
Discovery of Raf Family Is a Milestone in Deciphering the Ras-Mediated Intracellular Signaling Pathway


Abstract
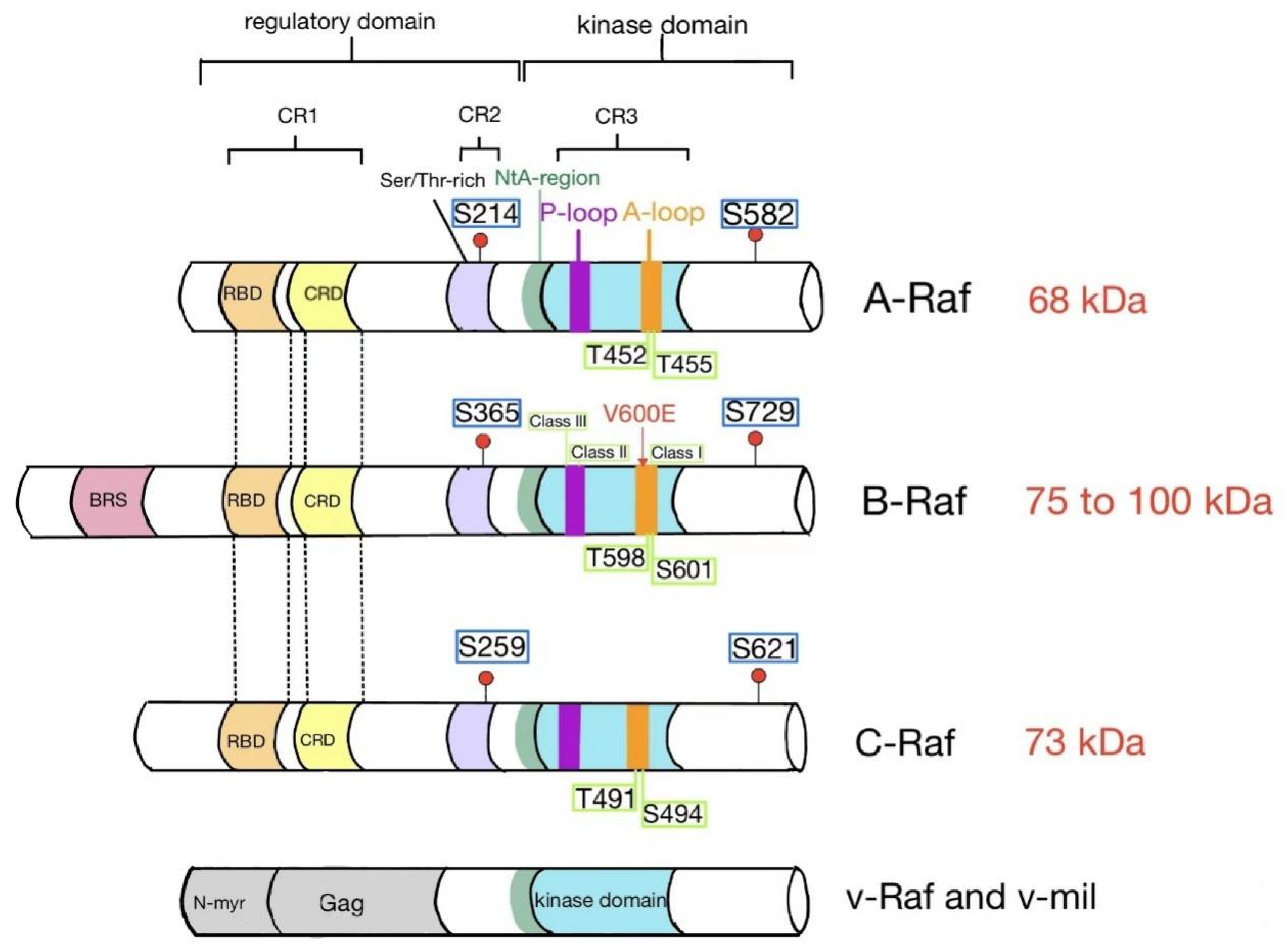
:1. Introduction
2. Discovery of the Raf/MEK/ERK Pathway
3. The Mechanism of Raf Activation
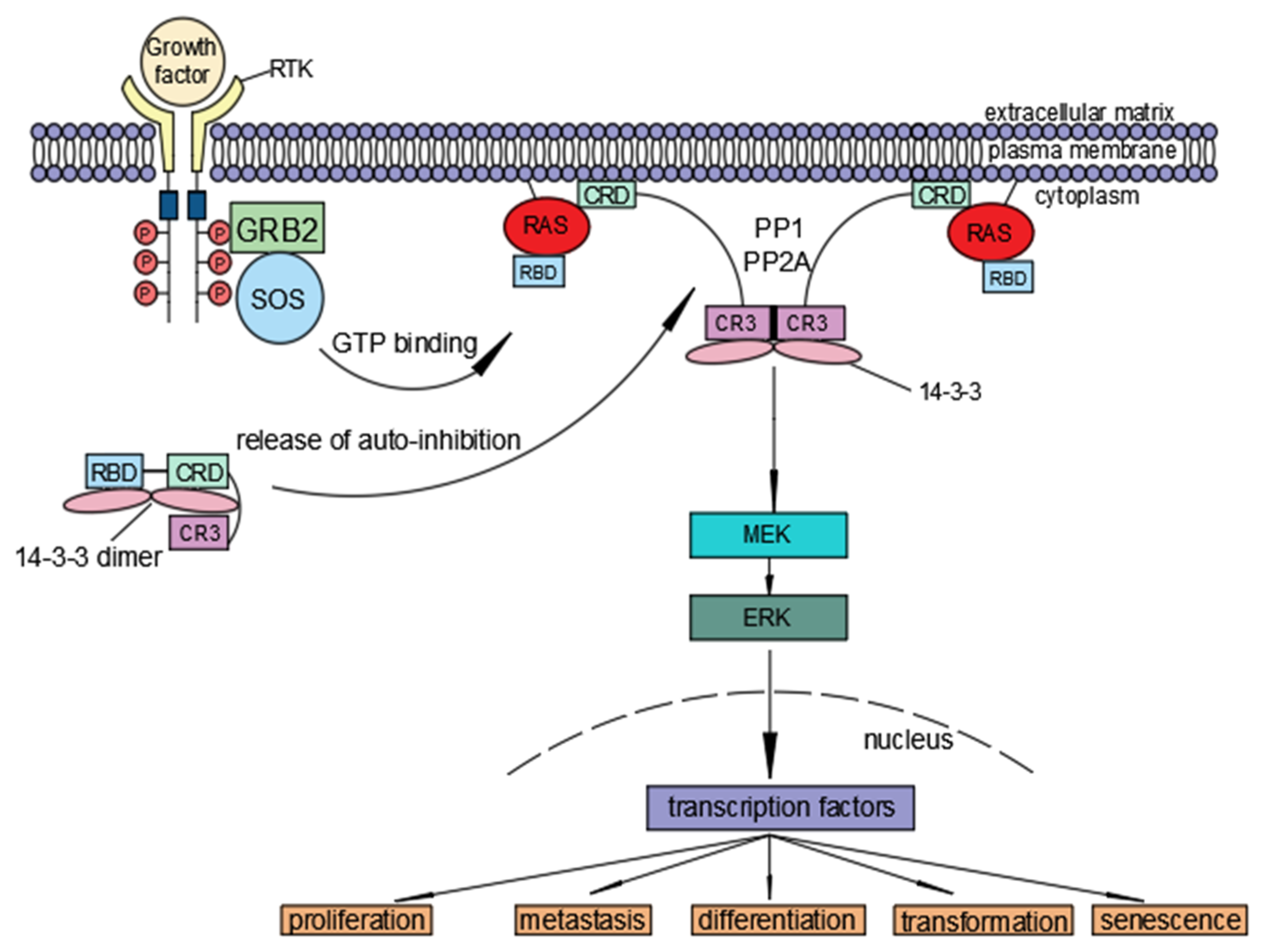
3.1. Ras and 14-3-3
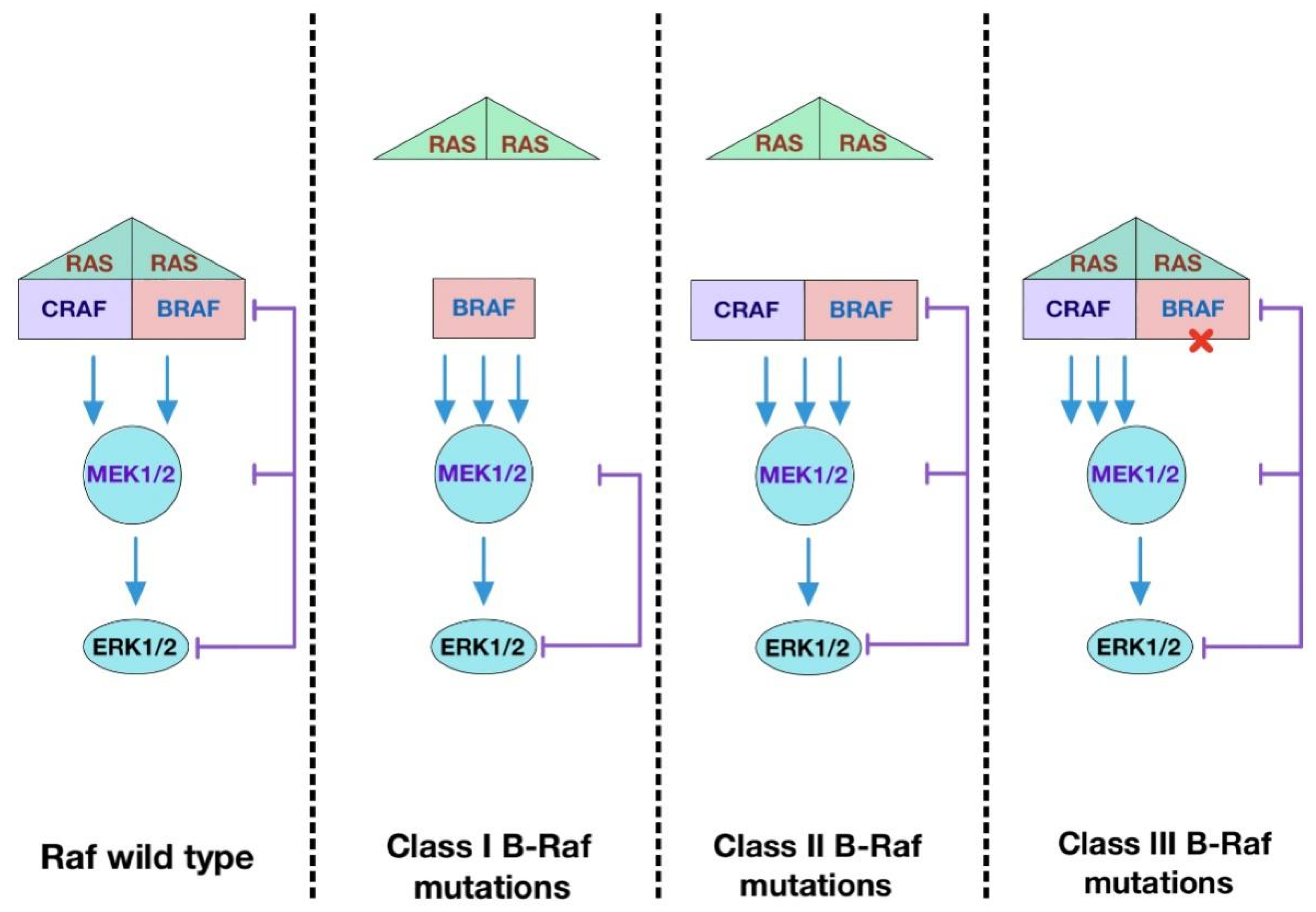
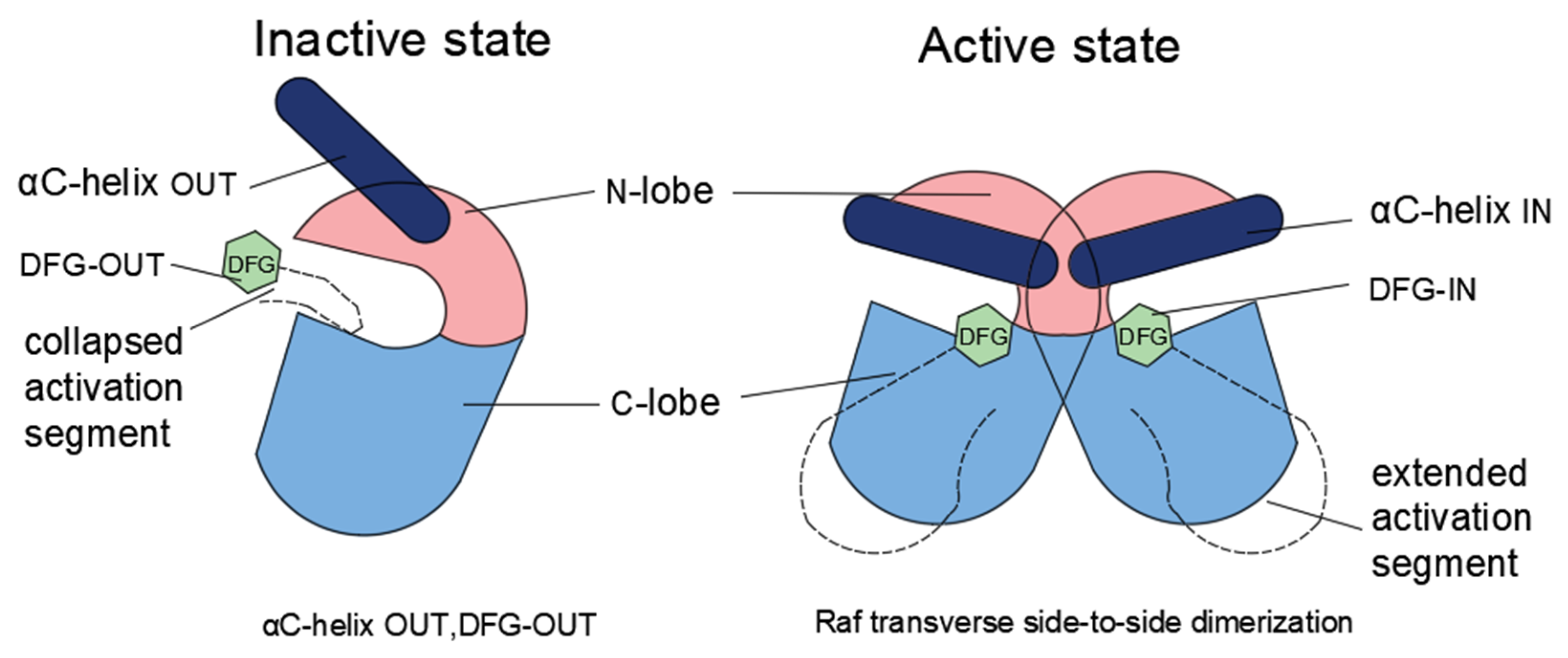
3.2. The Role of Dimerization
3.3. Regulation of Raf Kinase by Phosphorylation
3.3.1. Positive Regulation
3.3.2. Negative Regulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Impact on Raf Kinase Activity | Kinase | References |
|---|---|---|---|
| S29 | Negative | Kinases downstream of MEK1/2 | [103] |
| S43 | Negative | PKA | [98,99,100,105] |
| S259 | Negative, 14-3-3 binding | PKB, PKA | [106,107,108,109,110] |
| S269 | Positive | KSR | [111,112] |
| S289 | Negative, positive | Kinases downstream of MEK1/2 | [103,104] |
| S296 | Negative, positive | Kinases downstream of MEK1/2 | [103,104] |
| S301 | Negative, positive | Kinases downstream of MEK1/2 | [103,104] |
| S338 | Positive | PAK3, Raf, MEK | [78,87,90,91] |
| Y341 | Positive | Src | [78,87,90,91] |
| S471 | Positive | [77,94,95] | |
| S497 | Positive | PKC | [96,97,113] |
| S499 | Positive | PKC | [96,97,113] |
| T491 | Positive | Raf or unclear | [49] |
| S494 | Positive | Raf or unclear | [49] |
| S621 | Negative or positive, 14-3-3 binding | Raf, PKA | [67,84,85] |
| S642 | Negative | Kinases downstream of MEK1/2 | [103] |
3.4. Scaffolds as Raf Regulators
4. Role of Raf in Biology
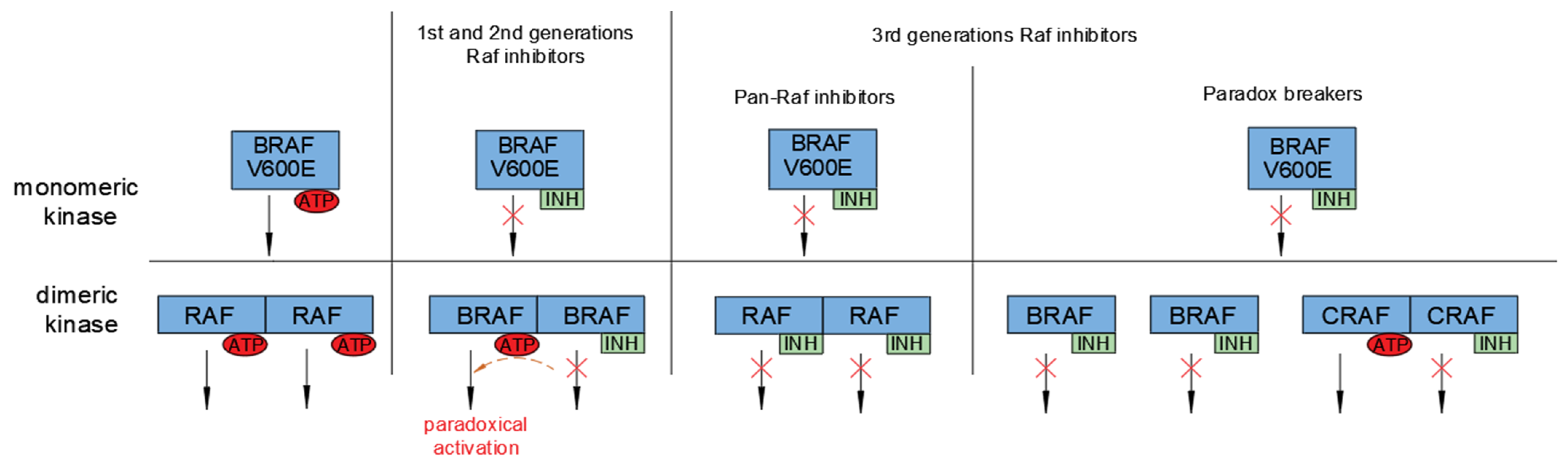
5. Development of Raf Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell. Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef]
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2011, 2, 232–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyriakis, J.M.; App, H.; Zhang, X.F.; Banerjee, P.; Brautigan, D.L.; Rapp, U.R.; Avruch, J. Raf-1 activates MAP kinase-kinase. Nature 1992, 358, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Settleman, J.; Kyriakis, J.M.; Takeuchi-Suzuki, E.; Elledge, S.J.; Marshall, M.S.; Bruder, J.T.; Rapp, U.R.; Avruch, J. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 1993, 364, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Rapp, U.R.; Goldsborough, M.D.; Mark, G.E.; Bonner, T.I.; Groffen, J.; Reynolds, F.H., Jr.; Stephenson, J.R. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc. Natl. Acad. Sci. USA 1983, 80, 4218–4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.; Quintanilla, M.; Ramsden, M.; Kerr, I.B.; Young, S.; Balmain, A. v-ras genes from Harvey and BALB murine sarcoma viruses can act as initiators of two-stage mouse skin carcinogenesis. Cell 1986, 46, 447–456. [Google Scholar] [CrossRef]
- Shimizu, K.; Goldfarb, M.; Suard, Y.; Perucho, M.; Li, Y.; Kamata, T.; Feramisco, J.; Stavnezer, E.; Fogh, J.; Wigler, M.H. Three human transforming genes are related to the viral ras oncogenes. Proc. Natl. Acad. Sci. USA 1983, 80, 2112–2116. [Google Scholar] [CrossRef] [Green Version]
- Ladeda, V.; Frankel, P.; Feig, L.A.; Foster, D.A.; Bal de Kier Joffe, E.; Aguirre-Ghiso, J.A. RalA mediates v-Src, v-Ras, and v-Raf regulation of CD44 and fibronectin expression in NIH3T3 fibroblasts. Biochem. Biophys. Res. Commun. 2001, 283, 854–861. [Google Scholar] [CrossRef]
- Pisapia, P.; Pepe, F.; Iaccarino, A.; Sgariglia, R.; Nacchio, M.; Russo, G.; Gragnano, G.; Malapelle, U.; Troncone, G. BRAF: A Two-Faced Janus. Cells 2020, 9, 2549. [Google Scholar] [CrossRef]
- Leevers, S.J.; Marshall, C.J. Activation of extracellular signal-regulated kinase, ERK2, by p21ras oncoprotein. EMBO J. 1992, 11, 569–574. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Ahn, N.G. The MAP kinase cascade. Discovery of a new signal transduction pathway. Mol. Cell. Biochem. 1993, 127–128, 201–209. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Kwong, L.N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17, 676–691. [Google Scholar] [CrossRef]
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell. Biol. 2003, 4, 373–384. [Google Scholar] [CrossRef]
- Garnett, M.J.; Marais, R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 2004, 6, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of BRAF mutations in thyroid cancer: Genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, H.; Bardelli, A.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.E. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 2002, 418, 934. [Google Scholar] [CrossRef]
- Never-smoker, N.E.S. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [Green Version]
- Rapp, U.R.; Reynolds, F.H., Jr.; Stephenson, J.R. New mammalian transforming retrovirus: Demonstration of a polyprotein gene product. J. Virol. 1983, 45, 914–924. [Google Scholar] [CrossRef] [Green Version]
- Rapp, U.R.; Cleveland, J.L.; Fredrickson, T.N.; Holmes, K.L.; Morse, H.C., 3rd; Jansen, H.W.; Patschinsky, T.; Bister, K. Rapid induction of hemopoietic neoplasms in newborn mice by a raf(mil)/myc recombinant murine retrovirus. J. Virol. 1985, 55, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Jansen, H.W.; Lurz, R.; Bister, K.; Bonner, T.I.; Mark, G.E.; Rapp, U.R. Homologous cell-derived oncogenes in avian carcinoma virus MH2 and murine sarcoma virus 3611. Nature 1984, 307, 281–284. [Google Scholar] [CrossRef]
- Rapp, U.R.; Todaro, C. Generation of new mouse sarcoma viruses in cell culture. Science 1978, 201, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Moelling, K.; Heimann, B.; Beimling, P.; Rapp, U.R.; Sander, T. Serine- and threonine-specific protein kinase activities of purified gag-mil and gag-raf proteins. Nature 1984, 312, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Bonner, T.I.; Kerby, S.B.; Sutrave, P.; Gunnell, M.A.; Mark, G.; Rapp, U.R. Structure and biological activity of human homologs of the raf/mil oncogene. Mol. Cell. Biol. 1985, 5, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Bonner, T.; O’Brien, S.J.; Nash, W.G.; Rapp, U.R.; Morton, C.C.; Leder, P. The human homologs of the raf (mil) oncogene are located on human chromosomes 3 and 4. Science 1984, 223, 71–74. [Google Scholar] [CrossRef]
- Ikawa, S.; Fukui, M.; Ueyama, Y.; Tamaoki, N.; Yamamoto, T.; Toyoshima, K. B-raf, a new member of the raf family, is activated by DNA rearrangement. Mol. Cell. Biol. 1988, 8, 2651–2654. [Google Scholar] [CrossRef]
- Huleihel, M.; Goldsborough, M.; Cleveland, J.; Gunnell, M.; Bonner, T.; Rapp, U.R. Characterization of murine A-raf, a new oncogene related to the v-raf oncogene. Mol. Cell. Biol. 1986, 6, 2655–2662. [Google Scholar] [CrossRef]
- Han, M.; Golden, A.; Han, Y.; Sternberg, P.W.C. C. elegans lin-45 raf gene participates in let-60 ras-stimulated vulval differentiation. Nature 1993, 363, 133–140. [Google Scholar] [CrossRef]
- Ambrosio, L.; Mahowald, A.P.; Perrimon, N. Requirement of the Drosophila raf homologue for torso function. Nature 1989, 342, 288–291. [Google Scholar] [CrossRef]
- Barnier, J.V.; Papin, C.; Eychène, A.; Lecoq, O.; Calothy, G. The mouse B-raf gene encodes multiple protein isoforms with tissue-specific expression. J. Biol. Chem. 1995, 270, 23381–23389. [Google Scholar] [CrossRef] [Green Version]
- Storm, S.M.; Cleveland, J.L.; Rapp, U.R. Expression of raf family proto-oncogenes in normal mouse tissues. Oncogene 1990, 5, 345–351. [Google Scholar] [CrossRef]
- Sturgill, T.W.; Ray, L.B.; Erikson, E.; Maller, J.L. Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature 1988, 334, 715–718. [Google Scholar] [CrossRef]
- Courchesne, W.E.; Kunisawa, R.; Thorner, J. A putative protein kinase overcomes pheromone-induced arrest of cell cycling in S. cerevisiae. Cell 1989, 58, 1107–1119. [Google Scholar] [CrossRef]
- Ray, L.B.; Sturgill, T.W. Insulin-stimulated microtubule-associated protein kinase is phosphorylated on tyrosine and threonine in vivo. Proc. Natl. Acad. Sci. USA 1988, 85, 3753–3757. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, R.A.; Quinn, A.M.; Hunter, T. Dual-specificity protein kinases: Will any hydroxyl do? Trends Biochem. Sci. 1992, 17, 114–119. [Google Scholar] [CrossRef]
- Anderson, N.G.; Maller, J.L.; Tonks, N.K.; Sturgill, T.W. Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature 1990, 343, 651–653. [Google Scholar] [CrossRef]
- Ullah, R.; Yin, Q.; Snell, A.H.; Wan, L. RAF-MEK-ERK pathway in cancer evolution and treatment. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef]
- Dent, P.; Haser, W.; Haystead, T.A.; Vincent, L.A.; Roberts, T.M.; Sturgill, T.W. Activation of mitogen-activated protein kinase kinase by v-Raf in NIH 3T3 cells and in vitro. Science 1992, 257, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Gómez, N.; Cohen, P. Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature 1991, 353, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Ahn, N.G.; Seger, R.; Bratlien, R.L.; Diltz, C.D.; Tonks, N.K.; Krebs, E.G. Multiple components in an epidermal growth factor-stimulated protein kinase cascade. In vitro activation of a myelin basic protein/microtubule-associated protein 2 kinase. J. Biol. Chem. 1991, 266, 4220–4227. [Google Scholar] [CrossRef]
- Crews, C.M.; Alessandrini, A.; Erikson, R.L. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science 1992, 258, 478–480. [Google Scholar] [CrossRef]
- Zheng, C.F.; Guan, K.L. Cloning and characterization of two distinct human extracellular signal-regulated kinase activator kinases, MEK1 and MEK2. J. Biol. Chem. 1993, 268, 11435–11439. [Google Scholar] [CrossRef]
- Vojtek, A.B.; Hollenberg, S.M.; Cooper, J.A. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 1993, 74, 205–214. [Google Scholar] [CrossRef]
- Warne, P.H.; Viciana, P.R.; Downward, J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 1993, 364, 352–355. [Google Scholar] [CrossRef]
- Heidecker, G.; Huleihel, M.; Cleveland, J.L.; Kolch, W.; Beck, T.W.; Lloyd, P.; Pawson, T.; Rapp, U.R. Mutational activation of c-raf-1 and definition of the minimal transforming sequence. Mol. Cell. Biol. 1990, 10, 2503–2512. [Google Scholar] [CrossRef]
- Chong, H.; Lee, J.; Guan, K.L. Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J. 2001, 20, 3716–3727. [Google Scholar] [CrossRef] [Green Version]
- Chuang, E.; Barnard, D.; Hettich, L.; Zhang, X.F.; Avruch, J.; Marshall, M.S. Critical binding and regulatory interactions between Ras and Raf occur through a small, stable N-terminal domain of Raf and specific Ras effector residues. Mol. Cell. Biol. 1994, 14, 5318–5325. [Google Scholar] [CrossRef]
- Beck, T.W.; Huleihel, M.; Gunnell, M.; Bonner, T.I.; Rapp, U.R. The complete coding sequence of the human A-raf-1 oncogene and transforming activity of a human A-raf carrying retrovirus. Nucleic Acids Res. 1987, 15, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Tran, N.H.; Wu, X.; Frost, J.A. B-Raf and Raf-1 are regulated by distinct autoregulatory mechanisms. J. Biol. Chem. 2005, 280, 16244–16253. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Diaz, B.; Marshall, M.S.; Avruch, J. An intact Raf zinc finger is required for optimal binding to processed Ras and for ras-dependent Raf activation in situ. Mol. Cell. Biol. 1997, 17, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, A.S.; Meikle, S.; Yazici, Z.; Eulitz, M.; Kolch, W. Regulation of Raf-1 activation and signalling by dephosphorylation. EMBO J. 2002, 21, 64–71. [Google Scholar] [CrossRef]
- Ishikawa, F.; Takaku, F.; Nagao, M.; Sugimura, T. The complete primary structure of the rat A-raf cDNA coding region: Conservation of the putative regulatory regions present in rat c-raf. Oncogene Res. 1987, 1, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell. Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef]
- Zhao, C.; Du, G.; Skowronek, K.; Frohman, M.A.; Bar-Sagi, D. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat. Cell Biol. 2007, 9, 706–712. [Google Scholar] [CrossRef]
- Tran, T.H.; Chan, A.H.; Young, L.C.; Bindu, L.; Neale, C.; Messing, S.; Dharmaiah, S.; Taylor, T.; Denson, J.P.; Esposito, D.; et al. KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat. Commun. 2021, 12, 1176. [Google Scholar] [CrossRef]
- Roy, S.; Lane, A.; Yan, J.; McPherson, R.; Hancock, J.F. Activity of plasma membrane-recruited Raf-1 is regulated by Ras via the Raf zinc finger. J. Biol. Chem. 1997, 272, 20139–20145. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Strum, J.C.; Sciorra, V.A.; Daniel, L.; Bell, R.M. Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-O-tetradecanoylphorbol-13-acetate-stimulated Madin-Darby canine kidney cells. J. Biol. Chem. 1996, 271, 8472–8480. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.J.; Zhang, X.F.; Rapp, U.; Avruch, J. Identification of the 14.3.3 zeta domains important for self-association and Raf binding. J. Biol. Chem. 1995, 270, 23681–23687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Janosch, P.; Tanji, M.; Rosenfeld, G.C.; Waymire, J.C.; Mischak, H.; Kolch, W.; Sedivy, J.M. Regulation of Raf-1 kinase activity by the 14-3-3 family of proteins. EMBO J. 1995, 14, 685–696. [Google Scholar] [CrossRef]
- Fantl, W.J.; Muslin, A.J.; Kikuchi, A.; Martin, J.A.; MacNicol, A.M.; Gross, R.W.; Williams, L.T. Activation of Raf-1 by 14-3-3 proteins. Nature 1994, 371, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.; Symons, M.; Macdonald, S.G.; McCormick, F.; Ruggieri, R. Binding of 14-3-3 proteins to the protein kinase Raf and effects on its activation. Science 1994, 265, 1713–1716. [Google Scholar] [CrossRef]
- Jia, S.; Flores-Saaib, R.D.; Courey, A.J. The Dorsal Rel homology domain plays an active role in transcriptional regulation. Mol. Cell. Biol. 2002, 22, 5089–5099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzivion, G.; Luo, Z.; Avruch, J. A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature 1998, 394, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Rommel, C.; Radziwill, G.; Lovrić, J.; Noeldeke, J.; Heinicke, T.; Jones, D.; Aitken, A.; Moelling, K. Activated Ras displaces 14-3-3 protein from the amino terminus of c-Raf-1. Oncogene 1996, 12, 609–619. [Google Scholar] [PubMed]
- Raabe, T.; Rapp, U.R. Ras signaling: PP2A puts Ksr and Raf in the right place. Curr. Biol. 2003, 13, R635–R637. [Google Scholar] [CrossRef] [Green Version]
- Jaumot, M.; Hancock, J.F. Protein phosphatases 1 and 2A promote Raf-1 activation by regulating 14-3-3 interactions. Oncogene 2001, 20, 3949–3958. [Google Scholar] [CrossRef]
- Plowman, S.J.; Muncke, C.; Parton, R.G.; Hancock, J.F. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2005, 102, 15500–15505. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Harding, A.; Inder, K.; Plowman, S.; Parton, R.G.; Hancock, J.F. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat. Cell Biol. 2007, 9, 905–914. [Google Scholar] [CrossRef]
- Farrar, M.A.; Alberol-Ila, J.; Perlmutter, R.M. Activation of the Raf-1 kinase cascade by coumermycin-induced dimerization. Nature 1996, 383, 178–181. [Google Scholar] [CrossRef]
- Luo, Z.; Tzivion, G.; Belshaw, P.J.; Vavvas, D.; Marshall, M.; Avruch, J. Oligomerization activates c-Raf-1 through a Ras-dependent mechanism. Nature 1996, 383, 181–185. [Google Scholar] [CrossRef]
- Weber, C.K.; Slupsky, J.R.; Kalmes, H.A.; Rapp, U.R. Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res. 2001, 61, 3595–3598. [Google Scholar]
- Rushworth, L.K.; Hindley, A.D.; O’Neill, E.; Kolch, W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol. Cell. Biol. 2006, 26, 2262–2272. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Stites, E.C.; Yu, H.; Germino, E.A.; Meharena, H.S.; Stork, P.J.S.; Kornev, A.P.; Taylor, S.S.; Shaw, A.S. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 2013, 154, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Zang, M.; Gong, J.; Luo, L.; Zhou, J.; Xiang, X.; Huang, W.; Huang, Q.; Luo, X.; Olbrot, M.; Peng, Y.; et al. Characterization of Ser338 phosphorylation for Raf-1 activation. J. Biol. Chem. 2008, 283, 31429–31437. [Google Scholar] [CrossRef] [Green Version]
- Marais, R.; Light, Y.; Paterson, H.F.; Mason, C.S.; Marshall, C.J. Differential regulation of Raf-1, A-Raf, and B-Raf by oncogenic ras and tyrosine kinases. J. Biol. Chem. 1997, 272, 4378–4383. [Google Scholar] [CrossRef] [Green Version]
- Mineo, C.; Anderson, R.G.; White, M.A. Physical association with ras enhances activation of membrane-bound raf (RafCAAX). J. Biol. Chem. 1997, 272, 10345–10348. [Google Scholar] [CrossRef]
- Weber, C.K.; Slupsky, J.R.; Herrmann, C.; Schuler, M.; Rapp, U.R.; Block, C. Mitogenic signaling of Ras is regulated by differential interaction with Raf isozymes. Oncogene 2000, 19, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, Y.H.; Pumiglia, K.; Jun, T.H.; Dent, P.; Sturgill, T.W.; Jove, R. Functional mapping of the N-terminal regulatory domain in the human Raf-1 protein kinase. J. Biol. Chem. 1995, 270, 14100–14106. [Google Scholar] [CrossRef] [Green Version]
- Marais, R.; Light, Y.; Paterson, H.F.; Marshall, C.J. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995, 14, 3136–3145. [Google Scholar] [CrossRef] [PubMed]
- Noble, C.; Mercer, K.; Hussain, J.; Carragher, L.; Giblett, S.; Hayward, R.; Patterson, C.; Marais, R.; Pritchard, C.A. CRAF autophosphorylation of serine 621 is required to prevent its proteasome-mediated degradation. Mol. Cell 2008, 31, 862–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mischak, H.; Seitz, T.; Janosch, P.; Eulitz, M.; Steen, H.; Schellerer, M.; Philipp, A.; Kolch, W. Negative regulation of Raf-1 by phosphorylation of serine 621. Mol. Cell. Biol. 1996, 16, 5409–5418. [Google Scholar] [CrossRef] [Green Version]
- Diaz, B.; Barnard, D.; Filson, A.; MacDonald, S.; King, A.; Marshall, M. Phosphorylation of Raf-1 serine 338-serine 339 is an essential regulatory event for Ras-dependent activation and biological signaling. Mol. Cell. Biol. 1997, 17, 4509–4516. [Google Scholar] [CrossRef] [Green Version]
- Mason, C.S.; Springer, C.J.; Cooper, R.G.; Superti-Furga, G.; Marshall, C.J.; Marais, R. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 1999, 18, 2137–2148. [Google Scholar] [CrossRef] [Green Version]
- Galabova-Kovacs, G.; Kolbus, A.; Matzen, D.; Meissl, K.; Piazzolla, D.; Rubiolo, C.; Steinitz, K.; Baccarini, M. ERK and beyond: Insights from B-Raf and Raf-1 conditional knockouts. Cell Cycle 2006, 5, 1514–1518. [Google Scholar] [CrossRef]
- Sun, H.; King, A.J.; Diaz, H.B.; Marshall, M.S. Regulation of the protein kinase Raf-1 by oncogenic Ras through phosphatidylinositol 3-kinase, Cdc42/Rac and Pak. Curr. Biol. 2000, 10, 281–284. [Google Scholar] [CrossRef] [Green Version]
- King, A.J.; Sun, H.; Diaz, B.; Barnard, D.; Miao, W.; Bagrodia, S.; Marshall, M.S. The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature 1998, 396, 180–183. [Google Scholar] [CrossRef]
- Chaudhary, A.; King, W.G.; Mattaliano, M.D.; Frost, J.A.; Diaz, B.; Morrison, D.K.; Cobb, M.H.; Marshall, M.S.; Brugge, J.S. Phosphatidylinositol 3-kinase regulates Raf1 through Pak phosphorylation of serine 338. Curr. Biol. 2000, 10, 551–554. [Google Scholar] [CrossRef] [Green Version]
- Zang, M.; Waelde, C.A.; Xiang, X.; Rana, A.; Wen, R.; Luo, Z. Microtubule integrity regulates Pak leading to Ras-independent activation of Raf-1. insights into mechanisms of Raf-1 activation. J. Biol. Chem. 2001, 276, 25157–25165. [Google Scholar] [CrossRef] [Green Version]
- Zang, M.; Hayne, C.; Luo, Z. Interaction between active Pak1 and Raf-1 is necessary for phosphorylation and activation of Raf-1. J. Biol. Chem. 2002, 277, 4395–4405. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Balan, V.; Bronisz, A.; Balan, K.; Sun, H.; Leicht, D.T.; Luo, Z.; Qin, J.; Avruch, J.; Tzivion, G. Identification of Raf-1 S471 as a novel phosphorylation site critical for Raf-1 and B-Raf kinase activities and for MEK binding. Mol. Biol. Cell 2005, 16, 4733–4744. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.H.; Guan, K.L. Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. EMBO J. 2000, 19, 5429–5439. [Google Scholar] [CrossRef] [Green Version]
- Carroll, M.P.; May, W.S. Protein kinase C-mediated serine phosphorylation directly activates Raf-1 in murine hematopoietic cells. J. Biol. Chem. 1994, 269, 1249–1256. [Google Scholar] [CrossRef]
- Kolch, W.; Heidecker, G.; Kochs, G.; Hummel, R.; Vahidi, H.; Mischak, H.; Finkenzeller, G.; Marmé, D.; Rapp, U.R. Protein kinase C alpha activates RAF-1 by direct phosphorylation. Nature 1993, 364, 249–252. [Google Scholar] [CrossRef]
- Cook, S.J.; McCormick, F. Inhibition by cAMP of Ras-dependent activation of Raf. Science 1993, 262, 1069–1072. [Google Scholar] [CrossRef]
- Wu, J.; Dent, P.; Jelinek, T.; Wolfman, A.; Weber, M.J.; Sturgill, T.W. Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3’,5’-monophosphate. Science 1993, 262, 1065–1069. [Google Scholar] [CrossRef]
- Schramm, K.; Niehof, M.; Radziwill, G.; Rommel, C.; Moelling, K. Phosphorylation of c-Raf-1 by protein kinase A interferes with activation. Biochem. Biophys. Res. Commun. 1994, 201, 740–747. [Google Scholar] [CrossRef]
- Häfner, S.; Adler, H.S.; Mischak, H.; Janosch, P.; Heidecker, G.; Wolfman, A.; Pippig, S.; Lohse, M.; Ueffing, M.; Kolch, W. Mechanism of inhibition of Raf-1 by protein kinase A. Mol. Cell. Biol. 1994, 14, 6696–6703. [Google Scholar] [CrossRef]
- Sidovar, M.F.; Kozlowski, P.; Lee, J.W.; Collins, M.A.; He, Y.; Graves, L.M. Phosphorylation of serine 43 is not required for inhibition of c-Raf kinase by the cAMP-dependent protein kinase. J. Biol. Chem. 2000, 275, 28688–28694. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, M.K.; Müller, J.; Ritt, D.A.; Zhou, M.; Zhou, X.Z.; Copeland, T.D.; Conrads, T.P.; Veenstra, T.D.; Lu, K.P.; Morrison, D.K. Regulation of Raf-1 by direct feedback phosphorylation. Mol. Cell 2005, 17, 215–224. [Google Scholar] [CrossRef]
- Balan, V.; Leicht, D.T.; Zhu, J.; Balan, K.; Kaplun, A.; Singh-Gupta, V.; Qin, J.; Ruan, H.; Comb, M.J.; Tzivion, G. Identification of novel in vivo Raf-1 phosphorylation sites mediating positive feedback Raf-1 regulation by extracellular signal-regulated kinase. Mol. Biol. Cell 2006, 17, 1141–1153. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.K.; Heidecker, G.; Rapp, U.R.; Copeland, T.D. Identification of the major phosphorylation sites of the Raf-1 kinase. J. Biol. Chem. 1993, 268, 17309–17316. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Pollock, C.; Steen, H.; Shaw, P.E.; Mischak, H.; Kolch, W. Cyclic AMP-dependent kinase regulates Raf-1 kinase mainly by phosphorylation of serine 259. Mol. Cell. Biol. 2002, 22, 3237–3246. [Google Scholar] [CrossRef] [Green Version]
- Michaud, N.R.; Fabian, J.R.; Mathes, K.D.; Morrison, D.K. 14-3-3 is not essential for Raf-1 function: Identification of Raf-1 proteins that are biologically activated in a 14-3-3- and Ras-independent manner. Mol. Cell. Biol. 1995, 15, 3390–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muslin, A.J.; Tanner, J.W.; Allen, P.M.; Shaw, A.S. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 1996, 84, 889–897. [Google Scholar] [CrossRef] [Green Version]
- Rommel, C.; Clarke, B.A.; Zimmermann, S.; Nuñez, L.; Rossman, R.; Reid, K.; Moelling, K.; Yancopoulos, G.D.; Glass, D.J. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science 1999, 286, 1738–1741. [Google Scholar] [CrossRef]
- Zimmermann, S.; Moelling, K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 1999, 286, 1741–1744. [Google Scholar] [CrossRef]
- Yao, B.; Zhang, Y.; Delikat, S.; Mathias, S.; Basu, S.; Kolesnick, R. Phosphorylation of Raf by ceramide-activated protein kinase. Nature 1995, 378, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yao, B.; Delikat, S.; Bayoumy, S.; Lin, X.H.; Basu, S.; McGinley, M.; Chan-Hui, P.Y.; Lichenstein, H.; Kolesnick, R. Kinase suppressor of Ras is ceramide-activated protein kinase. Cell 1997, 89, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Smola, U.; Wixler, V.; Eisenmann-Tappe, I.; Diaz-Meco, M.T.; Moscat, J.; Rapp, U.; Cooper, G.M. Role of diacylglycerol-regulated protein kinase C isotypes in growth factor activation of the Raf-1 protein kinase. Mol. Cell. Biol. 1997, 17, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Li, Y.; Dillon, T.J.; Kariya, Y.; Stork, P.J.S. Phosphorylation of the C-Raf N Region Promotes Raf Dimerization. Mol. Cell. Biol. 2017, 37, e00132-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leicht, D.T.; Balan, V.; Zhu, J.; Kaplun, A.; Bronisz, A.; Rana, A.; Tzivion, G. MEK-1 activates C-Raf through a Ras-independent mechanism. Biochim. Biophys. Acta 2013, 1833, 976–986. [Google Scholar] [CrossRef] [Green Version]
- Unni, A.M.; Harbourne, B.; Oh, M.H.; Wild, S.; Ferrarone, J.R.; Lockwood, W.W.; Varmus, H. Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. Elife 2018, 7, e33718. [Google Scholar] [CrossRef]
- Leung, G.P.; Feng, T.; Sigoillot, F.D.; Geyer, F.C.; Shirley, M.D.; Ruddy, D.A.; Rakiec, D.P.; Freeman, A.K.; Engelman, J.A.; Jaskelioff, M.; et al. Hyperactivation of MAPK Signaling Is Deleterious to RAS/RAF-mutant Melanoma. Mol. Cancer Res. 2019, 17, 199–211. [Google Scholar] [CrossRef] [Green Version]
- Sale, M.J.; Balmanno, K.; Saxena, J.; Ozono, E.; Wojdyla, K.; McIntyre, R.E.; Gilley, R.; Woroniuk, A.; Howarth, K.D.; Hughes, G.; et al. MEK1/2 inhibitor withdrawal reverses acquired resistance driven by BRAF(V600E) amplification whereas KRAS(G13D) amplification promotes EMT-chemoresistance. Nat. Commun. 2019, 10, 2030. [Google Scholar] [CrossRef] [Green Version]
- Yeung, K.; Janosch, P.; McFerran, B.; Rose, D.W.; Mischak, H.; Sedivy, J.M.; Kolch, W. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the raf kinase inhibitor protein. Mol. Cell. Biol. 2000, 20, 3079–3085. [Google Scholar] [CrossRef] [Green Version]
- Yeung, K.; Seitz, T.; Li, S.; Janosch, P.; McFerran, B.; Kaiser, C.; Fee, F.; Katsanakis, K.D.; Rose, D.W.; Mischak, H.; et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature 1999, 401, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Wakioka, T.; Sasaki, A.; Kato, R.; Shouda, T.; Matsumoto, A.; Miyoshi, K.; Tsuneoka, M.; Komiya, S.; Baron, R.; Yoshimura, A. Spred is a Sprouty-related suppressor of Ras signalling. Nature 2001, 412, 647–651. [Google Scholar] [CrossRef]
- Mason, J.M.; Morrison, D.J.; Basson, M.A.; Licht, J.D. Sprouty proteins: Multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006, 16, 45–54. [Google Scholar] [CrossRef]
- Leicht, D.T.; Balan, V.; Kaplun, A.; Singh-Gupta, V.; Kaplun, L.; Dobson, M.; Tzivion, G. Raf kinases: Function, regulation and role in human cancer. Biochim. Biophys. Acta 2007, 1773, 1196–1212. [Google Scholar] [CrossRef] [Green Version]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell. Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Noh, S.J.; Zhou, G.; Dixon, J.E.; Guan, K.L. Selective activation of MEK1 but not MEK2 by A-Raf from epidermal growth factor-stimulated Hela cells. J. Biol. Chem. 1996, 271, 3265–3271. [Google Scholar] [CrossRef] [Green Version]
- Frost, J.A.; Steen, H.; Shapiro, P.; Lewis, T.; Ahn, N.; Shaw, P.E.; Cobb, M.H. Cross-cascade activation of ERKs and ternary complex factors by Rho family proteins. EMBO J. 1997, 16, 6426–6438. [Google Scholar] [CrossRef]
- Coles, L.C.; Shaw, P.E. PAK1 primes MEK1 for phosphorylation by Raf-1 kinase during cross-cascade activation of the ERK pathway. Oncogene 2002, 21, 2236–2244. [Google Scholar] [CrossRef] [Green Version]
- Kholodenko, B.N.; Hancock, J.F.; Kolch, W. Signalling ballet in space and time. Nat. Rev. Mol. Cell. Biol. 2010, 11, 414–426. [Google Scholar] [CrossRef] [Green Version]
- Kornfeld, K.; Hom, D.B.; Horvitz, H.R. The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell 1995, 83, 903–913. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, M.; Han, M. The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell 1995, 83, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Therrien, M.; Chang, H.C.; Solomon, N.M.; Karim, F.D.; Wassarman, D.A.; Rubin, G.M. KSR, a novel protein kinase required for RAS signal transduction. Cell 1995, 83, 879–888. [Google Scholar] [CrossRef] [Green Version]
- Therrien, M.; Wong, A.M.; Rubin, G.M. CNK, a RAF-binding multidomain protein required for RAS signaling. Cell 1998, 95, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Sieburth, D.S.; Sun, Q.; Han, M. SUR-8, a conserved Ras-binding protein with leucine-rich repeats, positively regulates Ras-mediated signaling in C. elegans. Cell 1998, 94, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc. Natl. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar] [CrossRef] [Green Version]
- Ishibe, S.; Joly, D.; Liu, Z.X.; Cantley, L.G. Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol. Cell 2004, 16, 257–267. [Google Scholar] [CrossRef]
- Sharma, C.; Vomastek, T.; Tarcsafalvi, A.; Catling, A.D.; Schaeffer, H.J.; Eblen, S.T.; Weber, M.J. MEK partner 1 (MP1): Regulation of oligomerization in MAP kinase signaling. J. Cell Biochem. 2005, 94, 708–719. [Google Scholar] [CrossRef] [PubMed]
- McKay, M.M.; Ritt, D.A.; Morrison, D.K. Signaling dynamics of the KSR1 scaffold complex. Proc. Natl. Acad. Sci. USA 2009, 106, 11022–11027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajakulendran, T.; Sahmi, M.; Lefrançois, M.; Sicheri, F.; Therrien, M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature 2009, 461, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Burack, W.R.; Stock, J.L.; Kortum, R.; Chaika, O.V.; Afkarian, M.; Muller, W.J.; Murphy, K.M.; Morrison, D.K.; Lewis, R.E.; et al. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol. Cell. Biol. 2002, 22, 3035–3045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, F.D.; Langeberg, L.K.; Cellurale, C.; Pawson, T.; Morrison, D.K.; Davis, R.J.; Scott, J.D. AKAP-Lbc enhances cyclic AMP control of the ERK1/2 cascade. Nat. Cell Biol. 2010, 12, 1242–1249. [Google Scholar] [CrossRef]
- Dougherty, M.K.; Ritt, D.A.; Zhou, M.; Specht, S.I.; Monson, D.M.; Veenstra, T.D.; Morrison, D.K. KSR2 is a calcineurin substrate that promotes ERK cascade activation in response to calcium signals. Mol. Cell 2009, 34, 652–662. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, C.A.; Bolin, L.; Slattery, R.; Murray, R.; McMahon, M. Post-natal lethality and neurological and gastrointestinal defects in mice with targeted disruption of the A-Raf protein kinase gene. Curr. Biol. 1996, 6, 614–617. [Google Scholar] [CrossRef] [Green Version]
- Wojnowski, L.; Zimmer, A.M.; Beck, T.W.; Hahn, H.; Bernal, R.; Rapp, U.R.; Zimmer, A. Endothelial apoptosis in Braf-deficient mice. Nat. Genet. 1997, 16, 293–297. [Google Scholar] [CrossRef]
- Wojnowski, L.; Stancato, L.F.; Zimmer, A.M.; Hahn, H.; Beck, T.W.; Larner, A.C.; Rapp, U.R.; Zimmer, A. Craf-1 protein kinase is essential for mouse development. Mech. Dev. 1998, 76, 141–149. [Google Scholar] [CrossRef]
- Wiese, S.; Pei, G.; Karch, C.; Troppmair, J.; Holtmann, B.; Rapp, U.R.; Sendtner, M. Specific function of B-Raf in mediating survival of embryonic motoneurons and sensory neurons. Nat. Neurosci. 2001, 4, 137–142. [Google Scholar] [CrossRef]
- De Iriarte Rodríguez, R.; Magariños, M.; Pfeiffer, V.; Rapp, U.R.; Varela-Nieto, I. C-Raf deficiency leads to hearing loss and increased noise susceptibility. Cell Mol. Life Sci. 2015, 72, 3983–3998. [Google Scholar] [CrossRef] [Green Version]
- Hüser, M.; Luckett, J.; Chiloeches, A.; Mercer, K.; Iwobi, M.; Giblett, S.; Sun, X.M.; Brown, J.; Marais, R.; Pritchard, C. MEK kinase activity is not necessary for Raf-1 function. EMBO J. 2001, 20, 1940–1951. [Google Scholar] [CrossRef] [Green Version]
- Mikula, M.; Schreiber, M.; Husak, Z.; Kucerova, L.; Rüth, J.; Wieser, R.; Zatloukal, K.; Beug, H.; Wagner, E.F.; Baccarini, M. Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. EMBO J. 2001, 20, 1952–1962. [Google Scholar] [CrossRef] [Green Version]
- Rebocho, A.P.; Marais, R. ARAF acts as a scaffold to stabilize BRAF:CRAF heterodimers. Oncogene 2013, 32, 3207–3212. [Google Scholar] [CrossRef] [Green Version]
- Dumaz, N.; Hayward, R.; Martin, J.; Ogilvie, L.; Hedley, D.; Curtin, J.A.; Bastian, B.C.; Springer, C.; Marais, R. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res. 2006, 66, 9483–9491. [Google Scholar] [CrossRef] [Green Version]
- McPhillips, F.; Mullen, P.; MacLeod, K.G.; Sewell, J.M.; Monia, B.P.; Cameron, D.A.; Smyth, J.F.; Langdon, S.P. Raf-1 is the predominant Raf isoform that mediates growth factor-stimulated growth in ovarian cancer cells. Carcinogenesis 2006, 27, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Angelini, S.; Czene, K.; Sauroja, I.; Hahka-Kemppinen, M.; Pyrhönen, S.; Hemminki, K. BRAF mutations in metastatic melanoma: A possible association with clinical outcome. Clin. Cancer Res. 2003, 9, 3362–3368. [Google Scholar]
- Dhomen, N.; Marais, R. New insight into BRAF mutations in cancer. Curr. Opin. Genet. Dev. 2007, 17, 31–39. [Google Scholar] [CrossRef]
- Garnett, M.J.; Rana, S.; Paterson, H.; Barford, D.; Marais, R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol. Cell 2005, 20, 963–969. [Google Scholar] [CrossRef]
- Tran, N.H.; Frost, J.A. Phosphorylation of Raf-1 by p21-activated kinase 1 and Src regulates Raf-1 autoinhibition. J. Biol. Chem. 2003, 278, 11221–11226. [Google Scholar] [CrossRef] [Green Version]
- Zebisch, A.; Staber, P.B.; Delavar, A.; Bodner, C.; Hiden, K.; Fischereder, K.; Janakiraman, M.; Linkesch, W.; Auner, H.W.; Emberger, W.; et al. Two transforming C-RAF germ-line mutations identified in patients with therapy-related acute myeloid leukemia. Cancer Res. 2006, 66, 3401–3408. [Google Scholar] [CrossRef] [Green Version]
- Pandit, B.; Sarkozy, A.; Pennacchio, L.A.; Carta, C.; Oishi, K.; Martinelli, S.; Pogna, E.A.; Schackwitz, W.; Ustaszewska, A.; Landstrom, A.; et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 2007, 39, 1007–1012. [Google Scholar] [CrossRef]
- Razzaque, M.A.; Nishizawa, T.; Komoike, Y.; Yagi, H.; Furutani, M.; Amo, R.; Kamisago, M.; Momma, K.; Katayama, H.; Nakagawa, M.; et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat. Genet. 2007, 39, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Rauen, K.A. The RASopathies. Annu. Rev. Genomics Hum. Genet. 2013, 14, 355–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, H.; Thevakumaran, N.; Gavory, G.; Li, J.J.; Padeganeh, A.; Guiral, S.; Duchaine, J.; Mao, D.Y.; Bouvier, M.; Sicheri, F.; et al. Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat. Chem. Biol. 2013, 9, 428–436. [Google Scholar] [CrossRef]
- Shaw, A.S.; Kornev, A.P.; Hu, J.; Ahuja, L.G.; Taylor, S.S. Kinases and pseudokinases: Lessons from RAF. Mol. Cell. Biol. 2014, 34, 1538–1546. [Google Scholar] [CrossRef] [Green Version]
- Karoulia, Z.; Wu, Y.; Ahmed, T.A.; Xin, Q.; Bollard, J.; Krepler, C.; Wu, X.; Zhang, C.; Bollag, G.; Herlyn, M.; et al. An Integrated Model of RAF Inhibitor Action Predicts Inhibitor Activity against Oncogenic BRAF Signaling. Cancer Cell 2016, 30, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Lyons, J.F.; Wilhelm, S.; Hibner, B.; Bollag, G. Discovery of a novel Raf kinase inhibitor. Endocr. Relat. Cancer 2001, 8, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Hall-Jackson, C.A.; Eyers, P.A.; Cohen, P.; Goedert, M.; Boyle, F.T.; Hewitt, N.; Plant, H.; Hedge, P. Paradoxical activation of Raf by a novel Raf inhibitor. Chem. Biol. 1999, 6, 559–568. [Google Scholar] [CrossRef]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [Green Version]
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015, 27, 240–256. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Man, R.J.; Zhang, Y.L.; Jiang, A.Q.; Zhu, H.L. A patent review of RAF kinase inhibitors (2010–2018). Expert Opin. Ther. Pat. 2019, 29, 675–688. [Google Scholar] [CrossRef]
- Henry, J.R.; Kaufman, M.D.; Peng, S.B.; Ahn, Y.M.; Caldwell, T.M.; Vogeti, L.; Telikepalli, H.; Lu, W.P.; Hood, M.M.; Rutkoski, T.J.; et al. Discovery of 1-(3,3-dimethylbutyl)-3-(2-fluoro-4-methyl-5-(7-methyl-2-(methylamino)pyrido [2,3-d]pyrimidin-6-yl)phenyl)urea (LY3009120) as a pan-RAF inhibitor with minimal paradoxical activation and activity against BRAF or RAS mutant tumor cells. J. Med. Chem. 2015, 58, 4165–4179. [Google Scholar] [CrossRef]
- Peng, S.B.; Henry, J.R.; Kaufman, M.D.; Lu, W.P.; Smith, B.D.; Vogeti, S.; Rutkoski, T.J.; Wise, S.; Chun, L.; Zhang, Y.; et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 2015, 28, 384–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noeparast, A.; Giron, P.; De Brakeleer, S.; Eggermont, C.; De Ridder, U.; Teugels, E.; De Grève, J. Type II RAF inhibitor causes superior ERK pathway suppression compared to type I RAF inhibitor in cells expressing different BRAF mutant types recurrently found in lung cancer. Oncotarget 2018, 9, 16110–16123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girotti, M.R.; Lopes, F.; Preece, N.; Niculescu-Duvaz, D.; Zambon, A.; Davies, L.; Whittaker, S.; Saturno, G.; Viros, A.; Pedersen, M.; et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell 2015, 27, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Alberta, J.A.; Pilarz, C.; Calligaris, D.; Chadwick, E.J.; Ramkissoon, S.H.; Ramkissoon, L.A.; Garcia, V.M.; Mazzola, E.; Goumnerova, L.; et al. A brain-penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low-grade astrocytomas. Neuro Oncol. 2017, 19, 774–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, R.J.; Hollebecque, A.; Flaherty, K.T.; Shapiro, G.I.; Rodon Ahnert, J.; Millward, M.J.; Zhang, W.; Gao, L.; Sykes, A.; Willard, M.D.; et al. A Phase I Study of LY3009120, a Pan-RAF Inhibitor, in Patients with Advanced or Metastatic Cancer. Mol. Cancer Ther. 2020, 19, 460–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koumaki, K.; Kontogianni, G.; Kosmidou, V.; Pahitsa, F.; Kritsi, E.; Zervou, M.; Chatziioannou, A.; Souliotis, V.L.; Papadodima, O.; Pintzas, A. BRAF paradox breakers PLX8394, PLX7904 are more effective against BRAFV600Ε CRC cells compared with the BRAF inhibitor PLX4720 and shown by detailed pathway analysis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166061. [Google Scholar] [CrossRef]
- Yao, Z.; Gao, Y.; Su, W.; Yaeger, R.; Tao, J.; Na, N.; Zhang, Y.; Zhang, C.; Rymar, A.; Tao, A.; et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat. Med. 2019, 25, 284–291. [Google Scholar] [CrossRef]
- Dumaz, N.; Lebbé, C. New perspectives on targeting RAF, MEK and ERK in melanoma. Curr. Opin. Oncol. 2021, 33, 120–126. [Google Scholar] [CrossRef]
- Turajlic, S.; Ali, Z.; Yousaf, N.; Larkin, J. Phase I/II RAF kinase inhibitors in cancer therapy. Expert Opin. Investig. Drugs 2013, 22, 739–749. [Google Scholar] [CrossRef]
- Basile, K.J.; Le, K.; Hartsough, E.J.; Aplin, A.E. Inhibition of mutant BRAF splice variant signaling by next-generation, selective RAF inhibitors. Pigment Cell Melanoma Res. 2014, 27, 479–484. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Kirkwood, J.M.; Chiarion Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; et al. Five-Year Analysis of Adjuvant Dabrafenib plus Trametinib in Stage III Melanoma. N. Engl. J. Med. 2020, 383, 1139–1148. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- Gogas, H.; Dréno, B.; Larkin, J.; Demidov, L.; Stroyakovskiy, D.; Eroglu, Z.; Francesco Ferrucci, P.; Pigozzo, J.; Rutkowski, P.; Mackiewicz, J.; et al. Cobimetinib plus atezolizumab in BRAF(V600) wild-type melanoma: Primary results from the randomized phase III IMspire170 study. Ann. Oncol. 2021, 32, 384–394. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]





| RAF Inhibitor | Mechanism | Clinical Stage | Features |
|---|---|---|---|
| First generation | |||
| Sorafenib | ‘αC-IN’/‘DFG-OUT’ inhibitor | Approved for advanced renal cell carcinoma and hepatocellular carcinoma | Transactivation of ERK1/2 pathway in WT B-Raf cells |
| Second generation | |||
| Vemurafenib | ‘αC-OUT’/‘DFG-IN’ inhibitor | Approved for B-Raf-V600E metastatic melanoma | Causes photosensitivity, development of drug resistance and tumor recurrence |
| Dabrafenib | ‘αC-OUT’/‘DFG-IN’ inhibitor | Approved for melanoma patients with B-Raf-V600E/K mutations | Causes fever, development of drug resistance and tumor recurrence |
| Third generation | |||
| CCT196969 | ‘αC-IN’/‘DFG-OUT’ inhibitor | Antitumor activity in preclinical studies against B-Raf-V600E melanomas, Ras-mutant melanomas and colorectal tumors | Dual pan-Raf and SRC kinase inhibitor, effective in patient-derived xenograft (PDX) models that included melanomas with intrinsic or acquired resistance to second-generation Raf and MEK inhibitors |
| CCT241161 | ‘αC-IN’/‘DFG-OUT’ inhibitor | Antitumor activity in preclinical studies against B-Raf-V600E melanomas, Ras-mutant melanomas and colorectal tumors | Dual pan-Raf and SRC kinase inhibitor, effective in patient-derived xenograft (PDX) models that included melanomas with intrinsic or acquired resistance to second-generation Raf and MEK inhibitors |
| LY3009120 | ‘αC-IN’/‘DFG-OUT’ inhibitor | Antitumor activity in Phase I clinical studies against NRas or KRas mutant tumors and B-Raf deletions in pancreatic and thyroid tumors | Effective in vemurafenib-resistant melanomas; inhibit monomeric and dimeric B-Raf with similar potency |
| TAK-580 (MLN2480) | ‘αC-IN’/‘DFG-OUT’ inhibitor | Antiproliferative activity in Phase I clinical studies against melanomas and other solid tumor cell lines harboring B-Raf, NRas or KRas mutations; delay emergence of resistance | Effective in vemurafenib-resistant melanomas harboring B-Raf or N-Ras mutations and B-Raf-V600E colorectal or thyroid tumors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Luo, Z. Discovery of Raf Family Is a Milestone in Deciphering the Ras-Mediated Intracellular Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 5158. https://doi.org/10.3390/ijms23095158
Zhao J, Luo Z. Discovery of Raf Family Is a Milestone in Deciphering the Ras-Mediated Intracellular Signaling Pathway. International Journal of Molecular Sciences. 2022; 23(9):5158. https://doi.org/10.3390/ijms23095158
Chicago/Turabian StyleZhao, Jingtong, and Zhijun Luo. 2022. "Discovery of Raf Family Is a Milestone in Deciphering the Ras-Mediated Intracellular Signaling Pathway" International Journal of Molecular Sciences 23, no. 9: 5158. https://doi.org/10.3390/ijms23095158
APA StyleZhao, J., & Luo, Z. (2022). Discovery of Raf Family Is a Milestone in Deciphering the Ras-Mediated Intracellular Signaling Pathway. International Journal of Molecular Sciences, 23(9), 5158. https://doi.org/10.3390/ijms23095158