
Dormancy in Breast Cancer, the Role of Autophagy, lncRNAs, miRNAs and Exosomes


Abstract
:1. The Molecular Characteristics of Dormant Breast Cancer Cells
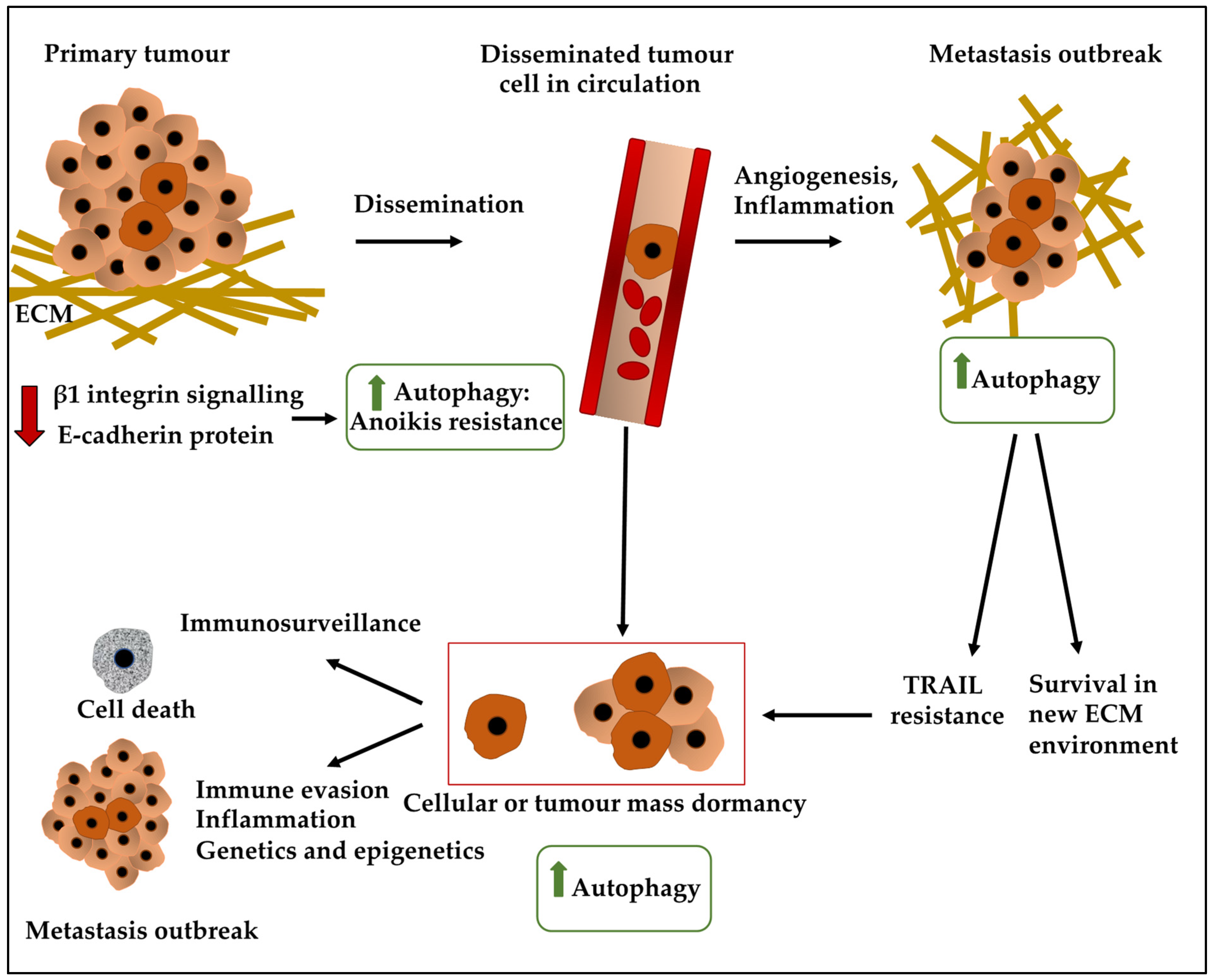
2. Dormancy in BC Cells
3. The Role of the Molecular Process of Autophagy in BC Dormancy and Treatment
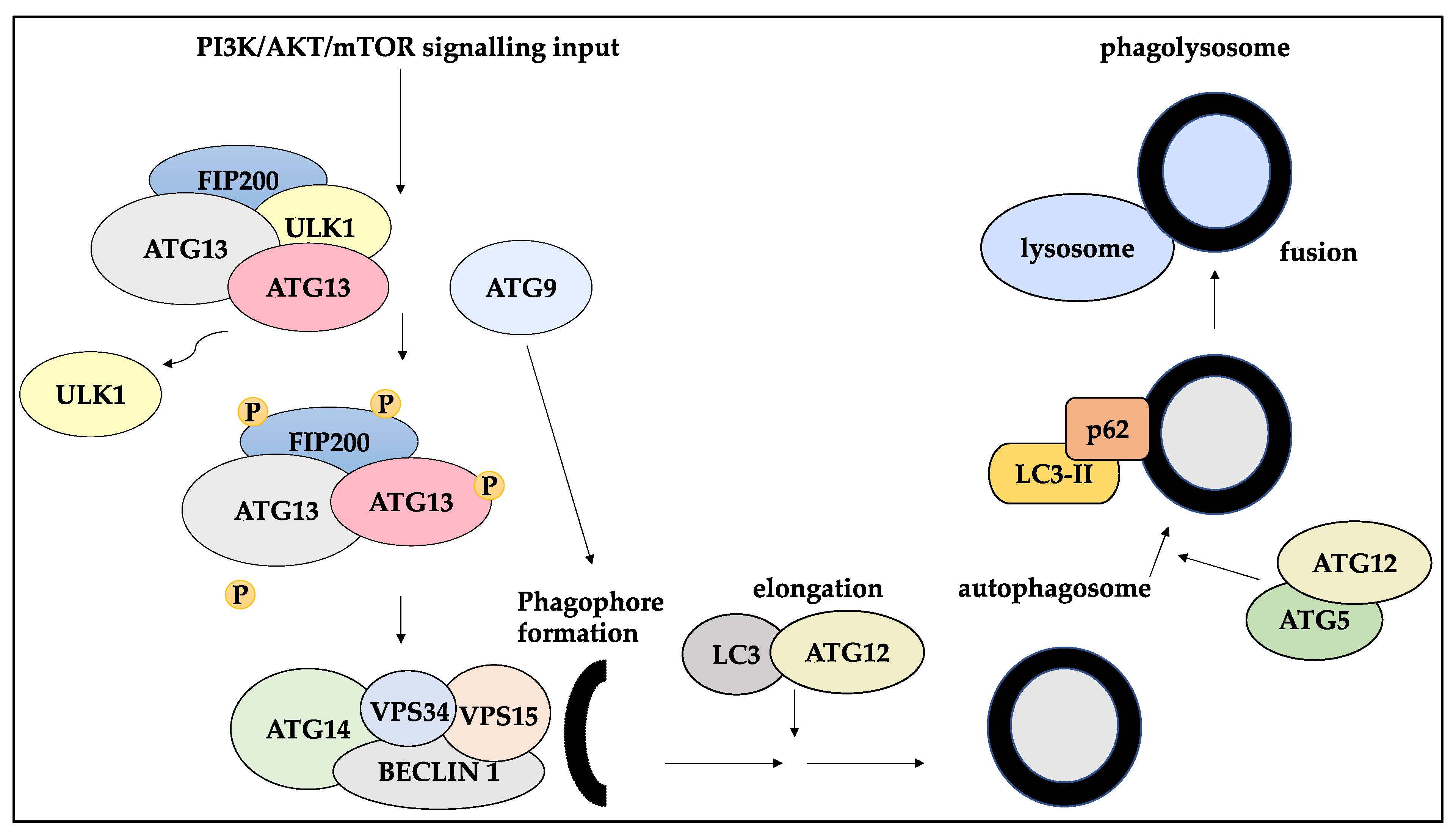
3.1. The Role of Autophagy and Autophagy-Related Proteins and Players in BC
3.2. Autophagy and Dormancy
3.3. Autophagy, Apoptosis, and Dormancy
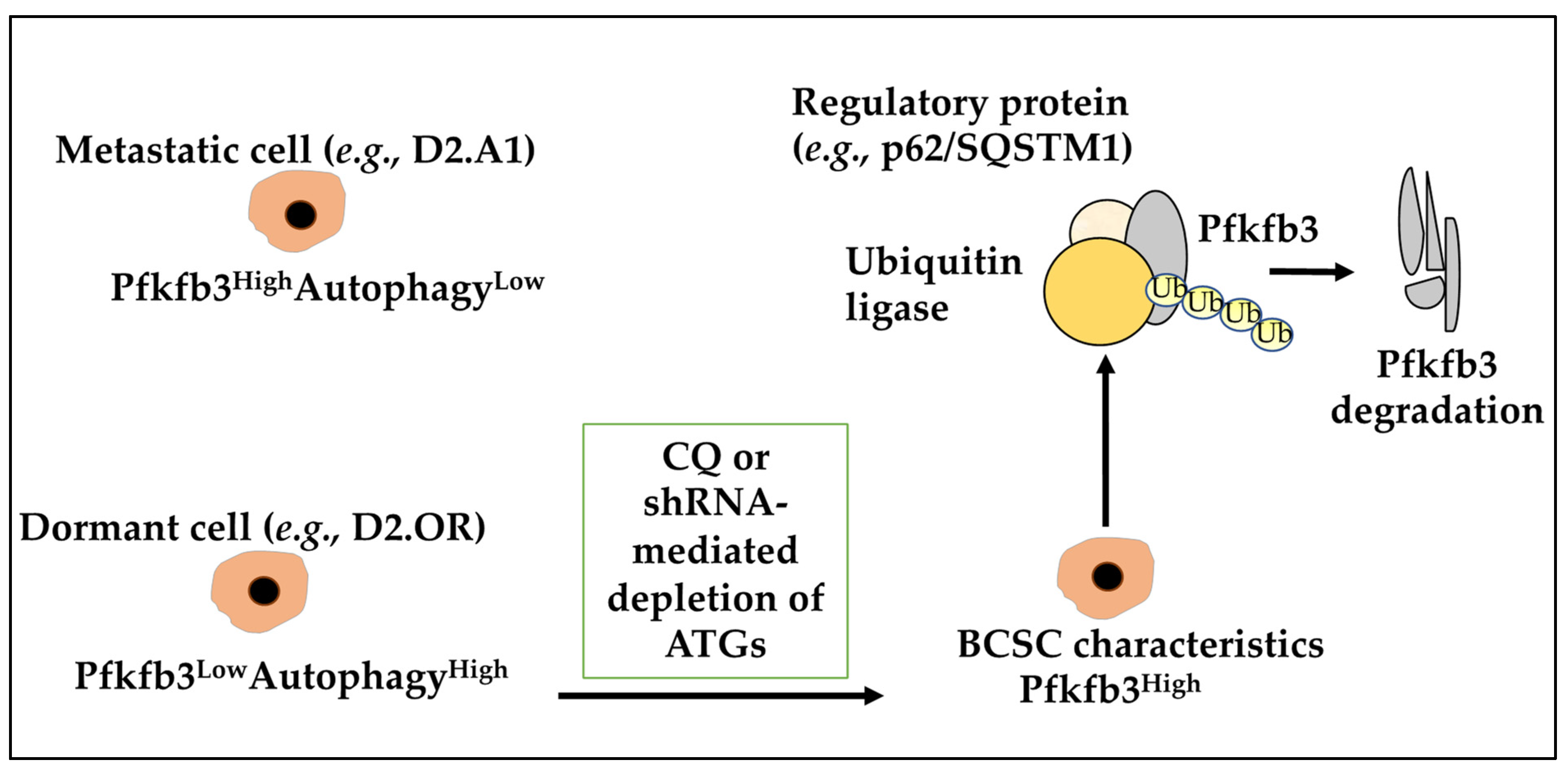
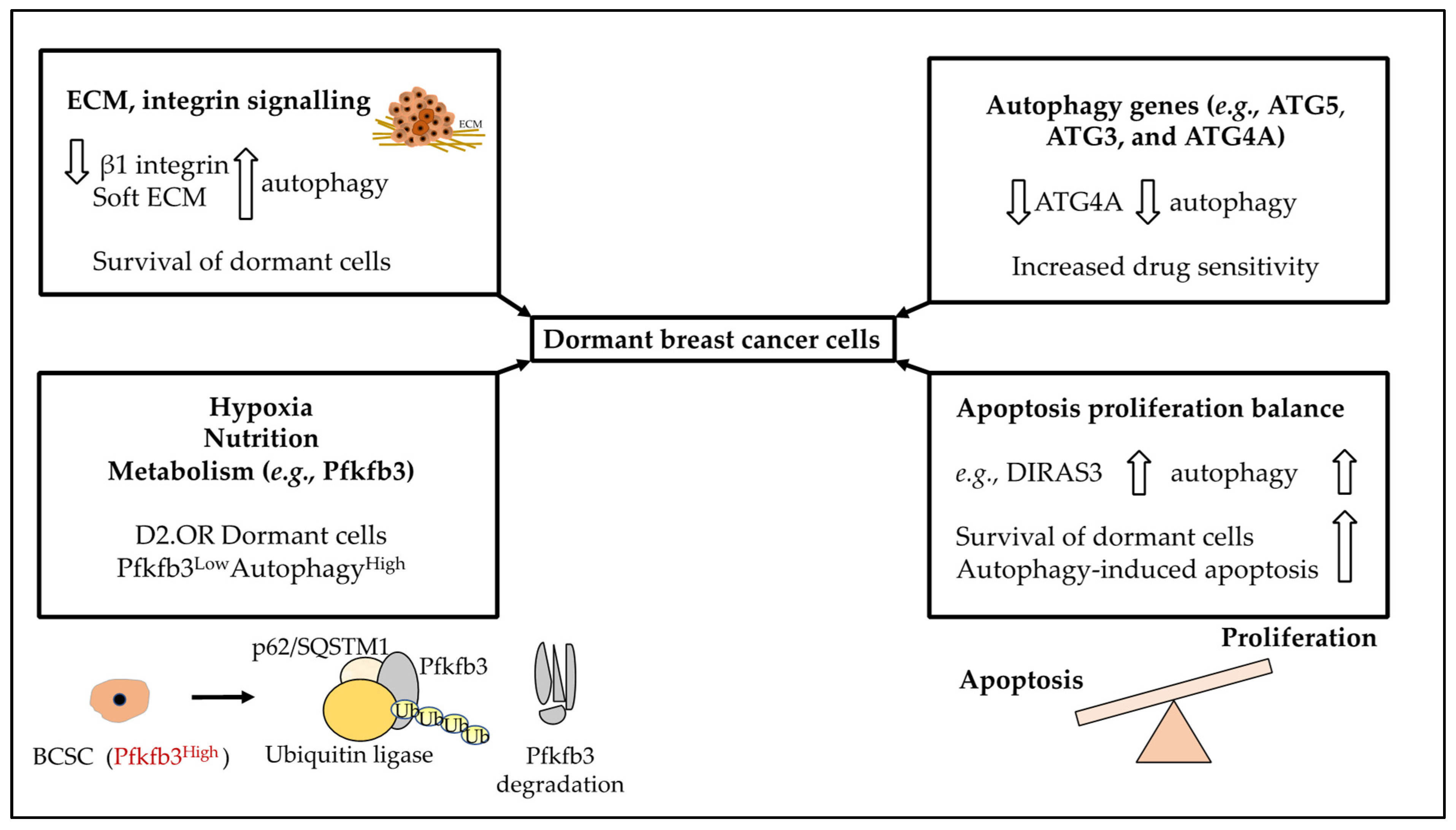
3.4. Autophagy, Metabolism, Nutrition, Hypoxia, and Dormancy
3.5. Autophagy, ECM, and Dormancy
3.6. Inhibiting Autophagy as a Therapeutic Strategy in BC
3.7. Autophagy, Dormancy and BC Treatment
4. The Role of lncRNAs, Exosomes and miRNAs in BC Dormancy
4.1. The Role of lncRNAs and miRNAs in BC Dormancy
4.2. The Role of lncRNAs and miRNAs in BC Dormant Cell Awakening
4.3. The Role of Exosomes in BC Dormancy
4.4. DNA Repair, Exosomes, and BC Dormancy
5. Discussion; Aspects of Autophagic Processes, lncRNAs and miRNAs for Targeted Therapy in BC
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALDH | Aldehyde dehydrogenase |
| AMPK | AMP-activated protein kinase |
| ARH1 | Aplasia Ras homolog member I |
| ATG | Autophagy-related |
| BER | Basic excision repair (BER) |
| BM | Bone marrow |
| BM-MSCs | Bone marrow mesenchymal stem cells |
| BC | Breast cancer |
| BCSC | Breast cancer stem cells |
| C.B-17/Icr-scid/scidJc1mice | Severe combined immunodeficient mice |
| CQ | Chloroquine |
| CSCs | Cancer stem cells |
| DMFS | Distant metastasis-free survival |
| DNMT1 | DNA methyltransferase 1 |
| DTCs | Disseminated tumour cells |
| EC | Endothelial cell |
| ECM | Extracellular matrix |
| EDG2 | Lysophosphatidic acid receptor |
| ELEANORS | ESR1 locus enhancing and activating non-coding RNAs |
| EMT | Epithelial to mesenchymal transition |
| EphA5 | Eph receptor 5 |
| ER | Oestrogen receptor |
| FTIs | Farnesyl transferase inhibitors |
| HER2 | Human epidermal growth factor receptor 2 |
| IGF-1 | Insulin growth factor-1 |
| LncRNAs | Long non-coding RNAs |
| MALAT1 | Metastasis-associated lung adenocarcinoma transcript 1 |
| MHC | Major histocompatibility complex |
| miRNAs | Micro RNAs |
| MMC | Mouse mammary carcinoma |
| MMTV-PyMT | mouse mammary tumour virus-polyoma middle tumour-antigen |
| MRD | Minimal residual disease |
| NETs | Neutrophil extracellular traps |
| OS | Overall survival |
| Pfkfb3 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 |
| PR | Progesterone receptor |
| PTX | Pentoxifylline |
| ROS | Reactive oxygen species |
| SIM | Simvastatin |
| TET | Ten-eleven translocation |
| TME | Tumour microenvironment |
| TNBC | Triple-negative breast cancer |
| TRAIL | TNF-alpha-Related Apoptosis-Inducing Ligand |
| TSP1 | Thrombospondin-1 |
References
- Maughan, K.L.; Lutterbie, M.A.; Ham, P.S. Treatment of breast cancer. Am. Fam. Physician 2010, 81, 1339–1346. [Google Scholar] [PubMed]
- Romond, E.H.; Perez, E.A.; Bryant, J.; Suman, V.J.; Geyer, C.E.J.; Davidson, N.E.; Tan-Chiu, E.; Martino, S.; Paik, S.; Kaufman, P.A.; et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1673–1684. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.K.; Guan, J.-L. Breast Cancer: Multiple Subtypes within a Tumor? Trends Cancer 2017, 3, 753–760. [Google Scholar] [CrossRef]
- Niklaus, N.J.; Tokarchuk, I.; Zbinden, M.; Schläfli, A.M.; Maycotte, P.; Tschan, M.P. The Multifaceted Functions of Autophagy in Breast Cancer Development and Treatment. Cells 2021, 10, 1447. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Muñoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. S2), S26–S35. [Google Scholar] [CrossRef] [Green Version]
- Rivenbark, A.G.; O’Connor, S.M.; Coleman, W.B. Molecular and Cellular Heterogeneity in Breast Cancer: Challenges for Personalized Medicine. Am. J. Pathol. 2013, 183, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Morrow, M.; Strom, E.A.; Bassett, L.W.; Dershaw, D.D.; Fowble, B.; Giuliano, A.; Harris, J.R.; O’Malley, F.; Schnitt, S.J.; Singletary, S.E.; et al. Standard for breast conservation therapy in the management of invasive breast carcinoma. CA Cancer J. Clin. 2002, 52, 277–300. [Google Scholar] [CrossRef]
- Darby, S.; McGale, P.; Correa, C.; Taylor, C.; Arriagada, R.; Clarke, M.; Cutter, D.; Davies, C.; Ewertz, M.; Godwin, J.; et al. Effect of radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year breast cancer death: Meta-analysis of individual patient data for 10,801 women in 17 randomised trials. Lancet 2011, 378, 1707–1716. [Google Scholar] [CrossRef] [Green Version]
- Buchholz, T.A.; Hill, B.S.; Tucker, S.L.; Frye, D.K.; Kuerer, H.M.; Buzdar, A.U.; McNeese, M.D.; Singletary, S.E.; Ueno, N.T.; Pusztai, L.; et al. Factors predictive of outcome in patients with breast cancer refractory to neoadjuvant chemotherapy. Cancer J. 2001, 7, 413–420. [Google Scholar]
- Mieog, J.S.D.; van der Hage, J.A.; van de Velde, C.J.H. Preoperative chemotherapy for women with operable breast cancer. Cochrane Database Syst. Rev. 2007, 2007, CD005002. [Google Scholar] [CrossRef]
- Beriwal, S.; Schwartz, G.F.; Komarnicky, L.; Garcia-Young, J.A. Breast-conserving therapy after neoadjuvant chemotherapy: Long-term results. Breast J. 2006, 12, 159–164. [Google Scholar] [CrossRef]
- Aguiar Bujanda, D.; Bohn Sarmiento, U.; Cabrera Suárez, M.A.; Pavcovich Ruiz, M.; Limeres González, M.A.; Aguiar Morales, J. Epirubicin, cyclophosphamide and weekly paclitaxel as neoadjuvant chemotherapy for stage II and III breast cancer. J. Cancer Res. Clin. Oncol. 2006, 132, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, T.A.; Mittendorf, E.A.; Hunt, K.K. Surgical Considerations after Neoadjuvant Chemotherapy: Breast Conservation Therapy. J. Natl. Cancer Inst. Monogr. 2015, 2015, 11–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, I.; Procter, M.; Gelber, R.D.; Guillaume, S.; Feyereislova, A.; Dowsett, M.; Goldhirsch, A.; Untch, M.; Mariani, G.; Baselga, J.; et al. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: A randomised controlled trial. Lancet 2007, 369, 29–36. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C.; Riethdorf, S. Cancer micrometastases. Nat. Rev. Clin. Oncol. 2009, 6, 339–351. [Google Scholar] [CrossRef]
- Dasgupta, A.; Lim, A.R.; Ghajar, C.M. Circulating and disseminated tumor cells: Harbingers or initiators of metastasis? Mol. Oncol. 2017, 11, 40–61. [Google Scholar] [CrossRef] [Green Version]
- Puig, I.; Tenbaum, S.P.; Chicote, I.; Arqués, O.; Martínez-Quintanilla, J.; Cuesta-Borrás, E.; Ramírez, L.; Gonzalo, P.; Soto, A.; Aguilar, S.; et al. TET2 controls chemoresistant slow-cycling cancer cell survival and tumor recurrence. J. Clin. Investig. 2018, 128, 3887–3905. [Google Scholar] [CrossRef]
- Santos-de-Frutos, K.; Djouder, N. When dormancy fuels tumour relapse. Commun. Biol. 2021, 4, 747. [Google Scholar] [CrossRef]
- Ruth, J.R.; Pant, D.K.; Pan, T.-C.; Seidel, H.E.; Baksh, S.C.; Keister, B.A.; Singh, R.; Sterner, C.J.; Bakewell, S.J.; Moody, S.E.; et al. Cellular dormancy in minimal residual disease following targeted therapy. Breast Cancer Res. 2021, 23, 63. [Google Scholar] [CrossRef]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Goncalves, F. Bone Metastases: An Overview. Oncol. Rev. 2017, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.A. Cancer progression and the invisible phase of metastatic colonization. Nat. Rev. Cancer 2020, 20, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, P.; Wu, Q.; Fang, H.; Wang, Y.; Xiao, Y.; Cong, M.; Wang, T.; He, Y.; Ma, C.; et al. Long non-coding RNA NR2F1-AS1 induces breast cancer lung metastatic dormancy by regulating NR2F1 and ΔNp63. Nat. Commun. 2021, 12, 5232. [Google Scholar] [CrossRef] [PubMed]
- Gomis, R.R.; Gawrzak, S. Tumor cell dormancy. Mol. Oncol. 2017, 11, 62–78. [Google Scholar] [CrossRef] [Green Version]
- Jahangiri, L.; Ishola, T.; Pucci, P.; Trigg, R.M.; Pereira, J.; Williams, J.A.; Cavanagh, M.L.; Gkoutos, G.V.; Tsaprouni, L.; Turner, S.D. The Role of Autophagy and lncRNAs in the Maintenance of Cancer Stem Cells. Cancers 2021, 13, 1239. [Google Scholar] [CrossRef]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. In Cancer Stem Cells; Academic Press: Cambridge, MA, USA, 2019; Volume 141, Chapter Two; pp. 43–84. [Google Scholar]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Fehm, T.; Banys, M.; Rack, B.; Janni, W.; Marth, C.; Blassl, C.; Hartkopf, A.; Trope, C.; Kimmig, R.; Krawczyk, N.; et al. Pooled analysis of the prognostic relevance of disseminated tumor cells in the bone marrow of patients with ovarian cancer. Int. J. Gynecol. Cancer 2013, 23, 839–845. [Google Scholar] [CrossRef]
- Schindlbeck, C.; Pfab, G.; Jueckstock, J.; Andergassen, U.; Sommer, H.; Janni, W.; Friese, K.; Rack, B. Prognostic relevance of disseminated tumor cells in the bone marrow of patients with primary breast cancer--results of a standardized follow-up. Anticancer Res. 2011, 31, 2749–2755. [Google Scholar]
- Folkman, J.; Kalluri, R. Cancer without disease. Nature 2004, 427, 787. [Google Scholar] [CrossRef]
- Manjili, M.H. Tumor Dormancy and Relapse: From a Natural Byproduct of Evolution to a Disease State. Cancer Res. 2017, 77, 2564–2569. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, A.B.; Schiemann, W.P. Autophagy in breast cancer metastatic dormancy: Tumor suppressing or tumor promoting functions? J. Cancer Metastasis Treat. 2019, 5, 43. [Google Scholar]
- Carcereri de Prati, A.; Butturini, E.; Rigo, A.; Oppici, E.; Rossin, M.; Boriero, D.; Mariotto, S. Metastatic Breast Cancer Cells Enter into Dormant State and Express Cancer Stem Cells Phenotype Under Chronic Hypoxia. J. Cell. Biochem. 2017, 118, 3237–3248. [Google Scholar] [CrossRef]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Whelan, K.A.; Chandramouleeswaran, P.M.; Tanaka, K.; Natsuizaka, M.; Guha, M.; Srinivasan, S.; Darling, D.S.; Kita, Y.; Natsugoe, S.; Winkler, J.D.; et al. Autophagy supports generation of cells with high CD44 expression via modulation of oxidative stress and Parkin-mediated mitochondrial clearance. Oncogene 2017, 36, 4843–4858. [Google Scholar] [CrossRef] [Green Version]
- Patel, L.R.; Camacho, D.F.; Shiozawa, Y.; Pienta, K.J.; Taichman, R.S. Mechanisms of cancer cell metastasis to the bone: A multistep process. Future Oncol. 2011, 7, 1285–1297. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.L.; Shajahan, A.N.; Clarke, R. Autophagy and endocrine resistance in breast cancer. Expert Rev. Anticancer Ther. 2011, 11, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Calle, A.; Kawamura, Y.; Yamamoto, Y.; Takeshita, F.; Ochiya, T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. 2018, 109, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Bobrie, A.; Krumeich, S.; Reyal, F.; Recchi, C.; Moita, L.F.; Seabra, M.C.; Ostrowski, M.; Théry, C. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012, 72, 4920–4930. [Google Scholar] [CrossRef] [Green Version]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, M.; Kosaka, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014, 7, ra63. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Fornace, A.J.J. p38 MAP kinase’s emerging role as a tumor suppressor. Adv. Cancer Res. 2004, 92, 95–118. [Google Scholar]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.-C.A.; Collins, J.W.; Nakayama, J.; Horak, C.E.; Liewehr, D.J.; Steinberg, S.M.; Albaugh, M.; Vidal-Vanaclocha, F.; Palmieri, D.; Barbier, M.; et al. Effect of Inhibition of the Lysophosphatidic Acid Receptor 1 on Metastasis and Metastatic Dormancy in Breast Cancer. JNCI J. Natl. Cancer Inst. 2012, 104, 1306–1319. [Google Scholar] [CrossRef]
- Gimbrone, M.A.J.; Leapman, S.B.; Cotran, R.S.; Folkman, J. Tumor dormancy in vivo by prevention of neovascularization. J. Exp. Med. 1972, 136, 261–276. [Google Scholar] [CrossRef]
- Clements, M.E.; Johnson, R.W. Breast Cancer Dormancy in Bone. Curr. Osteoporos. Rep. 2019, 17, 353–361. [Google Scholar] [CrossRef]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; de Stanchina, E.; Massagué, J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agudo, J.; Park, E.S.; Rose, S.A.; Alibo, E.; Sweeney, R.; Dhainaut, M.; Kobayashi, K.S.; Sachidanandam, R.; Baccarini, A.; Merad, M.; et al. Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity 2018, 48, 271–285.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, A.; Roser, C.T.; El-Far, M.H.; Savanur, V.H.; Eljarrah, A.; Gergues, M.; Kra, J.A.; Etchegaray, J.-P.; Rameshwar, P. Hypoxia-mediated changes in bone marrow microenvironment in breast cancer dormancy. Cancer Lett. 2020, 488, 9–17. [Google Scholar] [CrossRef]
- Naumov, G.N.; Bender, E.; Zurakowski, D.; Kang, S.-Y.; Sampson, D.; Flynn, E.; Watnick, R.S.; Straume, O.; Akslen, L.A.; Folkman, J.; et al. A Model of Human Tumor Dormancy: An Angiogenic Switch from the Nonangiogenic Phenotype. JNCI J. Natl. Cancer Inst. 2006, 98, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Almog, N.; Ma, L.; Raychowdhury, R.; Schwager, C.; Erber, R.; Short, S.; Hlatky, L.; Vajkoczy, P.; Huber, P.E.; Folkman, J.; et al. Transcriptional switch of dormant tumors to fast-growing angiogenic phenotype. Cancer Res. 2009, 69, 836–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. Immunogenic, cellular, and angiogenic drivers of tumor dormancy--a melanoma view. Pigment Cell Melanoma Res. 2016, 29, 27–42. [Google Scholar] [CrossRef] [Green Version]
- McGrath, J.; Panzica, L.; Ransom, R.; Withers, H.G.; Gelman, I.H. Identification of Genes Regulating Breast Cancer Dormancy in 3D Bone Endosteal Niche Cultures. Mol. Cancer Res. 2019, 17, 860–869. [Google Scholar] [CrossRef]
- Smid, M.; Wang, Y.; Zhang, Y.; Sieuwerts, A.M.; Yu, J.; Klijn, J.G.M.; Foekens, J.A.; Martens, J.W.M. Subtypes of Breast Cancer Show Preferential Site of Relapse. Cancer Res. 2008, 68, 3108–3114. [Google Scholar] [CrossRef] [Green Version]
- Spencer, V.A.; Costes, S.; Inman, J.L.; Xu, R.; Chen, J.; Hendzel, M.J.; Bissell, M.J. Depletion of nuclear actin is a key mediator of quiescence in epithelial cells. J. Cell Sci. 2011, 124, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Rubin, M.; Fenig, E.; DeBlasio, A.; Mendelsohn, J.; Yahalom, J.; Wieder, R. Basic fibroblast growth factor causes growth arrest in MCF-7 human breast cancer cells while inducing both mitogenic and inhibitory G1 events. Cancer Res. 1997, 57, 1750–1757. [Google Scholar] [PubMed]
- Korah, R.M.; Sysounthone, V.; Golowa, Y.; Wieder, R. Basic fibroblast growth factor confers a less malignant phenotype in MDA-MB-231 human breast cancer cells. Cancer Res. 2000, 60, 733–740. [Google Scholar]
- Korah, R.; Boots, M.; Wieder, R. Integrin α5β1 Promotes Survival of Growth-Arrested Breast Cancer Cells: An in Vitro Paradigm for Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2004, 64, 4514–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.; Zheng, Y.; Lam, H.-M.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat. Commun. 2015, 6, 6170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez Calle, A.; Yamamoto, T.; Kawamura, Y.; Hironaka-Mitsuhashi, A.; Ono, M.; Tsuda, H.; Shimomura, A.; Tamura, K.; Takeshita, F.; Ochiya, T.; et al. Long non-coding NR2F1-AS1 is associated with tumor recurrence in estrogen receptor-positive breast cancers. Mol. Oncol. 2020, 14, 2271–2287. [Google Scholar] [CrossRef] [PubMed]
- Boral, D.; Liu, H.N.; Kenney, S.R.; Marchetti, D. Molecular Interplay between Dormant Bone Marrow-Resident Cells (BMRCs) and CTCs in Breast Cancer. Cancers 2020, 12, 1626. [Google Scholar] [CrossRef]
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573–590. [Google Scholar] [CrossRef]
- Adam, A.P.; George, A.; Schewe, D.; Bragado, P.; Iglesias, B.V.; Ranganathan, A.C.; Kourtidis, A.; Conklin, D.S.; Aguirre-Ghiso, J.A. Computational Identification of a p38SAPK-Regulated Transcription Factor Network Required for Tumor Cell Quiescence. Cancer Res. 2009, 69, 5664–5672. [Google Scholar] [CrossRef] [Green Version]
- Onder, T.T.; Kara, N.; Cherry, A.; Sinha, A.U.; Zhu, N.; Bernt, K.M.; Cahan, P.; Marcarci, B.O.; Unternaehrer, J.; Gupta, P.B.; et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature 2012, 483, 598–602. [Google Scholar] [CrossRef]
- Kim, R.S.; Avivar-Valderas, A.; Estrada, Y.; Bragado, P.; Sosa, M.S.; Aguirre-Ghiso, J.A.; Segall, J.E. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS ONE 2012, 7, e35569. [Google Scholar] [CrossRef] [PubMed]
- Sowder, M.E.; Johnson, R.W. Bone as a Preferential Site for Metastasis. JBMR Plus 2019, 3, e10126. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient Low Doses of DNA-Demethylating Agents Exert Durable Antitumor Effects on Hematological and Epithelial Tumor Cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29 (Suppl. S16), 15–18. [Google Scholar] [CrossRef]
- Park, S.-Y.; Nam, J.-S. The force awakens: Metastatic dormant cancer cells. Exp. Mol. Med. 2020, 52, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Di Martino, J.S.; Akhter, T.; Bravo-Cordero, J.J. Remodeling the ECM: Implications for Metastasis and Tumor Dormancy. Cancers 2021, 13, 4916. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [Green Version]
- Zou, C.-F.; Jia, L.; Jin, H.; Yao, M.; Zhao, N.; Huan, J.; Lu, Z.; Bast, R.C.; Feng, Y.; Yu, Y. Re-expression of ARHI (DIRAS3) induces autophagy in breast cancer cells and enhances the inhibitory effect of paclitaxel. BMC Cancer 2011, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 2008, 118, 3917–3929. [Google Scholar] [CrossRef] [Green Version]
- Ovadia, E.M.; Pradhan, L.; Sawicki, L.A.; Cowart, J.E.; Huber, R.E.; Polson, S.W.; Chen, C.; van Golen, K.L.; Ross, K.E.; Wu, C.H.; et al. Understanding ER+ Breast Cancer Dormancy Using Bioinspired Synthetic Matrices for Long-Term 3D Culture and Insights into Late Recurrence. Adv. Biosyst. 2020, 4, e2000119. [Google Scholar] [CrossRef] [PubMed]
- La Belle Flynn, A.; Calhoun, B.C.; Sharma, A.; Chang, J.C.; Almasan, A.; Schiemann, W.P. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat. Commun. 2019, 10, 3668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, D.E.; Kurpios, N.A.; Zuo, D.; Hassell, J.A.; Blaess, S.; Mueller, U.; Muller, W.J. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell 2004, 6, 159–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Guadamillas, M.C.; Cerezo, A.; Del Pozo, M.A. Overcoming anoikis—Pathways to anchorage-independent growth in cancer. J. Cell Sci. 2011, 124 Pt 19, 3189–3197. [Google Scholar] [CrossRef] [Green Version]
- Anlaş, A.A.; Nelson, C.M. Soft Microenvironments Induce Chemoresistance by Increasing Autophagy Downstream of Integrin-Linked Kinase. Cancer Res. 2020, 80, 4103–4113. [Google Scholar] [CrossRef]
- Bildik, G.; Liang, X.; Sutton, M.N.; Bast, R.C.J.; Lu, Z. DIRAS3: An Imprinted Tumor Suppressor Gene that Regulates RAS and PI3K-driven Cancer Growth, Motility, Autophagy, and Tumor Dormancy. Mol. Cancer Ther. 2022, 21, 25–37. [Google Scholar] [CrossRef]
- Aqbi, H.F.; Tyutyunyk-Massey, L.; Keim, R.C.; Butler, S.E.; Thekkudan, T.; Joshi, S.; Smith, T.M.; Bandyopadhyay, D.; Idowu, M.O.; Bear, H.D.; et al. Autophagy-deficient breast cancer shows early tumor recurrence and escape from dormancy. Oncotarget 2018, 9, 22113–22122. [Google Scholar] [CrossRef] [Green Version]
- Gooding, A.J.; Zhang, B.; Jahanbani, F.K.; Gilmore, H.L.; Chang, J.C.; Valadkhan, S.; Schiemann, W.P. The lncRNA BORG Drives Breast Cancer Metastasis and Disease Recurrence. Sci. Rep. 2017, 7, 12698. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Gurrapu, S.; Han, H.; Wang, Y.; Bae, S.; Chen, H.; Wu, C.-J.; Giancotti, F. 750 Malat1 lncRNA controls metastatic reactivation of dormant breast cancer by immune evasion. J. Immunother. Cancer 2020, 8, A450–A451. [Google Scholar]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal stem cell-derived exosomes stimulate cycling quiescence and early breast cancer dormancy in bone marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef] [Green Version]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef] [Green Version]
- Vallabhaneni, K.C.; Penfornis, P.; Xing, F.; Hassler, Y.; Adams, K.V.; Mo, Y.-Y.; Watabe, K.; Pochampally, R. Stromal cell extracellular vesicular cargo mediated regulation of breast cancer cell metastasis via ubiquitin conjugating enzyme E2 N pathway. Oncotarget 2017, 8, 109861–109876. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Ghiso, J.A.; Liu, D.; Mignatti, A.; Kovalski, K.; Ossowski, L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol. Biol. Cell 2001, 12, 863–879. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.-H.; Liu, F.; Yang, Z.-L.; Fu, X.-H.; Yang, Z.-H.; Liu, Q.; Wang, L.; Wan, X.-B.; Fan, X.-J. Prognostic value of autophagy related proteins ULK1, Beclin 1, ATG3, ATG5, ATG7, ATG9, ATG10, ATG12, LC3B and p62/SQSTM1 in gastric cancer. Am. J. Transl. Res. 2016, 8, 3831–3847. [Google Scholar]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef]
- Tyutyunyk-Massey, L.; Gewirtz, D.A. Roles of autophagy in breast cancer treatment: Target, bystander or benefactor. Semin. Cancer Biol. 2020, 66, 155–162. [Google Scholar] [CrossRef]
- He, Y.; Zhao, X.; Subahan, N.R.; Fan, L.; Gao, J.; Chen, H. The prognostic value of autophagy-related markers beclin-1 and microtubule-associated protein light chain 3B in cancers: A systematic review and meta-analysis. Tumor Biol. 2014, 35, 7317–7326. [Google Scholar] [CrossRef]
- Grandvallet, C.; Feugeas, J.P.; Monnien, F.; Despouy, G.; Valérie, P.; Michaël, G.; Hervouet, E.; Peixoto, P. Autophagy is associated with a robust specific transcriptional signature in breast cancer subtypes. Genes Cancer 2020, 11, 154–168. [Google Scholar] [CrossRef]
- Tang, H.; Sebti, S.; Titone, R.; Zhou, Y.; Isidoro, C.; Ross, T.S.; Hibshoosh, H.; Xiao, G.; Packer, M.; Xie, Y.; et al. Decreased BECN1 mRNA Expression in Human Breast Cancer is Associated with Estrogen Receptor-Negative Subtypes and Poor Prognosis. EBioMedicine 2015, 2, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Deng, R.; Luo, R.-Z.; Shen, G.-P.; Cai, M.-Y.; Du, Z.-M.; Jiang, S.; Yang, M.-T.; Fu, J.-H.; Zhu, X.-F. Low expression of ULK1 is associated with operable breast cancer progression and is an adverse prognostic marker of survival for patients. Breast Cancer Res. Treat. 2012, 134, 549–560. [Google Scholar] [CrossRef]
- Lefort, S.; Joffre, C.; Kieffer, Y.; Givel, A.-M.; Bourachot, B.; Zago, G.; Bieche, I.; Dubois, T.; Meseure, D.; Vincent-Salomon, A.; et al. Inhibition of autophagy as a new means of improving chemotherapy efficiency in high-LC3B triple-negative breast cancers. Autophagy 2014, 10, 2122–2142. [Google Scholar] [CrossRef]
- Li, Q.; Zan, L. Knockdown of ATG4A inhibits breast cancer progression and promotes tamoxifen chemosensitivity by suppressing autophagy. Mol. Med. Rep. 2022, 25, 101. [Google Scholar] [CrossRef]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Hou, W.; Goldstein, L.A.; Lu, C.; Stolz, D.B.; Yin, X.-M.; Rabinowich, H. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J. Biol. Chem. 2008, 283, 19665–19677. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K. Autophagy-induced tumor dormancy in ovarian cancer. J. Clin. Investig. 2008, 118, 3837–3840. [Google Scholar] [CrossRef] [Green Version]
- Lock, R.; Debnath, J. Extracellular matrix regulation of autophagy. Curr. Opin. Cell Biol. 2008, 20, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy 2009, 5, 1232–1234. [Google Scholar] [CrossRef] [Green Version]
- Atsumi, T.; Chesney, J.; Metz, C.; Leng, L.; Donnelly, S.; Makita, Z.; Mitchell, R.; Bucala, R. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002, 62, 5881–5887. [Google Scholar]
- Yang, L.; Licastro, D.; Cava, E.; Veronese, N.; Spelta, F.; Rizza, W.; Bertozzi, B.; Villareal, D.T.; Hotamisligil, G.S.; Holloszy, J.O.; et al. Long-Term Calorie Restriction Enhances Cellular Quality-Control Processes in Human Skeletal Muscle. Cell Rep. 2016, 14, 422–428. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Mitchell, S.E.; Wang, B.; Tosti, V.; van Vliet, T.; Veronese, N.; Bertozzi, B.; Early, D.S.; Maissan, P.; Speakman, J.R.; et al. The effects of graded caloric restriction: XII. Comparison of mouse to human impact on cellular senescence in the colon. Aging Cell 2018, 17, e12746. [Google Scholar] [CrossRef]
- Flötotto, T.; Djahansouzi, S.; Gläser, M.; Hanstein, B.; Niederacher, D.; Brumm, C.; Beckmann, M.W. Hormones and hormone antagonists: Mechanisms of action in carcinogenesis of endometrial and breast cancer. Horm. Metab. Res. 2001, 33, 451–457. [Google Scholar] [CrossRef]
- Longo, V.D.; Fontana, L. Calorie restriction and cancer prevention: Metabolic and molecular mechanisms. Trends Pharmacol. Sci. 2010, 31, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, L.M.; Lavigne, J.A.; Chandramouli, G.V.R.; Lui, H.; Barrett, J.C.; Hursting, S.D. Dose-dependent effects of calorie restriction on gene expression, metabolism, and tumor progression are partially mediated by insulin-like growth factor-1. Cancer Med. 2012, 1, 275–288. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Wang, P.; Du, Y.; Wang, J. Indentification of breast cancer subtypes sensitive to HCQ-induced autophagy inhibition. Pathol. Res. Pract. 2019, 215, 152609. [Google Scholar] [CrossRef]
- Gong, C.; Hu, C.; Gu, F.; Xia, Q.; Yao, C.; Zhang, L.; Qiang, L.; Gao, S.; Gao, Y. Co-delivery of autophagy inhibitor ATG7 siRNA and docetaxel for breast cancer treatment. J. Control. Release 2017, 266, 272–286. [Google Scholar] [CrossRef]
- Orend, G.; Chiquet-Ehrismann, R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006, 244, 143–163. [Google Scholar] [CrossRef]
- Li, Z.-L.; Zhang, H.-L.; Huang, Y.; Huang, J.-H.; Sun, P.; Zhou, N.-N.; Chen, Y.-H.; Mai, J.; Wang, Y.; Yu, Y.; et al. Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-C degradation. Nat. Commun. 2020, 11, 3806. [Google Scholar] [CrossRef]
- Akkoc, Y.; Peker, N.; Akcay, A.; Gozuacik, D. Autophagy and Cancer Dormancy. Front. Oncol. 2021, 11, 627023. [Google Scholar] [CrossRef]
- Jahangiri, L.; Pucci, P.; Ishola, T.; Trigg, R.M.; Williams, J.A.; Pereira, J.; Cavanagh, M.L.; Turner, S.D.; Gkoutos, G.V.; Tsaprouni, L. The Contribution of Autophagy and LncRNAs to MYC-Driven Gene Regulatory Networks in Cancers. Int. J. Mol. Sci. 2021, 22, 8527. [Google Scholar] [CrossRef]
- Jadaliha, M.; Zong, X.; Malakar, P.; Ray, T.; Singh, D.K.; Freier, S.M.; Jensen, T.; Prasanth, S.G.; Karni, R.; Ray, P.S.; et al. Functional and prognostic significance of long non-coding RNA MALAT1 as a metastasis driver in ER negative lymph node negative breast cancer. Oncotarget 2016, 7, 40418–40436. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef]
- Ke, H.; Zhao, L.; Feng, X.; Xu, H.; Zou, L.; Yang, Q.; Su, X.; Peng, L.; Jiao, B. NEAT1 is Required for Survival of Breast Cancer Cells Through FUS and miR-548. Gene Regul. Syst. Bio. 2016, 10 (Suppl. S1), 11–17. [Google Scholar] [CrossRef] [Green Version]
- Yndestad, S.; Austreid, E.; Skaftnesmo, K.O.; Lønning, P.E.; Eikesdal, H.P. Divergent Activity of the Pseudogene PTENP1 in ER-Positive and Negative Breast Cancer. Mol. Cancer Res. 2018, 16, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Fridrichova, I.; Zmetakova, I. MicroRNAs Contribute to Breast Cancer Invasiveness. Cells 2019, 8, 1361. [Google Scholar] [CrossRef] [Green Version]
- Gregory, R.I.; Chendrimada, T.P.; Cooch, N.; Shiekhattar, R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 2005, 123, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Kleffel, S.; Schatton, T. Tumor dormancy and cancer stem cells: Two sides of the same coin? Adv. Exp. Med. Biol. 2013, 734, 145–179. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Abdalla, M.O.A.; Yamamoto, T.; Maehara, K.; Nogami, J.; Ohkawa, Y.; Miura, H.; Poonperm, R.; Hiratani, I.; Nakayama, H.; Nakao, M.; et al. The Eleanor ncRNAs activate the topological domain of the ESR1 locus to balance against apoptosis. Nat. Commun. 2019, 10, 3778. [Google Scholar] [CrossRef] [Green Version]
- Fukuoka, M.; Ichikawa, Y.; Osako, T.; Fujita, T.; Baba, S.; Takeuchi, K.; Tsunoda, N.; Ebata, T.; Ueno, T.; Ohno, S.; et al. The ELEANOR non-coding RNA expression contributes to cancer dormancy and predicts late recurrence of ER-positive breast cancer. Cancer Sci. 2022; Online ahead of print. [Google Scholar] [CrossRef]
- Papadaki, C.; Stratigos, M.; Markakis, G.; Spiliotaki, M.; Mastrostamatis, G.; Nikolaou, C.; Mavroudis, D.; Agelaki, S. Circulating microRNAs in the early prediction of disease recurrence in primary breast cancer. Breast Cancer Res. 2018, 20, 72. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [Green Version]
- Greco, S.J.; Rameshwar, P. Analysis of the transfer of circulating microRNA between cells mediated by gap junction. Methods Mol. Biol. 2013, 1024, 87–96. [Google Scholar]
- Mohd Ali, N.; Yeap, S.K.; Ho, W.Y.; Boo, L.; Ky, H.; Satharasinghe, D.A.; Tan, S.W.; Cheong, S.K.; Huang, H.D.; Lan, K.C.; et al. Adipose MSCs Suppress MCF7 and MDA-MB-231 Breast Cancer Metastasis and EMT Pathways Leading to Dormancy via Exosomal-miRNAs Following Co-Culture Interaction. Pharmaceuticals 2020, 14, 8. [Google Scholar] [CrossRef]
- Sandiford, O.A.; Donnelly, R.J.; El-Far, M.H.; Burgmeyer, L.M.; Sinha, G.; Pamarthi, S.H.; Sherman, L.S.; Ferrer, A.I.; DeVore, D.E.; Patel, S.A.; et al. Mesenchymal Stem Cell-Secreted Extracellular Vesicles Instruct Stepwise Dedifferentiation of Breast Cancer Cells into Dormancy at the Bone Marrow Perivascular Region. Cancer Res. 2021, 81, 1567–1582. [Google Scholar] [CrossRef]
- Jayaramayya, K.; Mahalaxmi, I.; Subramaniam, M.D.; Raj, N.; Dayem, A.A.; Lim, K.M.; Kim, S.J.; An, J.Y.; Lee, Y.; Choi, Y.; et al. Immunomodulatory effect of mesenchymal stem cells and mesenchymal stem-cell-derived exosomes for COVID-19 treatment. BMB Rep. 2020, 53, 400–412. [Google Scholar] [CrossRef]
- Dutta, S.; Warshall, C.; Bandyopadhyay, C.; Dutta, D.; Chandran, B. Interactions between exosomes from breast cancer cells and primary mammary epithelial cells leads to generation of reactive oxygen species which induce DNA damage response, stabilization of p53 and autophagy in epithelial cells. PLoS ONE 2014, 9, e97580. [Google Scholar] [CrossRef] [Green Version]
- Rossari, F.; Zucchinetti, C.; Buda, G.; Orciuolo, E. Tumor dormancy as an alternative step in the development of chemoresistance and metastasis—Clinical implications. Cell. Oncol. 2020, 43, 155–176. [Google Scholar] [CrossRef]
- Chu, P.-M.; Chen, L.-H.; Chen, M.-T.; Ma, H.-I.; Su, T.-L.; Hsieh, P.-C.; Chien, C.-S.; Jiang, B.-H.; Chen, Y.-C.; Lin, Y.-H.; et al. Targeting autophagy enhances BO-1051-induced apoptosis in human malignant glioma cells. Cancer Chemother. Pharmacol. 2012, 69, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Esparza Cristina, Y.; Wu, S.; Huang, L.; Buquet, C.; Shen, R.; Sanchez-Gonzalez, B.; García Latorre Awilda, E.; Boyer, O.; Varin, R.; Jiménez-Zamudio Antonio, L.; et al. Synergistic promoting effects of pentoxifylline and simvastatin on the apoptosis of triple-negative MDA-MB-231 breast cancer cells. Int. J. Oncol. 2018, 52, 1246–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Li, G.; Zheng, Y.; Shen, H.M.; Hu, X.; Ming, Q.L.; Huang, C.; Li, P.; Gao, N. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 2015, 11, 1259–1279. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yao, X.; Huang, J. New tricks for human farnesyltransferase inhibitor: Cancer and beyond. Medchemcomm 2017, 8, 841–854. [Google Scholar] [CrossRef]
- Chaterjee, M.; van Golen, K.L. Breast cancer stem cells survive periods of farnesyl-transferase inhibitor-induced dormancy by undergoing autophagy. Bone Marrow Res. 2011, 2011, 362938. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yu, Y.; Jiang, Z.; Cao, W.-M.; Wang, Z.; Dou, J.; Zhao, Y.; Cui, Y.; Zhang, H. Next-generation proteasome inhibitor MLN9708 sensitizes breast cancer cells to doxorubicin-induced apoptosis. Sci. Rep. 2016, 6, 26456. [Google Scholar] [CrossRef]
- Gavilán, E.; Giráldez, S.; Sánchez-Aguayo, I.; Romero, F.; Ruano, D.; Daza, P. Breast cancer cell line MCF7 escapes from G1/S arrest induced by proteasome inhibition through a GSK-3β dependent mechanism. Sci. Rep. 2015, 5, 10027. [Google Scholar] [CrossRef] [Green Version]
- Chery, J. RNA therapeutics: RNAi and antisense mechanisms and clinical applications. Postdoc J. 2016, 4, 35–50. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef]
- Sekhon, H.S.; London, C.A.; Sekhon, M.; Iversen, P.L.; Devi, G.R. c-MYC antisense phosphosphorodiamidate morpholino oligomer inhibits lung metastasis in a murine tumor model. Lung Cancer 2008, 60, 347–354. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors Influencing the Induction of Dormancy | Example of Effect and Mechanism Involved | Source |
|---|---|---|
| p38 MAPK/ERK TGFβ2 (and TGFβ-RIII) | TGFβ2 (and TGFβ-RIII) and EDG2 can lead to an enhanced ratio of the p38 MAPK/ERK levels and dormancy | [24,48,49] |
| Immunosurveillance | Loss of MHC class I or loss of tumour antigens leading to tumour cell survival | [53] |
| Angiogenic dormancy | Downregulation of pro-angiogenesis factors and the production of suppressors of angiogenesis impacting dormancy | [56,57] |
| FGF-2 | Growth and proliferation inhibition by inducing p21, leading to G1 cyclin complex inactivation in BC | [62,63] |
| α5β1 integrin-fibronectin | Survival of FGF2-responsive BC cell in the bone marrow | [64] |
| LIFR/STAT3/SOCS3 | The loss of STAT3 and LIFR in BC cells reduced dormancy and CSC markers and promoted proliferation | [65] |
| mTORC1/mTORC2, TGFβ2, BMP, NR2F1 and DEC2 (BHLHB3) | Induced dormancy | [48,66,67,68] |
| DNMT1 and CDKN1A | 5-Azacytidine induced p38-induced dormancy signature (e.g., reduced DNMT1, and increased CDKN1A) | [70,73,74] |
| Tumour microenvironment stromal cells | BC cell interaction with E-selectin+ ECs leading to maintenance of dormant state | [51,57,69] |
| Chronic inflammation, smoking, remodelling of the extracellular matrix, and signalling cascades converging on ERK | Promoted growth and awakening of dormant cells | [75,76,77,78,79] |
| ARHI (DIRAS3) re-expression | Induced autophagy by blocking PI3K/AKT/mTOR and enabled cells to remain dormant | [80,81] |
| Autophagy-related gene 7 (ATG7) knockout | Inhibited mitophagy alongside autophagy, leading to an accumulation of damaged mitochondria and reactive oxygen species, resulting in apoptotic cell death in dormant cells | [37] |
| ATG9B and LC3B | Expressed at significantly higher levels in dormant cells than in proliferating cells | [82] |
| Inhibiting autophagy in D2.OR cells, either with chloroquine (CQ) or shRNA-mediated depletion of ATGs | Induced BCSC expression of Pfkfb3 and emergence from metastatic dormancy | [83] |
| Inhibiting β1 integrin signalling | Prompted dormancy in the MMTV-PyMT model of BC, which can induce autophagy to give cancer cells time to establish cell-ECM contacts necessary to survive at secondary sites | [64,84,85,86] |
| Autophagy upregulation in ECM-detached cells or BC cells in soft substrata | Maintenance of the dormant state | [87] |
| DIRAS3 | DIRAS3 triggered autophagy both in vitro and in mice xenograft models and facilitated the survival of dormant cells | [88] |
| ATG5 | The in vitro and in vivo models of shRNA-mediated knockdown of ATG5, demonstrated reduced sensitivity to the chemotherapy treatment, inducing early escape from dormancy | [89] |
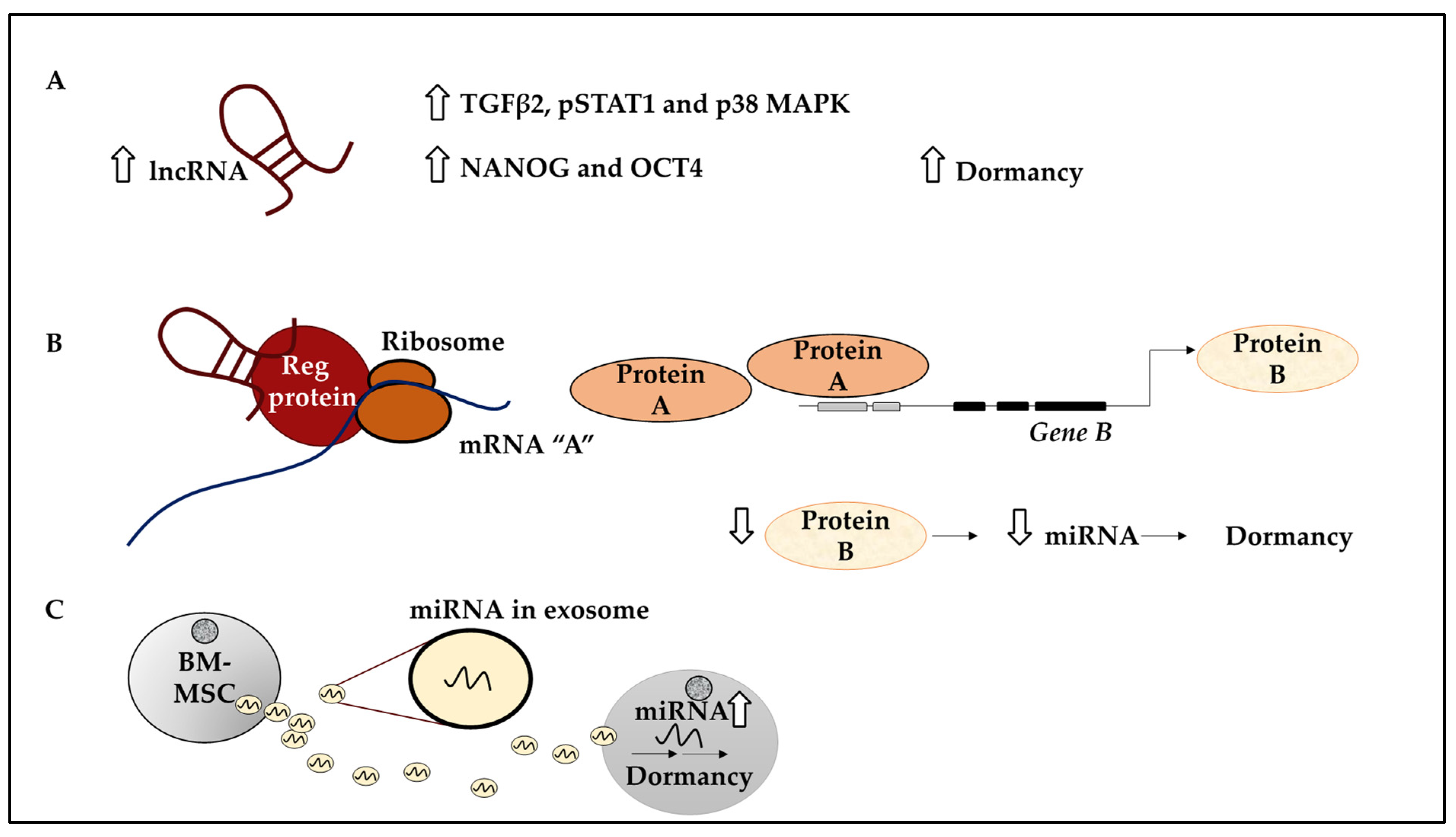
| Var1 NR2F1-AS1 expressing BT474 cells | Upregulation of markers of dormancy including TGFβ2, pSTAT1 and p38 MAPK and markers of pluripotency such as NANOG and OCT4 | [67] |
| NR2F1-AS1 | NR2F1-AS1 binding to NR2F1, leading to ΔNp63 transcription and increased dormancy of BC cells | [23] |
| BORG | Promoting transcriptional activity of TRIM28 leading to suppression of p21, TRIM28/BORG axes drove the reactivation of latent disseminated BC cells | [90] |
| MALAT1- Serpinb6b axis | Expressing Serpinb6b in MALAT1 knockout cells led to metastatic outbreak, since Serpinb6b could protect cells against T cells | [91] |
| miRNA-23b | Exosome generated by BM-MSCs enriched for miRNA-23b, suppresses MARCKS (a cell cycle gene) resulting in dormancy | [46] |
| miR-222/223 | BC cells primed BM-MSC to produce miR-222/223 containing exosomes which per se increased dormancy in BC cells | [92] |
| Stroma generated miRNA-containing exosomes including miR-222/223, mir-127 and mir-197 | Induced dormancy in BC cells through suppressing CXCL2 | [93] |
| Exosome-enclosed miRNAs including miR-205 and miR-31 | Induced dormancy in MDA-MB-231 cells by downregulating ubiquitin-conjugating enzyme E2N (UBE2N/Ubc13) and the resulting suppression of invasion and proliferation | [94] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahangiri, L.; Ishola, T. Dormancy in Breast Cancer, the Role of Autophagy, lncRNAs, miRNAs and Exosomes. Int. J. Mol. Sci. 2022, 23, 5271. https://doi.org/10.3390/ijms23095271
Jahangiri L, Ishola T. Dormancy in Breast Cancer, the Role of Autophagy, lncRNAs, miRNAs and Exosomes. International Journal of Molecular Sciences. 2022; 23(9):5271. https://doi.org/10.3390/ijms23095271
Chicago/Turabian StyleJahangiri, Leila, and Tala Ishola. 2022. "Dormancy in Breast Cancer, the Role of Autophagy, lncRNAs, miRNAs and Exosomes" International Journal of Molecular Sciences 23, no. 9: 5271. https://doi.org/10.3390/ijms23095271
APA StyleJahangiri, L., & Ishola, T. (2022). Dormancy in Breast Cancer, the Role of Autophagy, lncRNAs, miRNAs and Exosomes. International Journal of Molecular Sciences, 23(9), 5271. https://doi.org/10.3390/ijms23095271