] [CrossRef]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front Cell Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Talib, W.H. Melatonin and Cancer Hallmarks. Molecules 2018, 23, 518. [Google Scholar] [CrossRef] [Green Version]
- Blask, D.E.; Hill, S.M. Effects of melatonin on cancer: Studies on MCF-7 human breast cancer cells in culture. J. Neural Transm. Suppl. 1986, 21, 433–449. [Google Scholar]
- Hill, S.M.; Blask, D.E. Effects of the pineal hormone melatonin on the proliferation and morphological characteristics of human breast cancer cells (MCF-7) in culture. Cancer Res. 1988, 48, 6121–6126. [Google Scholar]
- Gonzalez, A.; Cos, S.; Martinez-Campa, C.; Alonso-Gonzalez, C.; Sanchez-Mateos, S.; Mediavilla, M.D.; Sanchez-Barcelo, E.J. Selective estrogen enzyme modulator actions of melatonin in human breast cancer cells. J. Pineal Res. 2008, 45, 86–92. [Google Scholar] [CrossRef]
- Martínez-Campa, C.; González, A.; Mediavilla, M.D.; Alonso-González, C.; Alvarez-García, V.; Sánchez-Barceló, E.J.; Cos, S. Melatonin inhibits aromatase promoter expression by regulating cyclooxygenases expression and activity in breast cancer cells. Br. J. Cancer 2009, 101, 1613–1619. [Google Scholar] [CrossRef] [Green Version]
- Somasundaram, A.; Rothenberger, N.J.; Stabile, L.P. The Impact of Estrogen in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1277, 33–52. [Google Scholar] [CrossRef]
- Arbeit, J.M.; Howley, P.M.; Hanahan, D. Chronic estrogen-induced cervical and vaginal squamous carcinogenesis in human papillomavirus type 16 transgenic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 2930–2935. [Google Scholar] [CrossRef] [Green Version]
- Brake, T.; Lambert, P.F. Estrogen contributes to the onset, persistence, and malignant progression of cervical cancer in a human papillomavirus-transgenic mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 2490–2495. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.H.; Lambert, P.F. Prevention and treatment of cervical cancer in mice using estrogen receptor antagonists. Proc. Natl. Acad. Sci. USA 2009, 106, 19467–19472. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, N.; Castellsagué, X.; Berrington de González, A.; Gissmann, L. Chapter 1: HPV in the etiology of human cancer. Vaccine 2006, 24 (Suppl. 3), S1–S10. [Google Scholar] [CrossRef]
- Liarte, S.; Alonso-Romero, J.L.; Nicolás, F.J. SIRT1 and Estrogen Signaling Cooperation for Breast Cancer Onset and Progression. Front. Endocrinol. 2018, 9, 552. [Google Scholar] [CrossRef]
- James, C.D.; Das, D.; Morgan, E.L.; Otoa, R.; Macdonald, A.; Morgan, I.M. Werner Syndrome Protein (WRN) Regulates Cell Proliferation and the Human Papillomavirus 16 Life Cycle during Epithelial Differentiation. mSphere 2020, 5, e00858-20. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.E.; Schmiedel, S.; Norrild, B.; Frederiksen, K.; Iftner, T.; Kjaer, S.K. Parity as a cofactor for high-grade cervical disease among women with persistent human papillomavirus infection: A 13-year follow-up. Br. J. Cancer 2013, 108, 234–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, V.; Bosch, F.X.; Muñoz, N.; Meijer, C.J.; Shah, K.V.; Walboomers, J.M.; Herrero, R.; Franceschi, S. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: The IARC multicentric case-control study. Lancet 2002, 359, 1085–1092. [Google Scholar] [CrossRef]
- Muñoz, N.; Franceschi, S.; Bosetti, C.; Moreno, V.; Herrero, R.; Smith, J.S.; Shah, K.V.; Meijer, C.J.; Bosch, F.X. Role of parity and human papillomavirus in cervical cancer: The IARC multicentric case-control study. Lancet 2002, 359, 1093–1101. [Google Scholar] [CrossRef]
- Rinaldi, S.; Plummer, M.; Biessy, C.; Castellsagué, X.; Overvad, K.; Krüger Kjær, S.; Tjønneland, A.; Clavel-Chapelon, F.; Chabbert-Buffet, N.; Mesrine, S.; et al. Endogenous sex steroids and risk of cervical carcinoma: Results from the EPIC study. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 2532–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roura, E.; Travier, N.; Waterboer, T.; de Sanjosé, S.; Bosch, F.X.; Pawlita, M.; Pala, V.; Weiderpass, E.; Margall, N.; Dillner, J.; et al. The Influence of Hormonal Factors on the Risk of Developing Cervical Cancer and Pre-Cancer: Results from the EPIC Cohort. PLoS ONE 2016, 11, e0147029. [Google Scholar] [CrossRef]
- Marks, M.; Klein, S.; Gravitt, P.; Feinstone, H. Hormonal contraception and HPV: A tale of differing and overlapping mechanisms. Open Access J. Contracept. 2011, 2, 161–174. [Google Scholar] [CrossRef] [Green Version]
- Marks, M.A.; Viscidi, R.P.; Chang, K.; Silver, M.; Burke, A.; Howard, R.; Gravitt, P.E. Differences in the concentration and correlation of cervical immune markers among HPV positive and negative perimenopausal women. Cytokine 2011, 56, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Eibye, S.; Krüger Kjær, S.; Nielsen, T.S.; Mellemkjær, L. Mortality Among Women With Cervical Cancer During or Shortly After a Pregnancy in Denmark 1968 to 2006. Int. J. Gynecol. Cancer 2016, 26, 951–958. [Google Scholar] [CrossRef]
- Lønning, P.E.; Haynes, B.P.; Straume, A.H.; Dunbier, A.; Helle, H.; Knappskog, S.; Dowsett, M. Exploring breast cancer estrogen disposition: The basis for endocrine manipulation. Clin. Cancer Res. 2011, 17, 4948–4958. [Google Scholar] [CrossRef] [Green Version]
- Adurthi, S.; Kumar, M.M.; Vinodkumar, H.S.; Mukherjee, G.; Krishnamurthy, H.; Acharya, K.K.; Bafna, U.D.; Uma, D.K.; Abhishekh, B.; Krishna, S.; et al. Oestrogen Receptor-α binds the FOXP3 promoter and modulates regulatory T-cell function in human cervical cancer. Sci. Rep. 2017, 7, 17289. [Google Scholar] [CrossRef]
- Kumar, M.M.; Davuluri, S.; Poojar, S.; Mukherjee, G.; Bajpai, A.K.; Bafna, U.D.; Devi, U.K.; Kallur, P.P.; Kshitish, A.K.; Jayshree, R.S. Role of estrogen receptor alpha in human cervical cancer-associated fibroblasts: A transcriptomic study. Tumor Biol. 2016, 37, 4409–4420. [Google Scholar] [CrossRef]
- Nair, H.B.; Luthra, R.; Kirma, N.; Liu, Y.G.; Flowers, L.; Evans, D.; Tekmal, R.R. Induction of aromatase expression in cervical carcinomas: Effects of endogenous estrogen on cervical cancer cell proliferation. Cancer Res. 2005, 65, 11164–11173. [Google Scholar] [CrossRef] [Green Version]
- Fournier, M.A.; Poirier, D. Estrogen formation in endometrial and cervix cancer cell lines: Involvement of aromatase, steroid sulfatase and 17beta-hydroxysteroid dehydrogenases (types 1, 5, 7 and 12). Mol. Cell. Endocrinol. 2009, 301, 142–145. [Google Scholar] [CrossRef]
- Chen, Y.H.; Huang, L.H.; Chen, T.M. Differential effects of progestins and estrogens on long control regions of human papillomavirus types 16 and 18. Biochem. Biophys. Res. Commun. 1996, 224, 651–659. [Google Scholar] [CrossRef]
- Kim, C.J.; Um, S.J.; Kim, T.Y.; Kim, E.J.; Park, T.C.; Kim, S.J.; Namkoong, S.E.; Park, J.S. Regulation of cell growth and HPV genes by exogenous estrogen in cervical cancer cells. Int. J. Gynecol. Cancer 2000, 10, 157–164. [Google Scholar] [CrossRef]
- Mitrani-Rosenbaum, S.; Tsvieli, R.; Tur-Kaspa, R. Oestrogen stimulates differential transcription of human papillomavirus type 16 in SiHa cervical carcinoma cells. J. Gen. Virol. 1989, 70 Pt 8, 2227–2232. [Google Scholar] [CrossRef]
- Ruutu, M.; Wahlroos, N.; Syrjänen, K.; Johansson, B.; Syrjänen, S. Effects of 17beta-estradiol and progesterone on transcription of human papillomavirus 16 E6/E7 oncogenes in CaSki and SiHa cell lines. Int. J. Gynecol. Cancer 2006, 16, 1261–1268. [Google Scholar] [CrossRef]
- Bristol, M.L.; James, C.D.; Wang, X.; Fontan, C.T.; Morgan, I.M. Estrogen Attenuates the Growth of Human Papillomavirus-Positive Epithelial Cells. mSphere 2020, 5, e00049-20. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Chen, J.; Ai, Y.; Gu, X.; Li, L.; Che, D.; Jiang, Z.; Chen, S.; Huang, H.; Wang, J.; et al. Estrogen-Related Hormones Induce Apoptosis by Stabilizing Schlafen-12 Protein Turnover. Mol. Cell 2019, 75, 1103–1116.e9. [Google Scholar] [CrossRef]
- Lamb, H.M.; Hardwick, J.M. The Dark Side of Estrogen Stops Translation to Induce Apoptosis. Mol. Cell 2019, 75, 1087–1089. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Rhyu, J.W.; Kim, C.J.; Kim, H.S.; Lee, S.Y.; Kwon, Y.I.; Namkoong, S.E.; Sin, H.S.; Um, S.J. Neoplastic change of squamo-columnar junction in uterine cervix and vaginal epithelium by exogenous estrogen in hpv-18 URR E6/E7 transgenic mice. Gynecol. Oncol. 2003, 89, 360–368. [Google Scholar] [CrossRef]
- Chung, S.H.; Franceschi, S.; Lambert, P.F. Estrogen and ERalpha: Culprits in cervical cancer? Trends Endocrinol. Metab. 2010, 21, 504–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.H.; Wiedmeyer, K.; Shai, A.; Korach, K.S.; Lambert, P.F. Requirement for estrogen receptor alpha in a mouse model for human papillomavirus-associated cervical cancer. Cancer Res. 2008, 68, 9928–9934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, R.R.; Duensing, S.; Brake, T.; Münger, K.; Lambert, P.F.; Arbeit, J.M. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003, 63, 4862–4871. [Google Scholar]
- Spurgeon, M.E.; den Boon, J.A.; Horswill, M.; Barthakur, S.; Forouzan, O.; Rader, J.S.; Beebe, D.J.; Roopra, A.; Ahlquist, P.; Lambert, P.F. Human papillomavirus oncogenes reprogram the cervical cancer microenvironment independently of and synergistically with estrogen. Proc. Natl. Acad. Sci. USA 2017, 114, E9076–E9085. [Google Scholar] [CrossRef] [Green Version]
- Coelho, F.R.; Prado, J.C.; Pereira Sobrinho, J.S.; Hamada, G.; Landman, G.; Pinto, C.A.; Nonogaki, S.; Villa, L.L. Estrogen and progesterone receptors in human papilloma virus-related cervical neoplasia. Braz. J. Med. Biol. Res. 2004, 37, 83–88. [Google Scholar] [CrossRef] [Green Version]
- den Boon, J.A.; Pyeon, D.; Wang, S.S.; Horswill, M.; Schiffman, M.; Sherman, M.; Zuna, R.E.; Wang, Z.; Hewitt, S.M.; Pearson, R.; et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E3255–E3264. [Google Scholar] [CrossRef] [Green Version]
- Konishi, I.; Fujii, S.; Nonogaki, H.; Nanbu, Y.; Iwai, T.; Mori, T. Immunohistochemical analysis of estrogen receptors, progesterone receptors, Ki-67 antigen, and human papillomavirus DNA in normal and neoplastic epithelium of the uterine cervix. Cancer 1991, 68, 1340–1350. [Google Scholar] [CrossRef]
- López-Romero, R.; Garrido-Guerrero, E.; Rangel-López, A.; Manuel-Apolinar, L.; Piña-Sánchez, P.; Lazos-Ochoa, M.; Mantilla-Morales, A.; Bandala, C.; Salcedo, M. The cervical malignant cells display a down regulation of ER-α but retain the ER-β expression. Int. J. Clin. Exp. Pathol. 2013, 6, 1594–1602. [Google Scholar]
- Nonogaki, H.; Fujii, S.; Konishi, I.; Nanbu, Y.; Ozaki, S.; Ishikawa, Y.; Mori, T. Estrogen receptor localization in normal and neoplastic epithelium of the uterine cervix. Cancer 1990, 66, 2620–2627. [Google Scholar] [CrossRef]
- Zhai, Y.; Bommer, G.T.; Feng, Y.; Wiese, A.B.; Fearon, E.R.; Cho, K.R. Loss of estrogen receptor 1 enhances cervical cancer invasion. Am. J. Pathol. 2010, 177, 884–895. [Google Scholar] [CrossRef]
- De Nola, R.; Menga, A.; Castegna, A.; Loizzi, V.; Ranieri, G.; Cicinelli, E.; Cormio, G. The Crowded Crosstalk between Cancer Cells and Stromal Microenvironment in Gynecological Malignancies: Biological Pathways and Therapeutic Implication. Int. J. Mol. Sci. 2019, 20, 2401. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Kozasa, K.; Mabuchi, S.; Matsumoto, Y.; Kuroda, H.; Yokoi, E.; Komura, N.; Kawano, M.; Takahashi, R.; Sasano, T.; Shimura, K.; et al. Estrogen stimulates female cancer progression by inducing myeloid-derived suppressive cells: Investigations on pregnant and non-pregnant experimental models. Oncotarget 2019, 10, 1887–1902. [Google Scholar] [CrossRef] [Green Version]
- Kwasniewska, A.; Postawski, K.; Gozdzicka-Jozefiak, A.; Kwasniewski, W.; Grywalska, E.; Zdunek, M.; Korobowicz, E. Estrogen and progesterone receptor expression in HPV-positive and HPV-negative cervical carcinomas. Oncol. Rep. 2011, 26, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Mosny, D.S.; Herholz, J.; Degen, W.; Bender, H.G. Immunohistochemical investigations of steroid receptors in normal and neoplastic squamous epithelium of the uterine cervix. Gynecol. Oncol. 1989, 35, 373–377. [Google Scholar] [CrossRef]
- Chung, S.H.; Shin, M.K.; Korach, K.S.; Lambert, P.F. Requirement for stromal estrogen receptor alpha in cervical neoplasia. Horm. Cancer 2013, 4, 50–59. [Google Scholar] [CrossRef]
- Spurgeon, M.E.; Lambert, P.F. Human Papillomavirus and the Stroma: Bidirectional Crosstalk during the Virus Life Cycle and Carcinogenesis. Viruses 2017, 9, 219. [Google Scholar] [CrossRef] [Green Version]
- Son, J.; Park, Y.; Chung, S.H. Epithelial oestrogen receptor α is dispensable for the development of oestrogen-induced cervical neoplastic diseases. J. Pathol. 2018, 245, 147–152. [Google Scholar] [CrossRef]
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2017, 7, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polanczyk, M.J.; Carson, B.D.; Subramanian, S.; Afentoulis, M.; Vandenbark, A.A.; Ziegler, S.F.; Offner, H. Cutting edge: Estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J. Immunol. 2004, 173, 2227–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.F. Factors regulating apoptosis and homeostasis of CD4+ CD25(high) FOXP3+ regulatory T cells are new therapeutic targets. Front. Biosci. 2008, 13, 1472–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Cai, S.F.; Fehniger, T.A.; Song, J.; Collins, L.I.; Piwnica-Worms, D.R.; Ley, T.J. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 2007, 27, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J. Neurosci. Res. 2006, 84, 370–378. [Google Scholar] [CrossRef]
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1). Int. Immunol. 2007, 19, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Prieto, G.A.; Rosenstein, Y. Oestradiol potentiates the suppressive function of human CD4 CD25 regulatory T cells by promoting their proliferation. Immunology 2006, 118, 58–65. [Google Scholar] [CrossRef]
- Valor, L.; Teijeiro, R.; Aristimuño, C.; Faure, F.; Alonso, B.; de Andrés, C.; Tejera, M.; López-Lazareno, N.; Fernández-Cruz, E.; Sánchez-Ramón, S. Estradiol-dependent perforin expression by human regulatory T-cells. Eur. J. Clin. Investig. 2011, 41, 357–364. [Google Scholar] [CrossRef]
- Yates, M.A.; Li, Y.; Chlebeck, P.J.; Offner, H. GPR30, but not estrogen receptor-alpha, is crucial in the treatment of experimental autoimmune encephalomyelitis by oral ethinyl estradiol. BMC Immunol. 2010, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Traboulsi, T.; El Ezzy, M.; Gleason, J.L.; Mader, S. Antiestrogens: Structure-activity relationships and use in breast cancer treatment. J. Mol. Endocrinol. 2017, 58, R15–R31. [Google Scholar] [CrossRef]
- Khan, M.S.; Singh, P.; Azhar, A.; Naseem, A.; Rashid, Q.; Kabir, M.A.; Jairajpuri, M.A. Serpin Inhibition Mechanism: A Delicate Balance between Native Metastable State and Polymerization. J. Amino Acids 2011, 2011, 606797. [Google Scholar] [CrossRef] [Green Version]
- Medema, J.P.; de Jong, J.; Peltenburg, L.T.; Verdegaal, E.M.; Gorter, A.; Bres, S.A.; Franken, K.L.; Hahne, M.; Albar, J.P.; Melief, C.J.; et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 11515–11520. [Google Scholar] [CrossRef] [Green Version]
- Munguía-Moreno, J.A.; Díaz-Chavéz, J.; García-Villa, E.; Albino-Sanchez, M.E.; Mendoza-Villanueva, D.; Ocadiz-Delgado, R.; Bonilla-Delgado, J.; Marín-Flores, A.; Cortés-Malagón, E.M.; Alvarez-Rios, E.; et al. Early synergistic interactions between the HPV16-E7 oncoprotein and 17β-oestradiol for repressing the expression of Granzyme B in a cervical cancer model. Int. J. Oncol. 2018, 53, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Pigeon, H.; Jean, C.; Charruyer, A.; Haure, M.J.; Baudouin, C.; Charveron, M.; Quillet-Mary, A.; Laurent, G. UVA induces granzyme B in human keratinocytes through MIF: Implication in extracellular matrix remodeling. J. Biol. Chem. 2007, 282, 8157–8164. [Google Scholar] [CrossRef] [Green Version]
- Parkinson, L.G.; Toro, A.; Zhao, H.; Brown, K.; Tebbutt, S.J.; Granville, D.J. Granzyme B mediates both direct and indirect cleavage of extracellular matrix in skin after chronic low-dose ultraviolet light irradiation. Aging Cell 2015, 14, 67–77. [Google Scholar] [CrossRef]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Orr, B.A.; Kranz, D.M.; Shapiro, D.J. Estrogen induction of the granzyme B inhibitor, proteinase inhibitor 9, protects cells against apoptosis mediated by cytotoxic T lymphocytes and natural killer cells. Endocrinology 2006, 147, 1419–1426. [Google Scholar] [CrossRef] [Green Version]
- Svensson, S.; Abrahamsson, A.; Rodriguez, G.V.; Olsson, A.K.; Jensen, L.; Cao, Y.; Dabrosin, C. CCL2 and CCL5 Are Novel Therapeutic Targets for Estrogen-Dependent Breast Cancer. Clin. Cancer Res. 2015, 21, 3794–3805. [Google Scholar] [CrossRef] [Green Version]
- deGraffenried, L.A.; Hilsenbeck, S.G.; Fuqua, S.A. Sp1 is essential for estrogen receptor alpha gene transcription. J. Steroid Biochem. Mol. Biol. 2002, 82, 7–18. [Google Scholar] [CrossRef]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen signaling multiple pathways to impact gene transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Webb, P.; Nguyen, P.; Valentine, C.; Lopez, G.N.; Kwok, G.R.; McInerney, E.; Katzenellenbogen, B.S.; Enmark, E.; Gustafsson, J.A.; Nilsson, S.; et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol. Endocrinol. 1999, 13, 1672–1685. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.J.; Hong, M.K.; Chen, P.C.; Wang, J.H.; Chu, T.Y. Antiestrogen use reduces risk of cervical neoplasia in breast cancer patients: A population-based study. Oncotarget 2017, 8, 29361–29369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Generali, D.; Bates, G.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Immunomodulation of FOXP3+ regulatory T cells by the aromatase inhibitor letrozole in breast cancer patients. Clin. Cancer Res. 2009, 15, 1046–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, A.; Ficek, M. Immunomodulatory effects of anti-estrogenic drugs. Acta Pharm. 2012, 62, 141–155. [Google Scholar] [CrossRef]
- Polese, B.; Gridelet, V.; Araklioti, E.; Martens, H.; Perrier d’Hauterive, S.; Geenen, V. The Endocrine Milieu and CD4 T-Lymphocyte Polarization during Pregnancy. Front. Endocrinol. 2014, 5, 106. [Google Scholar] [CrossRef] [Green Version]
- Purohit, A.; Foster, P.A. Steroid sulfatase inhibitors for estrogen- and androgen-dependent cancers. J. Endocrinol. 2012, 212, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Secky, L.; Svoboda, M.; Klameth, L.; Bajna, E.; Hamilton, G.; Zeillinger, R.; Jäger, W.; Thalhammer, T. The sulfatase pathway for estrogen formation: Targets for the treatment and diagnosis of hormone-associated tumors. J. Drug Deliv. 2013, 2013, 957605. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens--Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Gheit, T. Mucosal and Cutaneous Human Papillomavirus Infections and Cancer Biology. Front. Oncol. 2019, 9, 355. [Google Scholar] [CrossRef] [Green Version]
- Fakhry, C.; Westra, W.H.; Wang, S.J.; van Zante, A.; Zhang, Y.; Rettig, E.; Yin, L.X.; Ryan, W.R.; Ha, P.K.; Wentz, A.; et al. The prognostic role of sex, race, and human papillomavirus in oropharyngeal and nonoropharyngeal head and neck squamous cell cancer. Cancer 2017, 123, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Goyal, A.; Sahu, R.K.; Kumar, M.; Sharma, S.; Qayyum, S.; Kaur, N.; Singh, U.R.; Mehrotra, R.; Hedau, S. p16 promoter methylation, expression, and its association with estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 subtype of breast carcinoma. J. Cancer Res. Ther. 2019, 15, 1147–1154. [Google Scholar] [CrossRef]
- Guidozzi, F. Estrogen therapy in gynecological cancer survivors. Climacteric 2013, 16, 611–617. [Google Scholar] [CrossRef]
- Hong, K.; Choi, Y. Role of estrogen and RAS signaling in repeated implantation failure. BMB Rep. 2018, 51, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, P.F.; Sorosky, J.I.; Wheelock, J.B.; Stevens, C.W., Jr. The significance of atypical cervical cytology in an older population. Obstet. Gynecol. 1989, 73, 13–15. [Google Scholar]
- Kano, M.; Kondo, S.; Wakisaka, N.; Wakae, K.; Aga, M.; Moriyama-Kita, M.; Ishikawa, K.; Ueno, T.; Nakanishi, Y.; Hatano, M.; et al. Expression of estrogen receptor alpha is associated with pathogenesis and prognosis of human papillomavirus-positive oropharyngeal cancer. Int. J. Cancer 2019, 145, 1547–1557. [Google Scholar] [CrossRef]
- Koenigs, M.B.; Lefranc-Torres, A.; Bonilla-Velez, J.; Patel, K.B.; Hayes, D.N.; Glomski, K.; Busse, P.M.; Chan, A.W.; Clark, J.R.; Deschler, D.G.; et al. Association of Estrogen Receptor Alpha Expression With Survival in Oropharyngeal Cancer Following Chemoradiation Therapy. J. Natl. Cancer Inst. 2019, 111, 933–942. [Google Scholar] [CrossRef]
- Piccoli, R.; Mandato, V.D.; Lavitola, G.; Acunzo, G.; Bifulco, G.; Tommaselli, G.A.; Attianese, W.; Nappi, C. Atypical squamous cells and low squamous intraepithelial lesions in postmenopausal women: Implications for management. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 140, 269–274. [Google Scholar] [CrossRef]
- Rhodes, H.E.; Chenevert, L.; Munsell, M. Vaginal intraepithelial neoplasia (VaIN 2/3): Comparing clinical outcomes of treatment with intravaginal estrogen. J. Low. Genit. Tract Dis. 2014, 18, 115–121. [Google Scholar] [CrossRef]
- Schön, H.J.; Grgurin, M.; Szekeres, T.; Schurz, B. A new mode of treatment of human papilloma virus associated anogenital lesions using a nonsteroid estrogen analogue. Wien. Klin. Wochenschr. 1996, 108, 45–47. [Google Scholar]
- Khan, D.; Ansar Ahmed, S. The Immune System Is a Natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front. Immunol. 2015, 6, 635. [Google Scholar] [CrossRef] [Green Version]
- Pagano, M.T.; Ortona, E.; Dupuis, M.L. A Role for Estrogen Receptor alpha36 in Cancer Progression. Front. Endocrinol. 2020, 11, 506. [Google Scholar] [CrossRef]
- Ranganathan, P.; Nadig, N.; Nambiar, S. Non-canonical Estrogen Signaling in Endocrine Resistance. Front. Endocrinol. 2019, 10, 708. [Google Scholar] [CrossRef] [PubMed]
- Tuong, Z.K.; Noske, K.; Kuo, P.; Bashaw, A.A.; Teoh, S.M.; Frazer, I.H. Murine HPV16 E7-expressing transgenic skin effectively emulates the cellular and molecular features of human high-grade squamous intraepithelial lesions. Papillomavirus Res. 2018, 5, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Hammes, L.S.; Tekmal, R.R.; Naud, P.; Edelweiss, M.I.; Kirma, N.; Valente, P.T.; Syrjänen, K.J.; Cunha-Filho, J.S. Macrophages, inflammation and risk of cervical intraepithelial neoplasia (CIN) progression--clinicopathological correlation. Gynecol. Oncol. 2007, 105, 157–165. [Google Scholar] [CrossRef]
- Lagenaur, L.A.; Hemmerling, A.; Chiu, C.; Miller, S.; Lee, P.P.; Cohen, C.R.; Parks, T.P. Connecting the Dots: Translating the Vaginal Microbiome Into a Drug. J. Infect. Dis. 2021, 223, S296–S306. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Chervin, C.S.; Gajewski, T.F. Cancer and the Microbiome-Influence of the Commensal Microbiota on Cancer, Immune Responses, and Immunotherapy. Gastroenterology 2021, 160, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Trinchieri, G. Microbiota: A key orchestrator of cancer therapy. Nat. Rev. Cancer 2017, 17, 271–285. [Google Scholar] [CrossRef]
- Draper, L.M.; Kwong, M.L.; Gros, A.; Stevanović, S.; Tran, E.; Kerkar, S.; Raffeld, M.; Rosenberg, S.A.; Hinrichs, C.S. Targeting of HPV-16+ Epithelial Cancer Cells by TCR Gene Engineered T Cells Directed against E6. Clin. Cancer Res. 2015, 21, 4431–4439. [Google Scholar] [CrossRef] [Green Version]
- Murad, J.P.; Tilakawardane, D.; Park, A.K.; Lopez, L.S.; Young, C.A.; Gibson, J.; Yamaguchi, Y.; Lee, H.J.; Kennewick, K.T.; Gittins, B.J.; et al. Pre-conditioning modifies the TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. Mol. Ther. 2021, 29, 2335–2349. [Google Scholar] [CrossRef]
- Nagarsheth, N.B.; Norberg, S.M.; Sinkoe, A.L.; Adhikary, S.; Meyer, T.J.; Lack, J.B.; Warner, A.C.; Schweitzer, C.; Doran, S.L.; Korrapati, S.; et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat Med. 2021, 27, 419–425. [Google Scholar] [CrossRef]
- Stevanović, S.; Draper, L.M.; Langhan, M.M.; Campbell, T.E.; Kwong, M.L.; Wunderlich, J.R.; Dudley, M.E.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J. Clin. Oncol. 2015, 33, 1543–1550. [Google Scholar] [CrossRef] [Green Version]
- Stevanović, S.; Pasetto, A.; Helman, S.R.; Gartner, J.J.; Prickett, T.D.; Howie, B.; Robins, H.S.; Robbins, P.F.; Klebanoff, C.A.; Rosenberg, S.A.; et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 2017, 356, 200–205. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Läsche, M.; Gallwas, J.; Gründker, C. Like Brothers in Arms: How Hormonal Stimuli and Changes in the Metabolism Signaling Cooperate, Leading HPV Infection to Drive the Onset of Cervical Cancer. Int. J. Mol. Sci. 2022, 23, 5050. https://doi.org/10.3390/ijms23095050
Läsche M, Gallwas J, Gründker C. Like Brothers in Arms: How Hormonal Stimuli and Changes in the Metabolism Signaling Cooperate, Leading HPV Infection to Drive the Onset of Cervical Cancer. International Journal of Molecular Sciences. 2022; 23(9):5050. https://doi.org/10.3390/ijms23095050
Chicago/Turabian StyleLäsche, Matthias, Julia Gallwas, and Carsten Gründker. 2022. "Like Brothers in Arms: How Hormonal Stimuli and Changes in the Metabolism Signaling Cooperate, Leading HPV Infection to Drive the Onset of Cervical Cancer" International Journal of Molecular Sciences 23, no. 9: 5050. https://doi.org/10.3390/ijms23095050
APA StyleLäsche, M., Gallwas, J., & Gründker, C. (2022). Like Brothers in Arms: How Hormonal Stimuli and Changes in the Metabolism Signaling Cooperate, Leading HPV Infection to Drive the Onset of Cervical Cancer. International Journal of Molecular Sciences, 23(9), 5050. https://doi.org/10.3390/ijms23095050