
Identification, Characterization and Expression Profiling of the RS Gene Family during the Withering Process of White Tea in the Tea Plant (Camellia sinensis) Reveal the Transcriptional Regulation of CsRS8

 and
and
Abstract
:1. Introduction
2. Results
2.1. Identification of RS Family Genes in Camellia sinensis
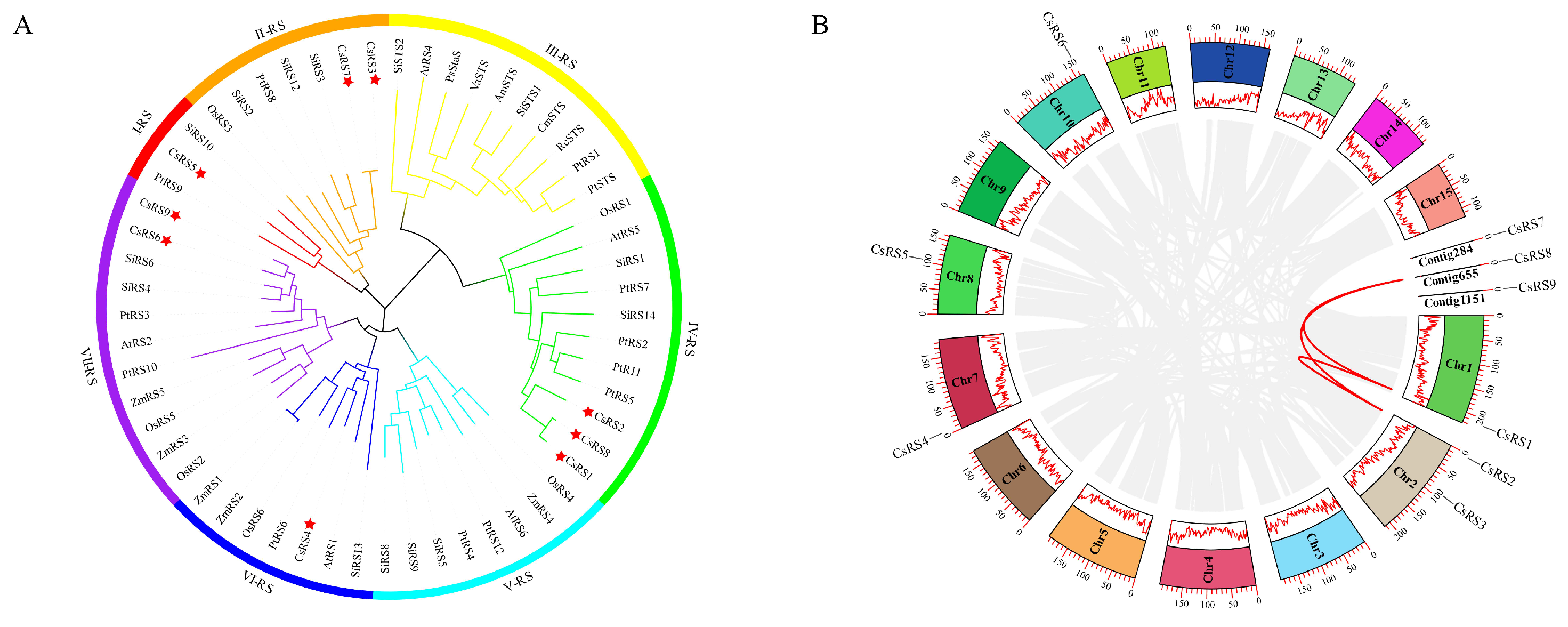
2.2. Phylogenetic Analysis of the CsRS Family
2.3. Chromosome Location and Synteny Analysis of CsRSs
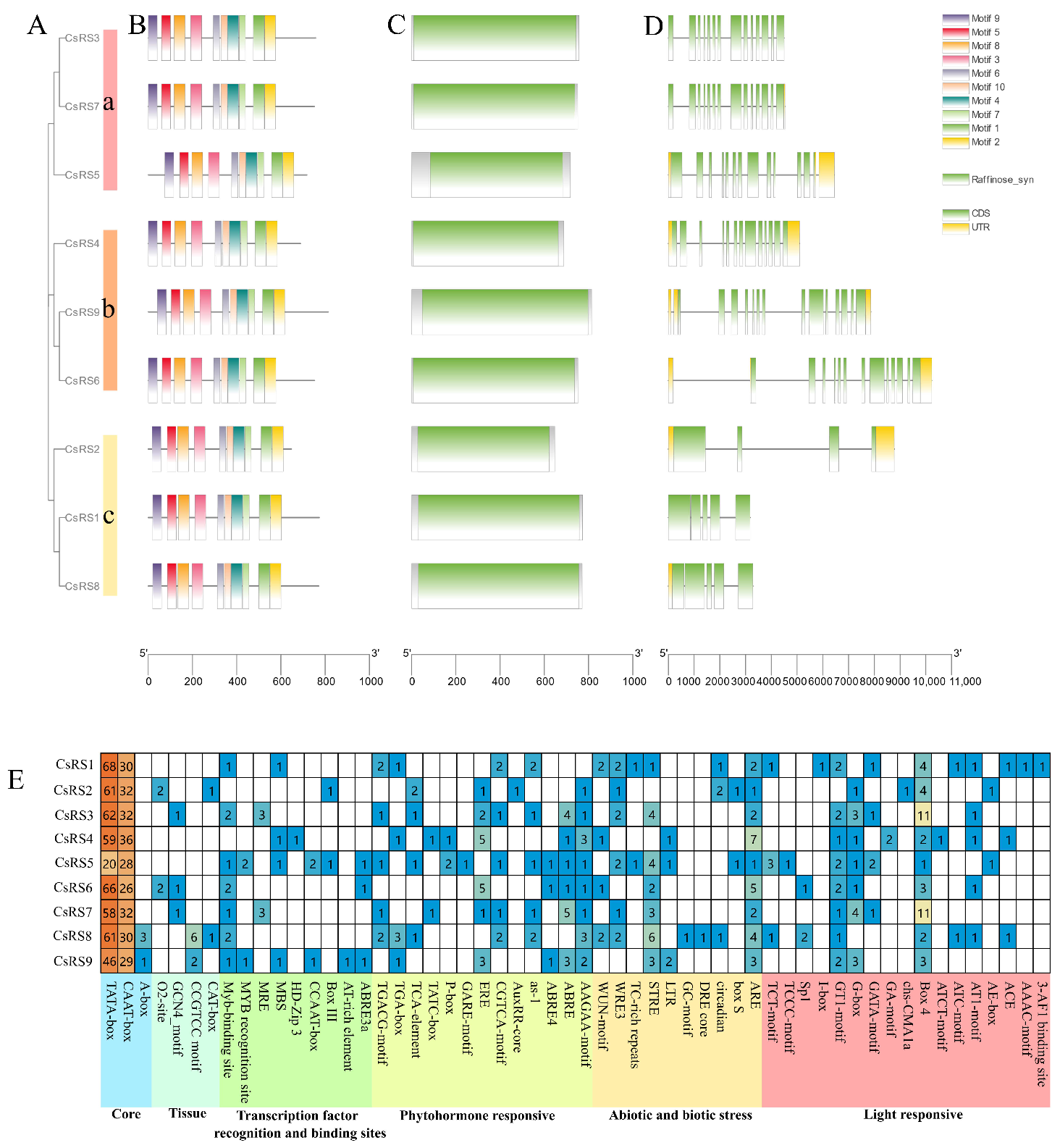
2.4. Gene Structure and Conserved Motifs of CsRSs
2.5. Secondary and Three-Dimensional Structures of CsRS
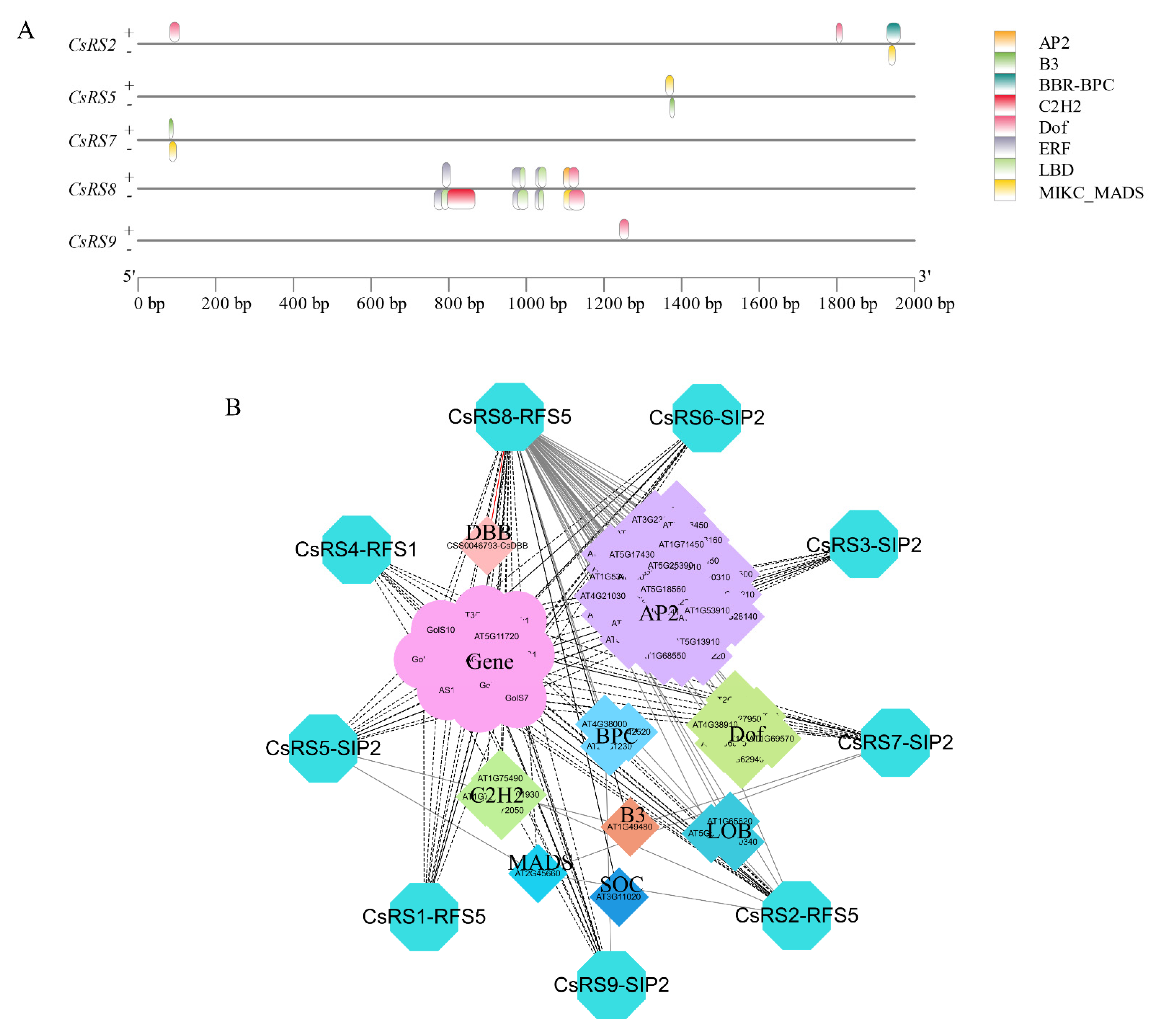
2.6. Cis-Elements in the Promoters of CsRSs
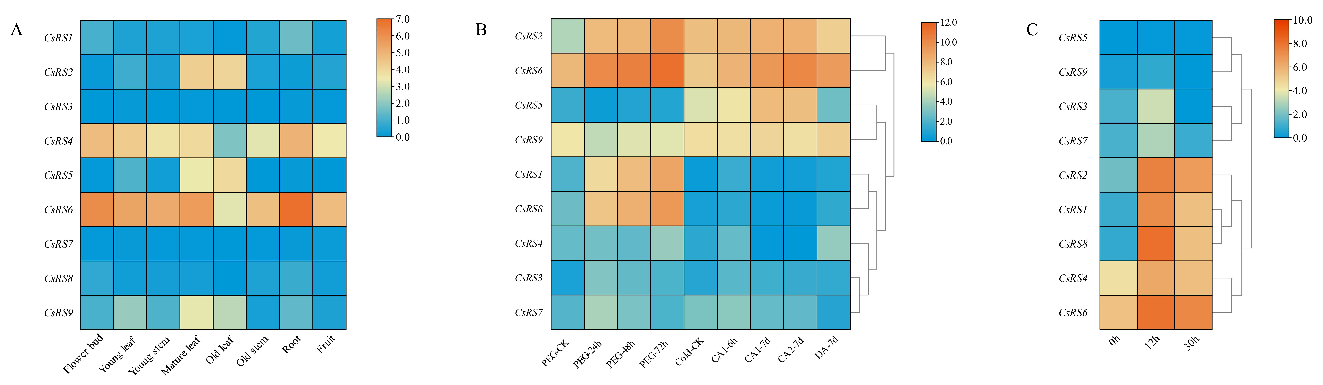
2.7. Expression Patterns of CsRSs in Different Organs
2.8. Expression Patterns of CsRSs during Continuous Drought Stress and Cold Acclimation
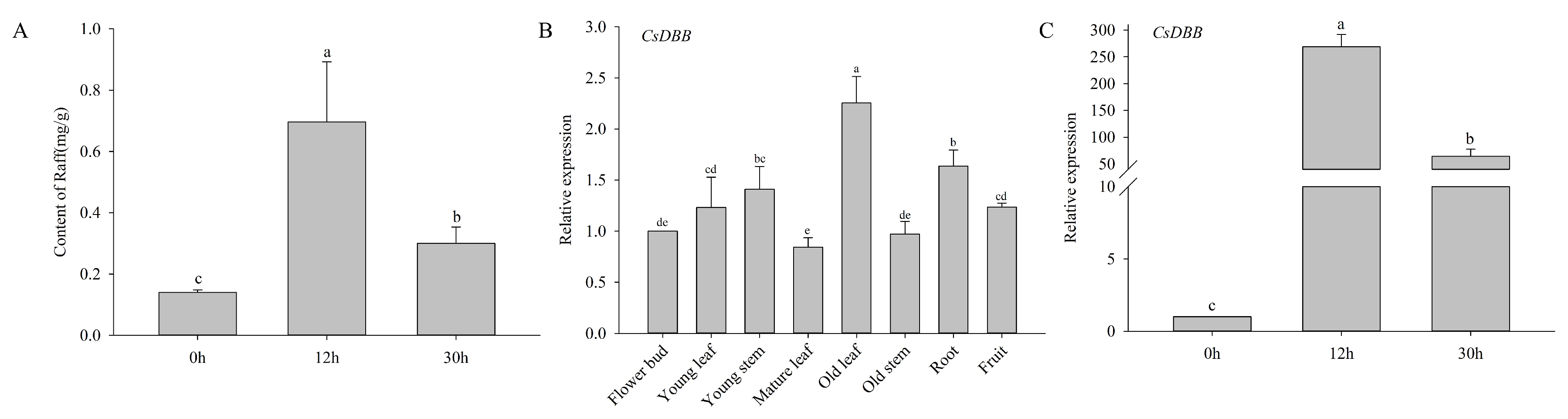
2.9. Expression Patterns of CsRSs and the Raf Content in the Withering Process of White Tea
2.10. Screening out Transcription Factors Regulating CsRS8
2.11. The Interaction and Regulation Network of CsRSs
3. Discussion
3.1. Evolution of the CsRS Gene Family
3.2. Potential Role of CsRS in Abiotic Stress and the Withering Process of White Tea
3.3. CsDBB Potentially Regulates CsRS8 to Affect Raffinose Accumulation during the Withering of White Tea
4. Materials and Methods
4.1. Identification of CsRSs from Tea Genome
4.2. Phylogenetic Tree Construction and Structural Analysis of CsRSs
4.3. Cis-Element Prediction for CsRSs Promoters
4.4. Plant Materials and Treatments
4.5. Analysis of Raffinose Content in the Withering Process of White Tea
4.6. Analysis of CsRSs Expression Patterns
4.7. Yeast One-Hybrid Screeing
4.8. Prediction of CsRSS Protein Interactions and Regulatory Networks
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sonali, S.; Sritama, M.; Papri, B.; Majumder, A.L. Significance of galactinol and raffinose family oligosaccharide synthesis in plants. Front. Plant Sci. 2015, 6, 656. [Google Scholar]
- Liu, J.J.; Odegard, W.; LumenM, B.O. Galactinol synthase from kidney bean cotyledon and zucchini leaf. Purification and N-terminal sequences. Plant Physiol. 1995, 109, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egert, A.; Keller, F.; Peters, S. Abiotic stress-induced accumulation of raffinose in Arabidopsis leaves is mediated by a single raffinose synthase (RS5, At5g40390). BMC Plant Biol. 2013, 13, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehle, L.; Tanner, W. The Function of myo-Inositol in the Biosynthesis of Raffinose. Eur. J. Biochem. 1973, 38, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Leopold, B.L.; LeopoldV, A.C. Changes in soluble carbohydrates during seed storage. Plant Physiol. 1992, 98, 1207–1210. [Google Scholar]
- Koster, K.L.; Leopold, A.C. Sugars and desiccation tolerance in seeds. Plant Physiol. 1988, 88, 829–832. [Google Scholar] [CrossRef] [Green Version]
- Smythe, B.M. Sucrose crystal growth. III. The relative growth rates of faces and their effect on sucrose crystal shape. Aust. J. Chem. 1967, 20, 1115–1131. [Google Scholar]
- Crowe, L.M.; Mouradian, R.; Crowe, J.H.; Jackson, S.A.; Womersley, C. Effects of carbohydrates on membrane stability at low water activities. Biochim. Biophys. Acta BBA Biomembr. 1984, 769, 141–150. [Google Scholar] [CrossRef]
- Mccarty, D.R. A core metabolome response of maize leaves subjected to long-duration abiotic stresses. Metabolites 2021, 11, 797. [Google Scholar]
- Rohde, P.; Hincha, D.K.; Heyer, A.G. Heterosis in the freezing tolerance of crosses between two Arabidopsis thaliana accessions (Columbia-0 and C24) that show differences in non-acclimated and acclimated freezing tolerance. Plant J. 2004, 38, 790–799. [Google Scholar] [CrossRef]
- Schneider, T.; Keller, F. Raffinose in chloroplasts is synthesized in the cytosol and transported across the chloroplast envelope. Plant Cell Physiol. 2009, 50, 2174–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayako, N.; Yukinori, Y.; Shigeru, S. Galactinol and raffinose constitute a novel function to protect plants from oxidative damage. Plant Physiol. 2008, 147, 1251–1263. [Google Scholar]
- Tortuero, F.; Fernández, E.; Rupérez, P.; Moreno, M. Raffinose and lactic acid bacteria influence caecal fermentation and serum cholesterol in rats. Nutr. Res. 1997, 17, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Collins, S.L.; Mcmillan, A.; Seney, S.; Veer, C.; Kort, R.; SumarahV, M.W.; Reid, K.; Mark, W.S.; Gregor, R. Promising prebiotic candidate established by evaluation of lactitol, lactulose, raffinose, and oligofructose for maintenance of a lactobacillus-dominated vaginal microbiota. Appl. Environ. Microb. 2017, 84, e2200–e2217. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Cha, E.; Kim, Y.H.; Jeon, Y.H.; Olson, B.H.; Byun, Y.; ParkV, H.D. Raffinose, a plant galactoside, inhibits Pseudomonas aeruginosa biofilm formation via binding to LecA and decreasing cellular cyclic diguanylate levels. Sci. Rep. 2016, 6, 25318. [Google Scholar] [CrossRef]
- Zhou, M.L.; Zhang, Q.; Zhou, M.; Sun, Z.M.; Zhu, X.M.; Shao, J.R.; Tang, Y.X.; Wu, Y.M. Genome-wide identification of genes involved in raffinose metabolism in Maize. Glycobiology 2012, 22, 1775–1785. [Google Scholar]
- Kannan, U.; Sharma, R.; Gangola, M.P.; Ganeshan, S.; Båga, M.; Chibbar, R.N. Sequential expression of raffinose synthase and stachyose synthase corresponds to successive accumulation of raffinose, stachyose and verbascose in developing seeds of Lens culinaris Medik. J. Plant Physiol. 2021, 265, 153494. [Google Scholar] [CrossRef]
- Cui, R.F.; Wang, X.G.; Malik, W.A.; Lu, X.K.; Chen, X.G.; Wang, D.L.; Wang, J.J.; Wang, S.; Chen, C.; Guo, L.X.; et al. Genome-wide identification and expression analysis of Raffinose synthetase family in cotton. BMC Bioinform. 2021, 22, 356. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, Y.; Yu, J.; Wang, L.K.; Yu, X.; Ohtani, M.; Kusano, M.; Saito, K.; Demura, T.; Zhuge, Q. Responses of Populus trichocarpa galactinol synthase genes to abiotic stresses. J. Plant Res. 2014, 127, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Koning, R.D.; Kiekens, R.; Toili, M.E.M.; Angenon, G. Identification and expression analysis of the genes involved in the raffinose family oligosaccharides pathway of phaseolus vulgaris and glycine max. Plants 2021, 10, 1465. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Y.M.; Wang, D.; Liu, Y.; Dirk, L.M.A.; Goodman, J.; Downie, A.B.; Wang, J.M.; Wang, G.Y.; Zhao, T.Y. Regulation of seed vigor by manipulation of raffinose family oligosaccharides in maize and Arabidopsis thaliana. Mol. Plant. 2017, 10, 1540–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, Y.; Lang, S.; Wang, D.M.; Xue, H.; Wang, X.F. Functional characterization of galactinol synthase and raffinose synthase in desiccation tolerance acquisition in developing Arabidopsis seeds. J. Plant Physiol. 2018, 230, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Yoshida, M. Expression analysis of the gene family associated with raffinose accumulation in rice seedlings under cold stress. J. Plant Physiol. 2011, 168, 2268–2271. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.Q.; Li, X.; Li, N.; Jameson, P.E.; Zhou, W.B. Transcription-associated metabolomic adjustments in maize occur during combined drought and cold stress. Plant Physiol. 2021, 186, 677–695. [Google Scholar] [CrossRef] [PubMed]
- Kito, K.; Yamane, K.; Yamamori, T.; Matsuhira, H.; Takabe, T. Isolation, functional characterization and stress responses of raffinose synthase genes in sugar beet. J. Plant Biochem. Biot. 2017, 27, 1–10. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Y.; Liu, Y.; Li, X.; Hao, G.; Han, Q.; Dirk, L.; Downie, A.B.; Ruan, Y.L.; Wang, J.M.; et al. Raffinose synthase enhances drought tolerance through raffinose synthesis or galactinol hydrolysis in maize and Arabidopsis plants. J. Biol. Chem. 2020, 295, 8064–8077. [Google Scholar] [CrossRef]
- Gu, H.; Lu, M.; Zhang, Z.P.; Xu, J.J.; Cao, W.H.; Miao, M.M. Metabolic process of raffinose family oligosaccharides during cold stress and recovery in cucumber leaves. J. Plant Physiol. 2017, 224–225, 112–120. [Google Scholar] [CrossRef]
- Gu, L.; Jiang, T.; Zhang, C.X. Maize HSFA2 and HSBP2 antagonistically modulate raffinose biosynthesis and heat tolerance in Arabidopsis. Plant J. 2019, 100, 128–142. [Google Scholar] [CrossRef]
- Wu, X.L.; Kishitani, S.; Ito, Y.; Toriyama, K. Accumulation of raffinose in rice seedlings overexpressing OsWRKY11 in relation to desiccation tolerance. Plant Bio. 2018, 26, 431–434. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.H.; Qi, J.L.; Hao, G.L.; Zhang, C.X.; Wang, C.M.; Dirk, L.M.A.; Bruce, D.A.; Zhao, T.Y. ZmDREB1A regulates raffinose synthase controlling raffinose accumulation and plant chilling stress tolerance in maize. Plant Cell Physiol. 2019, 61, 331–341. [Google Scholar] [CrossRef]
- Khan, M.; Hu, J.; Dahro, B.; Ming, R.; Zhang, Y.; Wang, Y.; Alhag, A.; Li, C.; Liu, J.H. ERF108 from Poncirus trifoliata (L.) Raf. functions in cold tolerance by modulating raffinose synthesis through transcriptional regulation of PtrRafS. Plant J. 2021, 108, 705–724. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Shen, S.S.; Wei, Y.M.; Jia, H.Y.; Li, T.H.; Yin, X.C.; Lu, C.Y.; Cui, Q.Q.; He, F.; Deng, W.W.; et al. Evaluation of the effects of solar withering on nonvolatile compounds in white tea through metabolomics and transcriptomics. Food Res. Int. 2022, 162, 112088. [Google Scholar] [CrossRef]
- Zhang, L.; Ho, C.T.; Zhou, J.; Santos, J.S.; Armstrong, L.; Granato, D. Chemistry and Biological Activities of Processed Camellia sinensis Teas: A Comprehensive Review. Compr. Rev. Food Sci. Food Saf. 2019, 18, 1474–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, T.R.; Tomás, G.; Teixeira, N.F.; Alves, M.G.; Silva, B.M. White tea (Camellia Sinensis (L.)): Antioxidant properties and beneficial health effects. Int. J. Food Sci. Nutr. 2013, 2, 19–26. [Google Scholar]
- Deng, X.M.; Shang, H.; Chen, J.J.; Wu, J.; Wang, T.; Wang, Y.Q.; Zhu, C.S.; Sun, W.J. Metabolomics combined with proteomics provide a novel interpretation of the changes in flavonoid glycosides during white tea processing. Foods 2022, 11, 1226. [Google Scholar] [CrossRef] [PubMed]
- Sharma, E.; Joshi, R.; Gulati, A. L-Theanine: An astounding sui generis integrant in tea. Food Chem. 2018, 242, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.Z.; Zhu, C.; Li, X.Z.; Chen, L.; Xie, S.Y.; Chen, G.W.; Zhang, H.; Lai, Z.X.; Lin, Y.L.; Guo, Y.Q. Transcriptome and phytochemical analyses reveal the roles of characteristic metabolites in the taste formation of white tea during the withering process. J. Integr. Agric. 2022, 21, 862–877. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Xia, E.H.; Li, F.D.; Tong, W.; Li, P.H.; Wu, Q.; Zhao, H.J.; Ge, R.H.; Li, R.P.; Li, Y.Y.; Zhang, Z.Z.; et al. Tea Plant Information Archive: A comprehensive genomics and bioinformatics platform for tea plant. Plant Biotechnol. J. 2019, 17, 1938–1953. [Google Scholar] [CrossRef]
- Zhang, Q.; Cai, M.C.; Yu, X.M.; Wang, L.S.; Guo, C.F.; Ming, R.; Zhang, J.S. Transcriptome dynamics of Camellia sinensis in response to continuous salinity and drought stress. Tree Genet. Genomes 2017, 13, 78. [Google Scholar] [CrossRef]
- Taji, T.; Ohsumi, C.; Iuchi, S.; Seki, M.; Kasuga, M.; Kobayashi, M.; Yamaguchi, S.K.; Shinozaki, K. Important roles of drought- and cold-inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. Plant J. 2002, 29, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Gang, R.; Behmüller, R.; Tenhaken, R. Molecular cloning of AtRS4, a seed specific multifunctional RFO synthase/galactosylhydrolase in Arabidopsis thaliana. Front. Plant Sci. 2015, 6, 789. [Google Scholar]
- Li, R.M.; Yuan, S.; He, Y.D.; Fan, J.; Zhou, Y.J.; Qiu, T.T.; Lin, X.J.; Yao, Y.; Liu, J.; Fu, S.P.; et al. Genome-wide identification and expression profiling analysis of the galactinol synthase gene family in Cassava (Manihot esculenta Crantz). Agron 2018, 8, 250. [Google Scholar] [CrossRef] [Green Version]
- You, J.; Wang, Y.Y.; Zhang, Y.J.; Dossa, K.; Li, D.H.; Zhou, R.; Wang, L.H.; Zhang, X.R. Genome-wide identification and expression analyses of genes involved in raffinose accumulation in sesame. Sci. Rep. 2018, 8, 4331. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and validation of promoters and cis-acting regulatory elements. Plant Sci. 2014, 217–218, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, X.L.; Meng, F.Z.; Wang, H.Y.; Wei, Y.X.; Li, R.F.; Wang, Z.Y.; Hu, L.P.; Wang, S.H.; Zhang, Z.X. Molecular cloning, characteristics and low temperature response of raffinose synthase gene in Cucumis sativus L. J. Plant Physiol. 2012, 169, 1883–1891. [Google Scholar] [CrossRef]
- Yue, C.; Cao, H.L.; Wang, L.; Zhou, Y.H.; Huang, Y.T.; Hao, X.Y.; Wang, Y.C.; Wang, B.; Yang, Y.J.; Wang, X.C. Effects of cold acclimation on sugar metabolism and sugar-related gene expression in tea plant during the winter season. Plant Mol. Biol. 2015, 88, 591–608. [Google Scholar] [CrossRef]
- Singh, K.B.; Foley, R.C.; Oñate, S.L. Transcription factors in plant defense and stress responses. Curr. Opin. Plant Biol. 2002, 5, 430–436. [Google Scholar] [CrossRef]
- Lang, S.R.; Liu, X.X.; Xue, H.; Li, X.; Wang, X.F. Functional characterization of BnHSFA4a as a heat shock transcription factor in controlling the re-establishment of desiccation tolerance in seeds. J. Exp. Bot. 2017, 68, 2361–2375. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.H.; Zhao, T.Y. ZmDREB2A regulates ZmGH3.2 and ZmRAFS, shifting metabolism towards seed aging tolerance over seedling growth. Plant J. 2020, 104, 268–282. [Google Scholar]
- Wei, W.; Zhang, Y.X.; Han, L.; Guan, Z.Q.; Chai, T.Y. A novel WRKY transcriptional factor from Thlaspi caerulescens negatively regulates the osmotic stress tolerance of transgenic tobacco. Plant Cell Rep. 2008, 27, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Khanna, R.; Kronmiller, B.; Maszle, D.R.; Coupland, G.; Holm, M.; Mizuno, T.; Wu, S.H. The Arabidopsis B-box zinc finger family. Plant Cell 2009, 21, 3416–3420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, E.H.; Tong, W.; Hou, Y.; An, Y.L.; Chen, L.B.; Wu, Q.; Liu, Y.L.; Yu, J.; Li, F.D.; Li, R.P.; et al. The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into its genome evolution and adaptation. Mol. Plant 2020, 13, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Jaina, M.; Sara, C.; Lowri, W.; Matloob, Q.; Gustavoa, S.; Sonnhammer, E.; Tosatto, S.; Lisanna, P.; Shriya, R.; Richardson, L.J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2020, 49, D412–D419. [Google Scholar]
- Xu, D.; Lu, Z.C.; Jin, K.M.; Qiu, W.M.; Qiao, G.R.; Han, X.J.; Zhuo, R.Y. Data and text mining SPDE: A multi-functional software for sequence processing and data extraction. Bioinformatics 2021, 37, 3686–3687. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Farideh, C.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Marc, G.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database. Nucleic Acids Res. 2019, 48, D265–D268. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools-an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. The Proteomics Protocols Handbook-Chapter 52: Protein Identification and Analysis Tools on the ExPASy Server. Proteom. Protoc. Handb. 2005, 571–607. [Google Scholar] [CrossRef]
- Xiong, E.H.; Zheng, C.Y.; Wu, X.L.; Wang, W. Protein Subcellular Location: The gap between prediction and experimentation. Plant Mol. Biol. Rep. 2015, 34, 1–10. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Thornton, J.M. PDBsum extras: SARS-CoV-2 and AlphaFold models. Protein Sci. 2022, 31, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Sun, L.L.; Sui, X.L.; Li, Y.X.; Chang, Y.W.; Fan, J.; Zhang, Z.X. Phloem loading in cucumber: Combined symplastic and apoplastic strategies. Plant J. 2019, 98, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.J.M. Analysis of relative gene expression data using real-time quantitative PCR and the 2-DDCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Alpha Helix | Extended Strand | Beta Turn | Random Coil |
|---|---|---|---|---|
| CsRS1 | 28.33% | 21.73% | 6.86% | 43.08% |
| CsRS2 | 32.77% | 16.85% | 6.34% | 44.05% |
| CsRS3 | 27.87% | 21.53% | 7% | 43.59% |
| CsRS4 | 28.92% | 19.62% | 6.25% | 45.20% |
| CsRS5 | 30.68% | 18.41% | 5.44% | 45.47% |
| CsRS6 | 26.99% | 21.68% | 7.31% | 44.02% |
| CsRS7 | 28.36% | 21.84% | 8.52% | 41.28% |
| CsRS8 | 27.89% | 20.62% | 6.23% | 45.27% |
| CsRS9 | 29.73% | 21.62% | 7% | 41.65% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Wang, Y.; Zhao, J.; Kong, J.; Zhang, L.; Qi, S.; Chen, J.; Chen, Z.; Zeng, W.; Sun, W. Identification, Characterization and Expression Profiling of the RS Gene Family during the Withering Process of White Tea in the Tea Plant (Camellia sinensis) Reveal the Transcriptional Regulation of CsRS8. Int. J. Mol. Sci. 2023, 24, 202. https://doi.org/10.3390/ijms24010202
Wang T, Wang Y, Zhao J, Kong J, Zhang L, Qi S, Chen J, Chen Z, Zeng W, Sun W. Identification, Characterization and Expression Profiling of the RS Gene Family during the Withering Process of White Tea in the Tea Plant (Camellia sinensis) Reveal the Transcriptional Regulation of CsRS8. International Journal of Molecular Sciences. 2023; 24(1):202. https://doi.org/10.3390/ijms24010202
Chicago/Turabian StyleWang, Tao, Yiqing Wang, Jiamin Zhao, Jiumei Kong, Lingzhi Zhang, Siyu Qi, Jiajia Chen, Zhidan Chen, Wen Zeng, and Weijiang Sun. 2023. "Identification, Characterization and Expression Profiling of the RS Gene Family during the Withering Process of White Tea in the Tea Plant (Camellia sinensis) Reveal the Transcriptional Regulation of CsRS8" International Journal of Molecular Sciences 24, no. 1: 202. https://doi.org/10.3390/ijms24010202
APA StyleWang, T., Wang, Y., Zhao, J., Kong, J., Zhang, L., Qi, S., Chen, J., Chen, Z., Zeng, W., & Sun, W. (2023). Identification, Characterization and Expression Profiling of the RS Gene Family during the Withering Process of White Tea in the Tea Plant (Camellia sinensis) Reveal the Transcriptional Regulation of CsRS8. International Journal of Molecular Sciences, 24(1), 202. https://doi.org/10.3390/ijms24010202