
Obesity as a Risk Factor for Dementia and Alzheimer’s Disease: The Role of Leptin

 , , ,
, , , 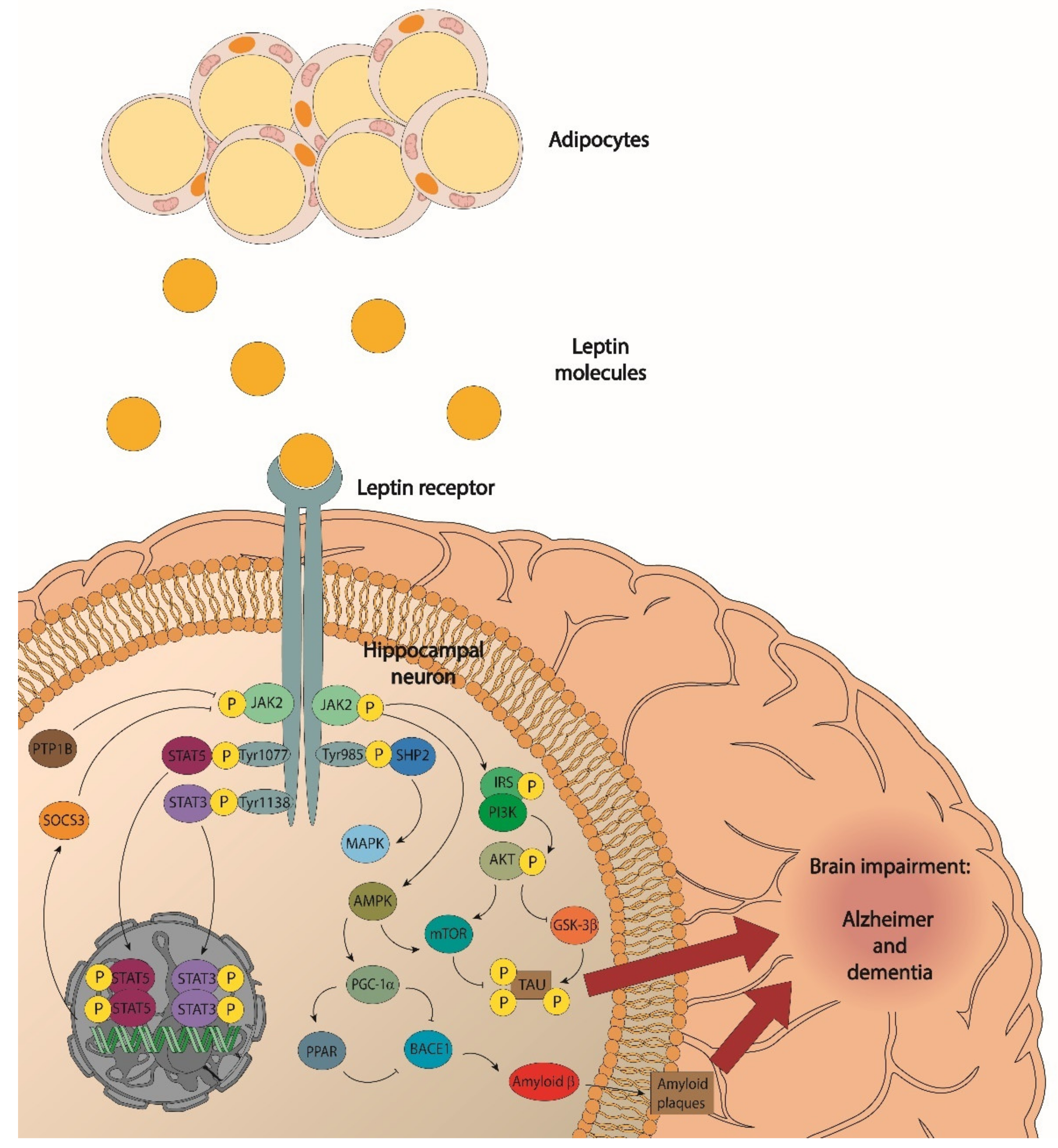{kind=link}
Abstract
:1. Introduction
2. Obesity and Dementia
3. Obesity and Leptin
- JAK/signal transducer activators of transcription 3 (STAT3);
- Phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt);
- Extracellular signalling-regulated kinases (ERK); and
- Signal transducer activators of transcription 5 (STAT5).
4. Leptin; Its Relationship with Cognition and Synaptic Function
5. Leptin Resistance: Mechanisms Involved
5.1. Pathways of Leptin Entry into the Brain and Leptin Resistance
5.2. Inflammation and Leptin Resistance
5.3. Hypothalamic Endoplasmic Reticulum Stress and Leptin Resistance
5.4. Reduced Sensitivity to Leptin (Receptors, Receptor Downstream Signaling, and Negative Feedback Signaling Pathways)
6. Leptin Signalling, Obesity, and Alzheimer’s Disease
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bradfield, N.I.; Ames, D. Mild cognitive impairment: Narrative review of taxonomies and systematic review of their prediction of incident Alzheimer’s disease dementia. BJPsych Bull. 2020, 44, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Chowen, J.A.; Garcia-Segura, L.M. Microglia, neurodegeneration and loss of neuroendocrine control. Prog. Neurobiol. 2020, 184, 101720. [Google Scholar] [CrossRef] [PubMed]
- Moser, V.A.; Christensen, A.; Liu, J.; Zhou, A.; Yagi, S.; Beam, C.R.; Galea, L.; Pike, C.J. Effects of aging, high-fat diet, and testosterone treatment on neural and metabolic outcomes in male brown Norway rats. Neurobiol. Aging 2019, 73, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Forny-Germano, L.; De Felice, F.G.; Do Nascimento Vieira, M.N. The Role of Leptin and Adiponectin in Obesity-Associated Cognitive Decline and Alzheimer’s Disease. Front. Neurosci. 2019, 12, 1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yam, K.Y.; Naninck, E.F.G.; Abbink, M.R.; la Fleur, S.E.; Schipper, L.; van den Beukel, J.C.; Grefhorst, A.; Oosting, A.; van der Beek, E.M.; Lucassen, P.J.; et al. Exposure to chronic early-life stress lastingly alters the adipose tissue, the leptin system and changes the vulnerability to western-style diet later in life in mice. Psychoneuroendocrinology 2017, 77, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Kiliaan, A.J.; Arnoldussen, I.A.C.; Gustafson, D.R. Adipokines: A link between obesity and dementia? Lancet Neurol. 2014, 13, 913–923. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.L.; Pan, C.Y.; Chen, F.C.; Huang, T.H.; Tsai, M.C.; Chuang, C.Y. Differences in neurocognitive performance and metabolic and inflammatory indices in male adults with obesity as a function of regular exercise. Exp. Physiol. 2019, 104, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Vamanu, E.; Rai, S.N. The Link between Obesity, Microbiota Dysbiosis, and Neurodegenerative Pathogenesis. Diseases 2021, 9, 45. [Google Scholar] [CrossRef]
- Abbott, R.D.; Ross, G.W.; White, L.R.; Nelson, J.S.; Masaki, K.H.; Tanner, C.M.; Curb, J.D.; Blanchette, P.L.; Popper, J.S.; Petrovitch, H. Midlife adiposity and the future risk of Parkinson’s disease. Neurology 2002, 59, 1051–1057. [Google Scholar] [CrossRef]
- Procaccini, C.; Santopaolo, M.; Faicchia, D.; Colamatteo, A.; Formisano, L.; De Candia, P.; Galgani, M.; De Rosa, V.; Matarese, G. Role of metabolism in neurodegenerative disorders. Metabolism 2016, 65, 1376–1390. [Google Scholar] [CrossRef]
- Gaba, A.M.; Zhang, K.; Marder, K.; Moskowitz, C.B.; Werner, P.; Boozer, C.N. Energy balance in early-stage Huntington disease. Am. J. Clin. Nutr. 2005, 81, 1335–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, F.; Masouleh, S.K.; Kratzsch, J.; Schroeter, M.L.; Röhr, S.; Riedel-Heller, S.G.; Villringer, A.; Veronica Witte, A. A Metabolic Obesity Profile Is Associated with Decreased Gray Matter Volume in Cognitively Healthy Older Adults. Front. Aging Neurosci. 2019, 11, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Q.; Guan, Y.; Yu, W.; Liu, X.; Wu, L.; Xiao, M.; Lü, Y. Associations between obesity and cognitive impairment in the Chinese elderly: An observational study. Clin. Interv. Aging 2019, 14, 367–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.P.; Moody, J.N.; Roca, J.G.; Hayes, S.M. Body mass index is associated with smaller medial temporal lobe volume in those at risk for Alzheimer’s disease. NeuroImage Clin. 2020, 25, 102156. [Google Scholar] [CrossRef]
- Nordestgaard, L.T.; Tybjærg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Body Mass Index and Risk of Alzheimer’s Disease: A Mendelian Randomization Study of 399,536 Individuals. J. Clin. Endocrinol. Metab. 2017, 102, 2310–2320. [Google Scholar] [CrossRef]
- Emmerzaal, T.L.; Kiliaan, A.J.; Gustafson, D.R. 2003–2013: A decade of body mass index, Alzheimer’s disease, and dementia. J. Alzheimers Dis. 2015, 43, 739–755. [Google Scholar] [CrossRef]
- García-Ptacek, S.; Faxén-Irving, G.; Čermáková, P.; Eriksdotter, M.; Religa, D. Body mass index in dementia. Eur. J. Clin. Nutr. 2014, 68, 1204–1209. [Google Scholar] [CrossRef] [Green Version]
- Qizilbash, N.; Gregson, J.; Johnson, M.E.; Pearce, N.; Douglas, I.; Wing, K.; Evans, S.J.W.; Pocock, S.J. BMI and risk of dementia in two million people over two decades: A retrospective cohort study. Lancet Diabetes Endocrinol. 2015, 3, 431–436. [Google Scholar] [CrossRef]
- Pannacciulli, N.; Del Parigi, A.; Chen, K.; Le, D.S.N.T.; Reiman, E.M.; Tataranni, P.A. Brain abnormalities in human obesity: A voxel-based morphometric study. Neuroimage 2006, 31, 1419–1425. [Google Scholar] [CrossRef]
- Gunstad, J.; Paul, R.H.; Cohen, R.A.; Tate, D.F.; Spitznagel, M.B.; Gordon, E. Elevated body mass index is associated with executive dysfunction in otherwise healthy adults. Compr. Psychiatry 2007, 48, 57–61. [Google Scholar] [CrossRef]
- Raji, C.A.; Ho, A.J.; Parikshak, N.N.; Becker, J.T.; Lopez, O.L.; Kuller, L.H.; Hua, X.; Leow, A.D.; Toga, A.W.; Thompson, P.M. Brain structure and obesity. Hum. Brain Mapp. 2010, 31, 353–364. [Google Scholar] [CrossRef]
- Marqués-Iturria, I.; Pueyo, R.; Garolera, M.; Segura, B.; Junqué, C.; García-García, I.; José Sender-Palacios, M.; Vernet-Vernet, M.; Narberhaus, A.; Ariza, M.; et al. Frontal cortical thinning and subcortical volume reductions in early adulthood obesity. Psychiatry Res. 2013, 214, 109–115. [Google Scholar] [CrossRef]
- Yokum, S.; Ng, J.; Stice, E. Relation of regional gray and white matter volumes to current BMI and future increases in BMI: A prospective MRI study. Int. J. Obes. 2012, 36, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Debette, S.; Wolf, C.; Lambert, J.C.; Crivello, F.; Soumaré, A.; Zhu, Y.C.; Schilling, S.; Dufouil, C.; Mazoyer, B.; Amouyel, P.; et al. Abdominal obesity and lower gray matter volume: A Mendelian randomization study. Neurobiol. Aging 2014, 35, 378–386. [Google Scholar] [CrossRef]
- Veit, R.; Kullmann, S.; Heni, M.; Machann, J.; Häring, H.U.; Fritsche, A.; Preissl, H. Reduced cortical thickness associated with visceral fat and BMI. NeuroImage Clin. 2014, 6, 307–311. [Google Scholar] [CrossRef] [Green Version]
- Sharkey, R.J.; Karama, S.; Dagher, A. Overweight is not associated with cortical thickness alterations in children. Front. Neurosci. 2015, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Van Boxtel, M.P.J.; Baars, L.; Jolles, J. Obesity, blood pressure and cognitive function: A reply to Waldstein and Katzel. Int. J. Obes. 2007, 31, 1186. [Google Scholar] [CrossRef] [Green Version]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Guillemot-Legris, O.; Muccioli, G.G. Obesity-Induced Neuroinflammation: Beyond the Hypothalamus. Trends Neurosci. 2017, 40, 237–253. [Google Scholar] [CrossRef]
- Cavaliere, G.; Trinchese, G.; Penna, E.; Cimmino, F.; Pirozzi, C.; Lama, A.; Annunziata, C.; Catapano, A.; Mattace Raso, G.; Meli, R.; et al. High-Fat Diet Induces Neuroinflammation and Mitochondrial Impairment in Mice Cerebral Cortex and Synaptic Fraction. Front. Cell. Neurosci. 2019, 13, 509. [Google Scholar] [CrossRef] [PubMed]
- Crispino, M.; Trinchese, G.; Penna, E.; Cimmino, F.; Catapano, A.; Villano, I.; Perrone-Capano, C.; Mollica, M.P. Interplay between Peripheral and Central Inflammation in Obesity-Promoted Disorders: The Impact on Synaptic Mitochondrial Functions. Int. J. Mol. Sci. 2020, 21, 5964. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.F.; Santos, A.E.; Moreira, P.I.; Pereira, A.C.; Sousa, F.J.; Cardoso, S.M.; Cruz, M.T. Is Alzheimer’s disease an inflammasomopathy? Ageing Res. Rev. 2019, 56, 100966. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Nuzzo, D.; Picone, P.; Caruana, L.; Vasto, S.; Barera, A.; Caruso, C.; Di Carlo, M. Inflammatory mediators as biomarkers in brain disorders. Inflammation 2014, 37, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Pistell, P.J.; Morrison, C.D.; Gupta, S.; Knight, A.G.; Keller, J.N.; Ingram, D.K.; Bruce-Keller, A.J. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J. Neuroimmunol. 2010, 219, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Winocur, G.; Greenwood, C.E. Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiol. Aging 2005, 26 (Suppl. 1), 46–49. [Google Scholar] [CrossRef]
- Sobesky, J.L.; Barrientos, R.M.; De May, H.S.; Thompson, B.M.; Weber, M.D.; Watkins, L.R.; Maier, S.F. High-fat diet consumption disrupts memory and primes elevations in hippocampal IL-1β, an effect that can be prevented with dietary reversal or IL-1 receptor antagonism. Brain Behav. Immun. 2014, 42, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.I.; Shen, C.F.; Hsu, T.H.; Lin, S.H. A High-Fructose-High-Coconut Oil Diet Induces Dysregulating Expressions of Hippocampal Leptin and Stearoyl-CoA Desaturase, and Spatial Memory Deficits in Rats. Nutrients 2017, 9, 619. [Google Scholar] [CrossRef]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, A.M.; Greene, M.W.; Geetha, T.; Babu, J.R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 499–508. [Google Scholar] [CrossRef]
- Ivanova, N.; Liu, Q.; Agca, C.; Agca, Y.; Noble, E.G.; Whitehead, S.N.; Cechetto, D.F. White matter inflammation and cognitive function in a co-morbid metabolic syndrome and prodromal Alzheimer’s disease rat model. J. Neuroinflammation 2020, 17, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sah, S.K.; Lee, C.; Jang, J.H.; Park, G.H. Effect of high-fat diet on cognitive impairment in triple-transgenic mice model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 493, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G. Overview of adipose tissue and its role in obesity and metabolic disorders. Methods Mol. Biol. 2008, 456, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Fietta and Delsante. Focus on Adipokines—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/24640423/ (accessed on 27 April 2022).
- Effect of Body Mass Index on Serum Leptin Levels—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/23272432/ (accessed on 27 April 2022).
- Kennedy, A.; Gettys, T.W.; Watson, P.; Wallace, P.; Ganaway, E.; Pan, Q.; Garvey, W.T. The metabolic significance of leptin in humans: Gender-based differences in relationship to adiposity, insulin sensitivity, and energy expenditure. J. Clin. Endocrinol. Metab. 1997, 82, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.K.; Ohannesian, J.P.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Magosin, S.; Marco, C.; Caro, J.F. Nocturnal rise of leptin in lean, obese, and non-insulin-dependent diabetes mellitus subjects. J. Clin. Investig. 1996, 97, 1344–1347. [Google Scholar] [CrossRef] [PubMed]
- Wada, N.; Hirako, S.; Takenoya, F.; Kageyama, H.; Okabe, M.; Shioda, S. Leptin and its receptors. J. Chem. Neuroanat. 2014, 61–62, 191–199. [Google Scholar] [CrossRef]
- Hsuchou, H.; Kastin, A.J.; Tu, H.; Markadakis, E.N.; Stone, K.P.; Wang, Y.; Heymsfield, S.B.; Chua, S.S.; Obici, S.; Magrisso, I.J.; et al. Effects of cell-type specific leptin receptor mutation on leptin transport across the BBB. Peptides 2011, 32, 1392–1399. [Google Scholar] [CrossRef] [Green Version]
- Lloret, A.; Monllor, P.; Esteve, D.; Cervera-Ferri, A.; Lloret, M.A. Obesity as a Risk Factor for Alzheimer’s Disease: Implication of Leptin and Glutamate. Front. Neurosci. 2019, 13, 508. [Google Scholar] [CrossRef]
- Li, S.; Li, X. Leptin in normal physiology and leptin resistance. Sci Bull. 2016, 61, 1480–1488. [Google Scholar] [CrossRef] [Green Version]
- Arnoldussen, I.A.C.; Kiliaan, A.J.; Gustafson, D.R. Obesity and dementia: Adipokines interact with the brain. Eur. Neuropsychopharmacol. 2014, 24, 1982–1999. [Google Scholar] [CrossRef] [Green Version]
- Aragonès, G.; Ardid-Ruiz, A.; Ibars, M.; Suárez, M.; Bladé, C. Modulation of leptin resistance by food compounds. Mol. Nutr. Food Res. 2016, 60, 1789–1803. [Google Scholar] [CrossRef]
- Fujita, Y.; Yamashita, T. The Effects of Leptin on Glial Cells in Neurological Diseases. Front. Neurosci. 2019, 13, 828. [Google Scholar] [CrossRef]
- Grizzanti, J.; Lee, H.G.; Camins, A.; Pallas, M.; Casadesus, G. The therapeutic potential of metabolic hormones in the treatment of age-related cognitive decline and Alzheimer’s disease. Nutr. Res. 2016, 36, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Paz-Filho, G.J. The Effects of Leptin Replacement on Neural Plasticity. Neural Plast. 2016, 2016, 8528934. [Google Scholar] [CrossRef] [Green Version]
- Parimisetty, A.; Dorsemans, A.C.; Awada, R.; Ravanan, P.; Diotel, N.; Lefebvre d’Hellencourt, C. Secret talk between adipose tissue and central nervous system via secreted factors-an emerging frontier in the neurodegenerative research. J. Neuroinflammation 2016, 13, 67. [Google Scholar] [CrossRef] [Green Version]
- Fruhwürth, S.; Vogel, H.; Schürmann, A.; Williams, K.J. Novel Insights into How Overnutrition Disrupts the Hypothalamic Actions of Leptin. Front. Endocrinol. 2018, 9, 89. [Google Scholar] [CrossRef]
- McGregor, G.; Harvey, J. Regulation of Hippocampal Synaptic Function by the Metabolic Hormone, Leptin: Implications for Health and Neurodegenerative Disease. Front. Cell. Neurosci. 2018, 12, 340. [Google Scholar] [CrossRef]
- Pimentel, G.D.; Ganeshan, K.; Carvalheira, J.B.C. Hypothalamic inflammation and the central nervous system control of energy homeostasis. Mol. Cell. Endocrinol. 2014, 397, 15–22. [Google Scholar] [CrossRef]
- Gruzdeva, O.; Borodkina, D.; Uchasova, E.; Dyleva, Y.; Barbarash, O. Leptin resistance: Underlying mechanisms and diagnosis. Diabetes Metab. Syndr. Obes. 2019, 12, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuire, M.J.; Ishii, M. Leptin Dysfunction and Alzheimer’s Disease: Evidence from Cellular, Animal, and Human Studies. Cell. Mol. Neurobiol. 2016, 36, 203–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejido, D.C.P.; Peny, J.A.; Vieira, M.N.N.; Ferreira, S.T.; De Felice, F.G. Insulin and leptin as potential cognitive enhancers in metabolic disorders and Alzheimer’s disease. Neuropharmacology 2020, 171, 108115. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Duval, G.T.; Cheng, C.Y.; Wong, T.Y.; Lamoureux, E.L.; Milea, D.; Sabanayagam, C. U-Shaped Relationship between Serum Leptin Concentration and Cognitive Performance in Older Asian Adults. Nutrients 2019, 11, 660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.; Mudd, J.; Hawkins, M. Neuroprotective effects of leptin in the context of obesity and metabolic disorders. Neurobiol. Dis. 2014, 72 Pt A, 61–71. [Google Scholar] [CrossRef]
- McGregor, G.; Harvey, J. Food for thought: Leptin regulation of hippocampal function and its role in Alzheimer’s disease. Neuropharmacology 2018, 136, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Harrison, L.; Schriever, S.C.; Feuchtinger, A.; Kyriakou, E.; Baumann, P.; Pfuhlmann, K.; Messias, A.C.; Walch, A.; Tschöp, M.H.; Pfluger, P.T. Fluorescent blood-brain barrier tracing shows intact leptin transport in obese mice. Int. J. Obes. 2019, 43, 1305–1318. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo, A.G.; Crujeiras, A.B.; Casanueva, F.F.; Carreira, M.C. Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later? Nutrients 2019, 11, 2704. [Google Scholar] [CrossRef] [Green Version]
- Sáinz, N.; Barrenetxe, J.; Moreno-Aliaga, M.J.; Martínez, J.A. Leptin resistance and diet-induced obesity: Central and peripheral actions of leptin. Metabolism 2015, 64, 35–46. [Google Scholar] [CrossRef]
- Roujeau, C.; Jockers, R.; Dam, J. New pharmacological perspectives for the leptin receptor in the treatment of obesity. Front. Endocrinol. 2014, 5, 167. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; Sánchez-Margalet, V. Role of leptin in inflammation and vice versa. Int. J. Mol. Sci. 2020, 21, 5887. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Vilariño-García, T.; Fernández-Riejos, P.; Martín-González, J.; Segura-Egea, J.J.; Sánchez-Margalet, V. Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev. 2017, 35, 71–84. [Google Scholar] [CrossRef]
- Sánchez-Margalet, V.; Martín-Romero, C.; Santos-Alvarez, J.; Goberna, R.; Najib, S.; Gonzalez-Yanes, C. Role of leptin as an immunomodulator of blood mononuclear cells: Mechanisms of action. Clin. Exp. Immunol. 2003, 133, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Margalet, V.; Fernández-Riejos, P.; Najib, S.; Santos-Alvarez, J.; Martín-Romero, C.; Pérez-Pérez, A.; González-Yanes, C. Role of leptin in the activation of immune cells. Mediat. Inflamm. 2010, 2010, 568343. [Google Scholar] [CrossRef]
- Santos-Alvarez, J.; Goberna, R.; Sánchez-Margalet, V. Human leptin stimulates proliferation and activation of human circulating monocytes. Cell. Immunol. 1999, 194, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Martín-Romero, C.; Santos-Alvarez, J.; Goberna, R.; Sánchez-Margalet, V. Human leptin enhances activation and proliferation of human circulating T lymphocytes. Cell. Immunol. 2000, 199, 15–24. [Google Scholar] [CrossRef]
- Di Spiezio, A.; Sandin, E.S.; Dore, R.; Müller-Fielitz, H.; Storck, S.E.; Bernau, M.; Mier, W.; Oster, H.; Jöhren, O.; Pietrzik, C.U.; et al. The LepR-mediated leptin transport across brain barriers controls food reward. Mol. Metab. 2018, 8, 13–22. [Google Scholar] [CrossRef]
- Banks, W.A. The blood-brain barrier as an endocrine tissue. Nat. Rev. Endocrinol. 2019, 15, 444–455. [Google Scholar] [CrossRef]
- Ardid-Ruiz, A.; Harazin, A.; Barna, L.; Walter, F.R.; Bladé, C.; Suárez, M.; Deli, M.A.; Aragonès, G. The effects of Vitis vinifera L. phenolic compounds on a blood-brain barrier culture model: Expression of leptin receptors and protection against cytokine-induced damage. J. Ethnopharmacol. 2020, 247, 112253. [Google Scholar] [CrossRef]
- Bartolome, F.; Antequera, D.; Tavares, E.; Pascual, C.; Maldonado, R.; Camins, A.; Carro, E. Obesity and neuroinflammatory phenotype in mice lacking endothelial megalin. J. Neuroinflammation 2017, 14, 26. [Google Scholar] [CrossRef] [Green Version]
- Rhea, E.M.; Salameh, T.S.; Logsdon, A.F.; Hanson, A.J.; Erickson, M.A.; Banks, W.A. Blood-Brain Barriers in Obesity. AAPS J. 2017, 19, 921–930. [Google Scholar] [CrossRef]
- López, M. Hypothalamic Leptin Resistance: From BBB to BBSome. PLoS Genet. 2016, 12, e1005980. [Google Scholar] [CrossRef]
- Baumgarner, K.M.; Setti, S.; Diaz, C.; Littlefield, A.; Jones, A.; Kohman, R.A. Diet-induced obesity attenuates cytokine production following an immune challenge. Behav. Brain Res. 2014, 267, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.; Oh, S.; Choi, J.; Jang, J.T.; Choi, C.H.; Park, K.Y.; Son, K.H.; Byun, K. Attenuation of Inflammation and Leptin Resistance by Pyrogallol-Phloroglucinol-6,6-Bieckol on in the Brain of Obese Animal Models. Nutrients 2019, 11, 2773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wei, D.; McCrory, M.A.; Szalai, A.J.; Yang, G.; Li, L.; Li, F.; Zha, A.Z. Human C-reactive protein impedes entry of leptin into the CNS and attenuates its physiological actions in the CNS. Biochem. J. 2016, 473, 1215–1224. [Google Scholar] [CrossRef]
- Liu, Z.; Gan, L.; Zhou, Z.; Jin, W.; Sun, C. SOCS3 promotes inflammation and apoptosis via inhibiting JAK2/STAT3 signaling pathway in 3T3-L1 adipocyte. Immunobiology 2015, 220, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Nakata, M.; Yamamoto, S.; Okada, T.; Gantulga, D.; Okano, H.; Ozawa, K.; Yada, T. IL-10 gene transfer upregulates arcuate POMC and ameliorates hyperphagia, obesity and diabetes by substituting for leptin. Int. J. Obes. 2016, 40, 425–433. [Google Scholar] [CrossRef]
- Cheng, L.; Hu, T.; Shi, H.; Chen, X.; Wang, H.; Zheng, K.; Huang, X.F.; Yu, Y. DHA reduces hypothalamic inflammation and improves central leptin signaling in mice. Life Sci. 2020, 257, 118036. [Google Scholar] [CrossRef]
- Wu, Y.; Huang, X.F.; Bell, C.; Yu, Y. Ginsenoside Rb1 improves leptin sensitivity in the prefrontal cortex in obese mice. CNS Neurosci. Ther. 2018, 24, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Yu, Y.; Szabo, A.; Han, M.; Huang, X.F. Central inflammation and leptin resistance are attenuated by ginsenoside Rb1 treatment in obese mice fed a high-fat diet. PLoS ONE 2014, 9, e92618. [Google Scholar] [CrossRef] [Green Version]
- Jeon, B.T.; Kim, K.E.; Heo, R.W.; Shin, H.J.; Yi, C.; Hah, Y.S.; Kim, W.H.; Lee, S.; Roh, G.S. Myeloid-specific deletion of SIRT1 increases hepatic steatosis and hypothalamic inflammation in mice fed a high-fat diet. Metab. Brain Dis. 2014, 29, 635–643. [Google Scholar] [CrossRef]
- Paz-Filho, G.; Mastronardi, C.A.; Licinio, J. Leptin treatment: Facts and expectations. Metabolism 2015, 64, 146–156. [Google Scholar] [CrossRef]
- Hosoi, T.; Ozawa, K. Possible Pharmacological Approach Targeting Endoplasmic Reticulum Stress to Ameliorate Leptin Resistance in Obesity. Front. Endocrinol. 2016, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Hosoi, T.; Kuwamura, A.; Thon, M.; Tsuchio, K.; El-Hafeez, A.A.A.; Ozawa, K. Possible involvement of 4-hydroxy-2-nonenal in the pathogenesis of leptin resistance in obesity. Am. J. Physiol. Cell Physiol. 2019, 316, C641–C648. [Google Scholar] [CrossRef]
- Ramírez, S.; Claret, M. Hypothalamic ER stress: A bridge between leptin resistance and obesity. FEBS Lett. 2015, 589, 1678–1687. [Google Scholar] [CrossRef]
- Hakim, F.; Wang, Y.; Carreras, A.; Hirotsu, C.; Zhang, J.; Peris, E.; Gozal, D. Chronic sleep fragmentation during the sleep period induces hypothalamic endoplasmic reticulum stress and PTP1b-mediated leptin resistance in male mice. Sleep 2015, 38, 31–40. [Google Scholar] [CrossRef]
- Yuan, X.W.; Han, S.F.; Zhang, J.W.; Xu, J.Y.; Qin, L.Q. Leucine supplementation improves leptin sensitivity in high-fat diet fed rats. Food Nutr. Res. 2015, 59, 27373. [Google Scholar] [CrossRef] [Green Version]
- Pedroso, J.A.B.; Silveira, M.A.; Lima, L.B.; Furigo, I.C.; Zampieri, T.T.; Ramos-Lobo, A.M.; Buonfiglio, D.C.; Teixeira, P.D.S.; Frazão, R.; Donato, J. Changes in Leptin Signaling by SOCS3 Modulate Fasting-Induced Hyperphagia and Weight Regain in Mice. Endocrinology 2016, 157, 3901–3914. [Google Scholar] [CrossRef]
- Pedroso, J.A.B.; Buonfiglio, D.C.; Cardinali, L.I.; Furigo, I.C.; Ramos-Lobo, A.M.; Tirapegui, J.; Elias, C.F.; Donato, J. Inactivation of SOCS3 in leptin receptor-expressing cells protects mice from diet-induced insulin resistance but does not prevent obesity. Mol. Metab. 2014, 3, 608–618. [Google Scholar] [CrossRef]
- Heldsinger, A.; Grabauskas, G.; Wu, X.; Zhou, S.; Lu, Y.; Song, I.; Owyang, C. Ghrelin induces leptin resistance by activation of suppressor of cytokine signaling 3 expression in male rats: Implications in satiety regulation. Endocrinology 2014, 155, 3956–3969. [Google Scholar] [CrossRef] [Green Version]
- Piao, L.; Park, J.; Li, Y.; Shin, S.; Shin, S.; Kong, G.; Shrestha, R.; Tran, Q.; Hur, G.M.; Kim, J.L.; et al. SOCS3 and SOCS6 are required for the risperidone-mediated inhibition of insulin and leptin signaling in neuroblastoma cells. Int. J. Mol. Med. 2014, 33, 1364–1370. [Google Scholar] [CrossRef]
- Pedroso, J.A.B.; Ramos-Lobo, A.M.; Donato, J. SOCS3 as a future target to treat metabolic disorders. Hormones 2019, 18, 127–136. [Google Scholar] [CrossRef]
- Ericson, E.; Wennberg Huldt, C.; Strömstedt, M.; Brodin, P. A novel role of the checkpoint kinase ATR in leptin signaling. Mol. Cell. Endocrinol. 2015, 412, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Dodd, G.T.; Xirouchaki, C.E.; Eramo, M.; Mitchell, C.A.; Andrews, Z.B.; Henry, B.A.; Cowley, M.A.; Tiganis, T. Intranasal Targeting of Hypothalamic PTP1B and TCPTP Reinstates Leptin and Insulin Sensitivity and Promotes Weight Loss in Obesity. Cell Rep. 2019, 28, 2905–2922.e5. [Google Scholar] [CrossRef] [PubMed]
- Tsou, R.C.; Rak, K.S.; Zimmer, D.J.; Bence, K.K. Improved metabolic phenotype of hypothalamic PTP1B-deficiency is dependent upon the leptin receptor. Mol. Metab. 2014, 3, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Ozek, C.; Kanoski, S.E.; Zhang, Z.Y.; Grill, H.J.; Bence, K.K. Protein-tyrosine phosphatase 1B (PTP1B) is a novel regulator of central brain-derived neurotrophic factor and tropomyosin receptor kinase B (TrkB) signaling. J. Biol. Chem. 2014, 289, 31682–31692. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Y.; Mu, S.; Zhang, S.P.; Guo, W.; Li, Q.F.; Xiao, X.Q.; Zhang, J.; Wang, Z.H. Roux-en-Y gastric bypass surgery suppresses hypothalamic PTP1B protein level and alleviates leptin resistance in obese rats. Exp. Ther. Med. 2017, 14, 2536–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alosco, M.L.; Spitznagel, M.B.; Strain, G.; Devlin, M.; Cohen, R.; Crosby, R.D.; Mitchell, J.E.; Gunstad, J. Improved serum leptin and ghrelin following bariatric surgery predict better postoperative cognitive function. J. Clin. Neurol. 2015, 11, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; Dodd, G.T.; Tiganis, T. Protein Tyrosine Phosphatases in Hypothalamic Insulin and Leptin Signaling. Trends Pharmacol. Sci. 2015, 36, 661–674. [Google Scholar] [CrossRef] [Green Version]
- Verma, M.; Gupta, S.J.; Chaudhary, A.; Garg, V.K. Protein tyrosine phosphatase 1B inhibitors as antidiabetic agents—A brief review. Bioorg. Chem. 2017, 70, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Konidaris, K.F.; Gasser, G.; Tonks, N.K. A potent, selective, and orally bioavailable inhibitor of the protein-tyrosine phosphatase PTP1B improves insulin and leptin signaling in animal models. J. Biol. Chem. 2018, 293, 1517–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derkach, K.V.; Zakharova, I.O.; Romanova, I.V.; Zorina, I.I.; Mikhrina, A.L.; Shpakov, A.O. Metabolic parameters and functional state of hypothalamic signaling systems in A Y/a mice with genetic predisposition to obesity and the effect of metformin. Dokl. Biochem. Biophys. 2017, 477, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Fukuda, S.; Sakata, S.; Morinaga, H.; Ohta, T. Pharmacological effects of JTT-551, a novel protein tyrosine phosphatase 1B inhibitor, in diet-induced obesity mice. J. Diabetes Res. 2014, 2014, 680348. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Fukui, M.; Kanda, M.; Morishita, K.; Shoji, Y.; Kitao, T.; Hinoi, E.; Shirahase, H. Therapeutic effects of the allosteric protein tyrosine phosphatase 1B inhibitor KY-226 on experimental diabetes and obesity via enhancements in insulin and leptin signaling in mice. J. Pharmacol. Sci. 2018, 137, 38–46. [Google Scholar] [CrossRef]
- Kaneko, K.; Fu, Y.; Lin, H.Y.; Cordonier, E.L.; Mo, Q.; Gao, Y.; Yao, T.; Naylor, J.; Howard, V.; Saito, K.; et al. Gut-derived GIP activates central Rap1 to impair neural leptin sensitivity during overnutrition. J. Clin. Investig. 2019, 129, 3786–3791. [Google Scholar] [CrossRef] [Green Version]
- Hurley, M.M.; Anderson, E.M.; Chen, C.; Maunze, B.; Hess, E.M.; Block, M.E.; Patel, N.; Cooper, Z.; McCoy, R.; Dabra, T.; et al. Acute Blockade of PACAP-Dependent Activity in the Ventromedial Nucleus of the Hypothalamus Disrupts Leptin-Induced Behavioral and Molecular Changes in Rats. Neuroendocrinology 2020, 110, 271–281. [Google Scholar] [CrossRef]
- Dhar-Mascareno, M.; Ramirez, S.N.; Rozenberg, I.; Rouille, Y.; Kral, J.G.; Mascareno, E.J. Hexim1, a Novel Regulator of Leptin Function, Modulates Obesity and Glucose Disposal. Mol. Endocrinol. 2016, 30, 314–324. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; McKnight, G.S. Hypothalamic PKA regulates leptin sensitivity and adiposity. Nat. Commun. 2015, 6, 8237. [Google Scholar] [CrossRef] [Green Version]
- Sahu, M.; Anamthathmakula, P.; Sahu, A. Phosphodiesterase-3B-cAMP pathway of leptin signalling in the hypothalamus is impaired during the development of diet-induced obesity in FVB/N mice. J. Neuroendocrinol. 2015, 27, 293–302. [Google Scholar] [CrossRef]
- Christensen, A.; Pike, C.J. APOE genotype affects metabolic and Alzheimer-related outcomes induced by Western diet in female EFAD mice. FASEB J. 2019, 33, 4054–4066. [Google Scholar] [CrossRef]
- Rai, S.N.; Singh, C.; Singh, A.; Singh, M.P.; Singh, B.K. Mitochondrial Dysfunction: A Potential Therapeutic Target to Treat Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 3075–3088. [Google Scholar] [CrossRef]
- Rai, Y.; Anita; Kumari, N.; Singh, S.; Kalra, N.; Soni, R.; Bhatt, A.N. Mild mitochondrial uncoupling protects from ionizing radiation induced cell death by attenuating oxidative stress and mitochondrial damage. Biochim. Biophys. Acta-Bioenerg. 2021, 1862, 148325. [Google Scholar] [CrossRef]
- Singh, A.K.; Rai, S.N.; Maurya, A.; Mishra, G.; Awasthi, R.; Shakya, A.; Chellappan, D.K.; Dua, K.; Vamanu, E.; Chaudhary, S.K.; et al. Therapeutic Potential of Phytoconstituents in Management of Alzheimer’s Disease. Evid. Based. Complement. Alternat. Med. 2021, 2021, 5578574. [Google Scholar] [CrossRef]
- Tripathi, P.N.; Srivastava, P.; Sharma, P.; Tripathi, M.K.; Seth, A.; Tripathi, A.; Rai, S.N.; Singh, S.P.; Shrivastava, S.K. Biphenyl-3-oxo-1,2,4-triazine linked piperazine derivatives as potential cholinesterase inhibitors with anti-oxidant property to improve the learning and memory. Bioorg. Chem. 2019, 85, 82–96. [Google Scholar] [CrossRef]
- Srivastava, P.; Tripathi, P.N.; Sharma, P.; Rai, S.N.; Singh, S.P.; Srivastava, R.K.; Shankar, S.; Shrivastava, S.K. Design and development of some phenyl benzoxazole derivatives as a potent acetylcholinesterase inhibitor with antioxidant property to enhance learning and memory. Eur. J. Med. Chem. 2019, 163, 116–135. [Google Scholar] [CrossRef]
- Kang, S.; Lee, Y.H.; Lee, J.E. Metabolism-Centric Overview of the Pathogenesis of Alzheimer’s Disease. Yonsei Med. J. 2017, 58, 479–488. [Google Scholar] [CrossRef]
- Folch, J.; Patraca, I.; Martínez, N.; Pedrós, I.; Petrov, D.; Ettcheto, M.; Abad, S.; Marin, M.; Beas-Zarate, C.; Camins, A. The role of leptin in the sporadic form of Alzheimer’s disease. Interactions with the adipokines amylin, ghrelin and the pituitary hormone prolactin. Life Sci. 2015, 140, 19–28. [Google Scholar] [CrossRef]
- Pedrós, I.; Petrov, D.; Artiach, G.; Abad, S.; Ramon-Duaso, C.; Sureda, F.; Pallàs, M.; Beas-Zarate, C.; Folch, J.; Camins, A. Adipokine pathways are altered in hippocampus of an experimental mouse model of Alzheimer’s disease. J. Nutr. Health Aging 2015, 19, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.N.N.; Lima-Filho, R.A.S.; De Felice, F.G. Connecting Alzheimer’s disease to diabetes: Underlying mechanisms and potential therapeutic targets. Neuropharmacology 2018, 136, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Maioli, S.; Lodeiro, M.; Merino-Serrais, P.; Falahati, F.; Khan, W.; Puerta, E.; Codita, A.; Rimondini, R.; Ramirez, M.J.; Simmons, A.; et al. Alterations in brain leptin signalling in spite of unchanged CSF leptin levels in Alzheimer’s disease. Aging Cell 2015, 14, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Stone, J.G.; Torres, S.L.; Siedlak, S.L.; Perry, G.; Kryscio, R.; Jicha, G.; Casadesus, G.; Smith, M.A.; Zhu, X.; et al. Dysregulation of leptin signaling in Alzheimer disease: Evidence for neuronal leptin resistance. J. Neurochem. 2014, 128, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Bik, A.; Bik, W.; Styczynska, M.; Chodakowska-Zebrowska, M.; Barcikowska, M.; Wolinska-Witort, E.; Kalisz, M.; Martynska, L.; Baranowska, B. Plasma leptin levels and free leptin index in women with Alzheimer’s disease. Neuropeptides 2015, 52, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Iadecola, C. Adipocyte-derived factors in age-related dementia and their contribution to vascular and Alzheimer pathology. Biochim. Biophys. Acta 2016, 1862, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Albala, C.; Angel, B.; Lera, L.; Sanchez, H.; Marquez, C.; Fuentes, P. Low Leptin Availability as a Risk Factor for Dementia in Chilean Older People. Dement. Geriatr. Cogn. Dis. Extra 2016, 6, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.; Hu, W.; Fardo, D.; Greco, S.; Perry, G.; Montine, T.; Trojanowski, J.; Shaw, L.; Ashford, J.; Tezapsidis, N. Low plasma leptin in cognitively impaired ADNI subjects: Gender differences and diagnostic and therapeutic potential. Curr. Alzheimer Res. 2014, 11, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, M.; Wang, G.; Racchumi, G.; Dyke, J.P.; Iadecola, C. Transgenic mice overexpressing amyloid precursor protein exhibit early metabolic deficits and a pathologically low leptin state associated with hypothalamic dysfunction in arcuate neuropeptide Y neurons. J. Neurosci. 2014, 34, 9096–9106. [Google Scholar] [CrossRef] [PubMed]
- Ülker, M.; Kenangil, G. The Relation of Circulating Levels of Leptin with Cognition in Patients with Alzheimer’s Disease. Noro Psikiyatr. Ars. 2018, 55, 211–214. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Van Der Flier, W.M.; Scheltens, P.; Duits, A.; Wijnstok, N.; Nijpels, G.; Dekker, J.M.; Blankenstein, R.M.A.; Heijboer, A.C. Serum leptin is not altered nor related to cognitive decline in Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Wan, Z.; Mah, D.; Simtchouk, S.; Kluftinger, A.; Little, J.P. Role of amyloid β in the induction of lipolysis and secretion of adipokines from human adipose tissue. Adipocyte 2014, 4, 212–216. [Google Scholar] [CrossRef] [Green Version]
- Yeh, S.H.H.; Shie, F.S.; Liu, H.K.; Yao, H.H.; Kao, P.C.; Lee, Y.H.; Chen, L.M.; Hsu, S.M.; Chao, L.J.; Wu, K.W.; et al. A high-sucrose diet aggravates Alzheimer’s disease pathology, attenuates hypothalamic leptin signaling, and impairs food-anticipatory activity in APPswe/PS1dE9 mice. Neurobiol. Aging 2020, 90, 60–74. [Google Scholar] [CrossRef]
- Meakin, P.J.; Jalicy, S.M.; Montagut, G.; Allsop, D.J.P.; Cavellini, D.L.; Irvine, S.W.; McGinley, C.; Liddell, M.K.; McNeilly, A.D.; Parmionova, K.; et al. Bace1-dependent amyloid processing regulates hypothalamic leptin sensitivity in obese mice. Sci. Rep. 2018, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Hsu, H.C.; Kao, P.C.; Shiao, Y.J.; Yeh, S.H.H.; Shie, F.S.; Hsu, S.M.; Yeh, C.W.; Liu, H.K.; Yang, S.B.; et al. Augmented Insulin and Leptin Resistance of High Fat Diet-Fed APPswe/PS1dE9 Transgenic Mice Exacerbate Obesity and Glycemic Dysregulation. Int. J. Mol. Sci. 2018, 19, 2333. [Google Scholar] [CrossRef] [Green Version]
- Vieira, M.N.N.; Lyra e Silva, N.M.; Ferreira, S.T.; De Felice, F.G. Protein Tyrosine Phosphatase 1B (PTP1B): A Potential Target for Alzheimer’s Therapy? Front. Aging Neurosci. 2017, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, M.G. Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog. Horm. Res. 2004, 59, 287–304. [Google Scholar] [CrossRef] [PubMed]
- Bjørbæk, C.; Elmquist, J.K.; Frantz, J.D.; Shoelson, S.E.; Flier, J.S. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol. Cell 1998, 1, 619–625. [Google Scholar] [CrossRef]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Ishida-Takahashi, R.; Villanueva, E.C.; Fingar, D.C.; Münzberg, H.; Myers, M.G. The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J. Biol. Chem. 2007, 282, 31019–31027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, L.R.; Farruggella, T.J.; Symes, A.; Karow, M.L.; Yancopoulos, G.D.; Stahl, N. Enhancing leptin response by preventing SH2-containing phosphatase 2 interaction with Ob receptor. Proc. Natl. Acad. Sci. USA 1998, 95, 6061–6066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Bereczki, E.; Zhang, H.; Wang, S.; Li, C.; Ji, X.; Branca, R.M.; Lehtiö, J.; Guan, Z.; Filipcik, P.; et al. Mammalian target of rapamycin (mTor) mediates tau protein dyshomeostasis: Implication for Alzheimer disease. J. Biol. Chem. 2013, 288, 15556–15570. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Cheng, S.; Yin, Z.; Xu, C.; Lu, S.; Hou, J.; Yu, T.; Zhu, X.; Zou, X.; Peng, Y.; et al. Conditional inactivation of Akt three isoforms causes tau hyperphosphorylation in the brain. Mol. Neurodegener. 2015, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Wang, Z.H.; Qu, M.; Gao, D.; Liu, X.P.; Zhu, L.Q.; Wang, J.Z. Stimulation of EphB2 attenuates tau phosphorylation through PI3K/Akt-mediated inactivation of glycogen synthase kinase-3β. Sci. Rep. 2015, 5, srep11765. [Google Scholar] [CrossRef] [Green Version]
- Uotani, S.; Abe, T.; Yamaguchi, Y. Leptin activates AMP-activated protein kinase in hepatic cells via a JAK2-dependent pathway. Biochem. Biophys. Res. Commun. 2006, 351, 171–175. [Google Scholar] [CrossRef]
- Greco, S.J.; Hamzelou, A.; Johnston, J.M.; Smith, M.A.; Ashford, J.W.; Tezapsidis, N. Leptin boosts cellular metabolism by activating AMPK and the sirtuins to reduce tau phosphorylation and β-amyloid in neurons. Biochem. Biophys. Res. Commun. 2011, 414, 170–174. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Li, J.J.; Diao, S.; Kwak, Y.D.; Liu, L.; Zhi, L.; Büeler, H.; Bhat, N.R.; Williams, R.W.; Park, E.A.; et al. Metabolic stress modulates Alzheimer’s β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metab. 2013, 17, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Marwarha, G.; Raza, S.; Meiers, C.; Ghribi, O. Leptin attenuates BACE1 expression and amyloid-β genesis via the activation of SIRT1 signaling pathway. Biochim. Biophys. Acta 2014, 1842, 1587–1595. [Google Scholar] [CrossRef] [Green Version]
- Cook, W.S.; Unger, R.H. Protein tyrosine phosphatase 1B: A potential leptin resistance factor of obesity. Dev. Cell 2002, 2, 385–387. [Google Scholar] [CrossRef] [Green Version]
- King, A.; Brain, A.; Hanson, K.; Dittmann, J.; Vickers, J.; Fernandez-Martos, C. Disruption of leptin signalling in a mouse model of Alzheimer’s disease. Metab. Brain Dis. 2018, 33, 1097–1110. [Google Scholar] [CrossRef]
- Dragano, N.R.V.; Haddad-Tovolli, R.; Velloso, L.A. Leptin, Neuroinflammation and Obesity. Front. Horm. Res. 2017, 48, 84–96. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Cordero, J.A.; Pérez-Pérez, A.; Jiménez-Cortegana, C.; Alba, G.; Flores-Barragán, A.; Sánchez-Margalet, V. Obesity as a Risk Factor for Dementia and Alzheimer’s Disease: The Role of Leptin. Int. J. Mol. Sci. 2022, 23, 5202. https://doi.org/10.3390/ijms23095202
Flores-Cordero JA, Pérez-Pérez A, Jiménez-Cortegana C, Alba G, Flores-Barragán A, Sánchez-Margalet V. Obesity as a Risk Factor for Dementia and Alzheimer’s Disease: The Role of Leptin. International Journal of Molecular Sciences. 2022; 23(9):5202. https://doi.org/10.3390/ijms23095202
Chicago/Turabian StyleFlores-Cordero, Juan Antonio, Antonio Pérez-Pérez, Carlos Jiménez-Cortegana, Gonzalo Alba, Alfonso Flores-Barragán, and Víctor Sánchez-Margalet. 2022. "Obesity as a Risk Factor for Dementia and Alzheimer’s Disease: The Role of Leptin" International Journal of Molecular Sciences 23, no. 9: 5202. https://doi.org/10.3390/ijms23095202
APA StyleFlores-Cordero, J. A., Pérez-Pérez, A., Jiménez-Cortegana, C., Alba, G., Flores-Barragán, A., & Sánchez-Margalet, V. (2022). Obesity as a Risk Factor for Dementia and Alzheimer’s Disease: The Role of Leptin. International Journal of Molecular Sciences, 23(9), 5202. https://doi.org/10.3390/ijms23095202