
Comprehensive Evaluation of Multiple Approaches Targeting ABCB1 to Resensitize Docetaxel-Resistant Prostate Cancer Cell Lines

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
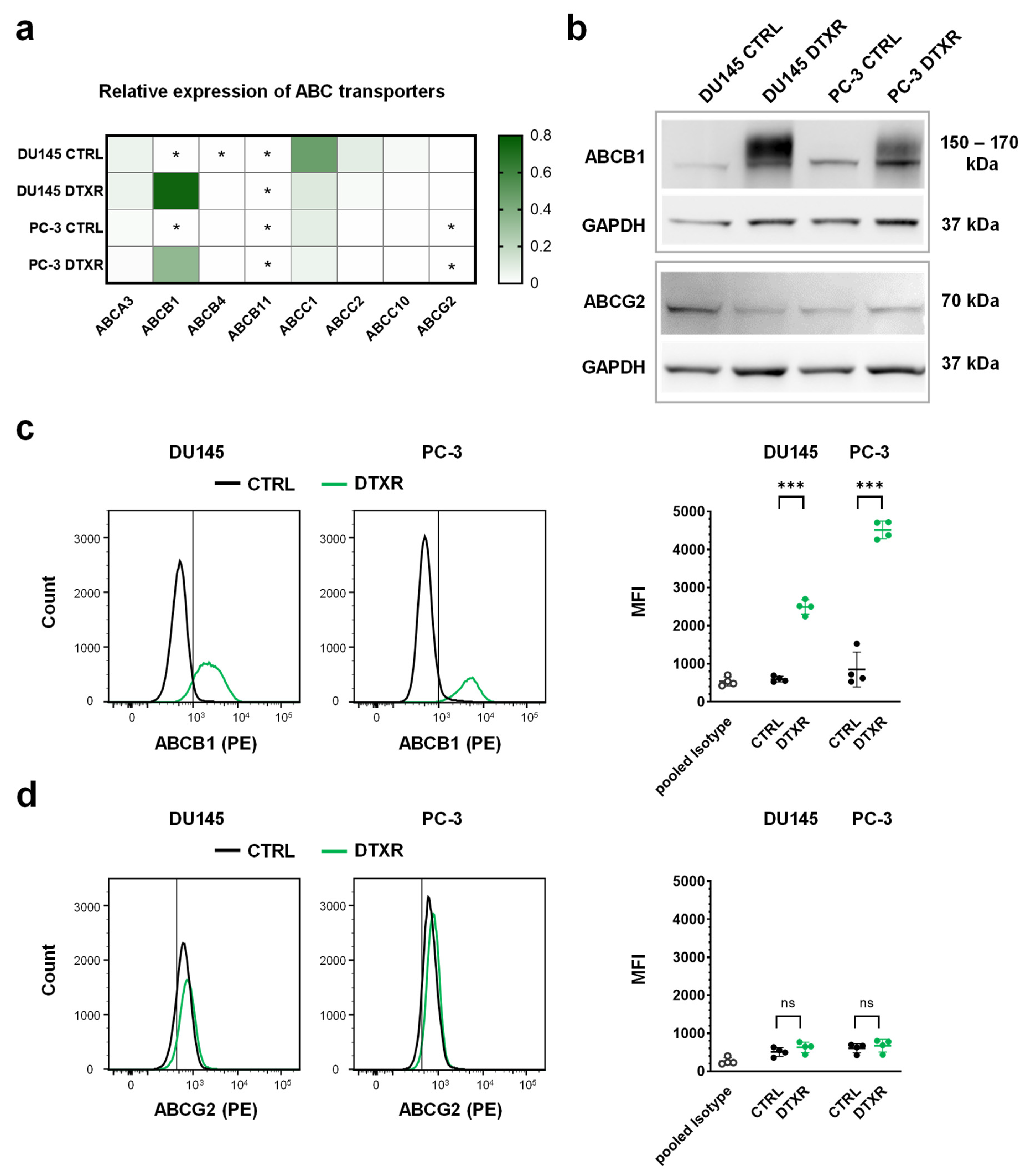
2.1. DTX-Resistant DU145 and PC-3 Cells Express High Levels of ABCB1
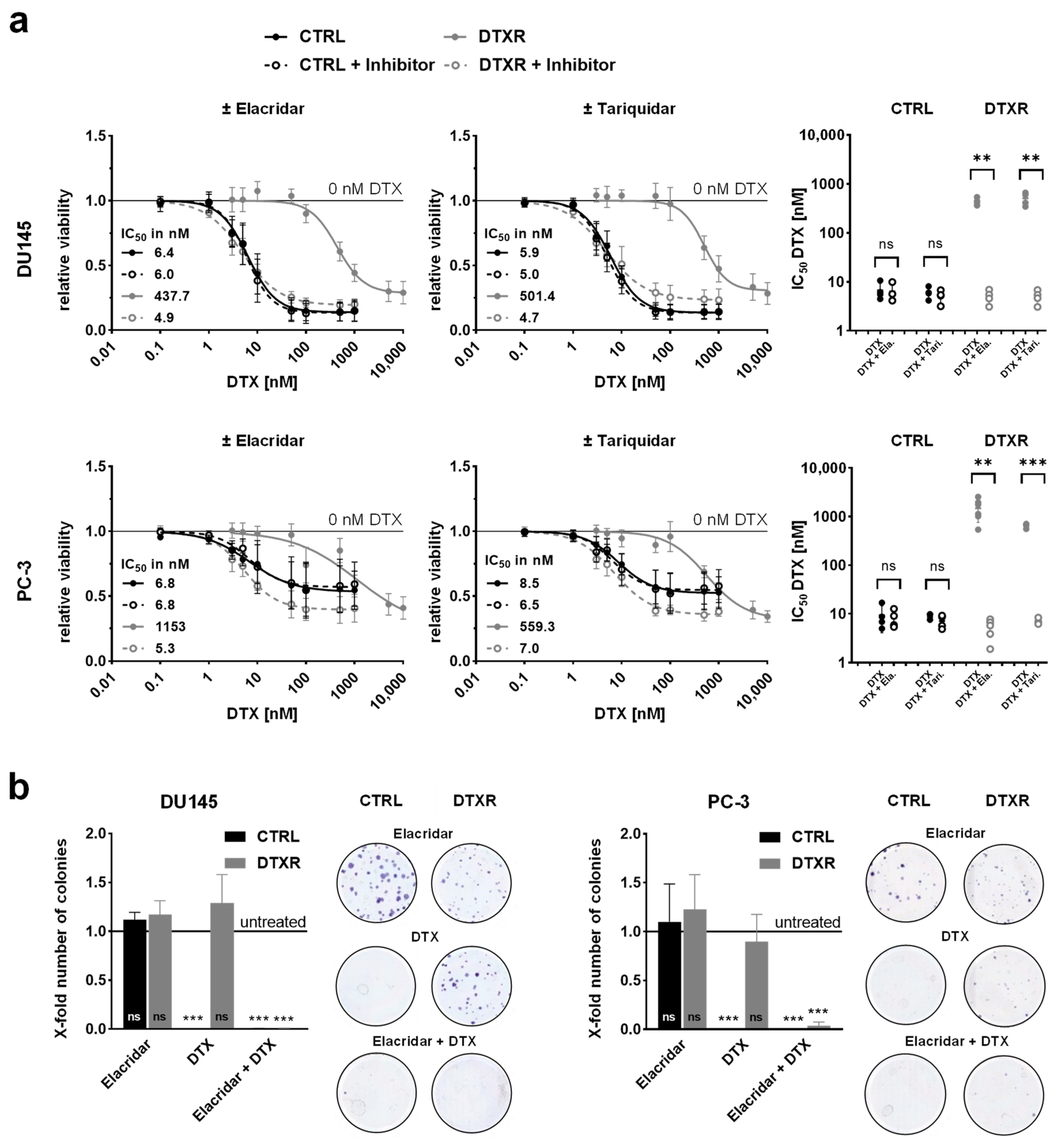
2.2. ABCB1 Inhibitors Reverse DTX Resistance in DU145 and PC-3 Cells
2.3. Specific Knockdown of ABCB1 Is Sufficient to Resensitize DTX-Resistant DU145 and PC-3 Cells
2.4. Tunicamycin Treatment Leads to Deglycosylation of the ABCB1 Protein in DU145 and PC-3 DTXR Cells
2.5. Deglycosylation by Tunicamycin Reduces DTX Tolerance in DU145 DTXR Cells but Not in PC-3 DTXR Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. RNA Isolation, cDNA Synthesis and Quantitative PCR Analysis
4.3. Western Blot Analysis
4.4. Flow Cytometry
4.5. Treatment Concentrations of ABCB1 Inhibitors, siRNA or Tunicamycin
4.6. Rhodamine 123 Efflux Assay
4.7. Treatment with ABCB1 Inhibitors
4.8. Treatment with ABCB1 siRNA
4.9. Treatment with Tunicamycin
4.10. WST-1 Assay to Assess Cellular Viability
4.11. Crystal Violet Assay to Assess Total Adherent Cell Mass
4.12. Colony Formation Assay to Assess Clonogenic Properties and Long-Term Proliferation
4.13. Digestion with PNGase F
4.14. Immunofluorescence Staining
4.15. Statistics, Data Visualization and Software
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cornford, P.; van den Bergh, R.C.N.; Briers, E.; Van den Broeck, T.; Cumberbatch, M.G.; De Santis, M.; Fanti, S.; Fossati, N.; Gandaglia, G.; Gillessen, S.; et al. EAU-EANM-ESTRO-ESUR-SIOG guidelines on prostate cancer. Part II—2020 update: Treatment of relapsing and metastatic prostate cancer. Eur. Urol. 2021, 79, 263–282. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.; Lara, P.N., Jr.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puhr, M.; Hoefer, J.; Schafer, G.; Erb, H.H.; Oh, S.J.; Klocker, H.; Heidegger, I.; Neuwirt, H.; Culig, Z. Epithelial-to-mesenchymal transition leads to docetaxel resistance in prostate cancer and is mediated by reduced expression of miR-200c and miR-205. Am. J. Pathol. 2012, 181, 2188–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donix, L.; Erb, H.H.H.; Peitzsch, C.; Dubrovska, A.; Pfeifer, M.; Thomas, C.; Fuessel, S.; Erdmann, K. Acquired resistance to irradiation or docetaxel is not associated with cross-resistance to cisplatin in prostate cancer cell lines. J. Cancer Res. Clin. Oncol. 2022, 148, 1313–1324. [Google Scholar] [CrossRef]
- Maloney, S.M.; Hoover, C.A.; Morejon-Lasso, L.V.; Prosperi, J.R. Mechanisms of taxane resistance. Cancers 2020, 12, 3323. [Google Scholar] [CrossRef]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281–290. [Google Scholar] [CrossRef]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Hodges, L.M.; Markova, S.M.; Chinn, L.W.; Gow, J.M.; Kroetz, D.L.; Klein, T.E.; Altman, R.B. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharm. Genom. 2011, 21, 152–161. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, I.S.; He, W.; Yin, L. Understanding of human ATP binding cassette superfamily and novel multidrug resistance modulators to overcome MDR. Biomed. Pharm. 2018, 100, 335–348. [Google Scholar] [CrossRef]
- Ween, M.P.; Armstrong, M.A.; Oehler, M.K.; Ricciardelli, C. The role of ABC transporters in ovarian cancer progression and chemoresistance. Crit. Rev. Oncol. Hematol. 2015, 96, 220–256. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Mizokami, A.; Mamiya, K.; Li, Y.Q.; Zhang, J.; Keller, E.T.; Namiki, M. The establishment of two paclitaxel-resistant prostate cancer cell lines and the mechanisms of paclitaxel resistance with two cell lines. Prostate 2007, 67, 955–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Liu, C.; Nadiminty, N.; Lou, W.; Tummala, R.; Evans, C.P.; Gao, A.C. Inhibition of ABCB1 expression overcomes acquired docetaxel resistance in prostate cancer. Mol. Cancer 2013, 12, 1829–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, H.K.; Lee, S.J.; Kwon, W.A.; Jeong, K.C. Docetaxel-resistant prostate cancer cells become sensitive to gemcitabine due to the upregulation of ABCB1. Prostate 2020, 80, 453–462. [Google Scholar] [CrossRef]
- Sabnis, N.G.; Miller, A.; Titus, M.A.; Huss, W.J. The efflux transporter ABCG2 maintains prostate stem cells. Mol. Cancer Res. 2017, 15, 128–140. [Google Scholar] [CrossRef] [Green Version]
- Cairo, M.S.; Siegel, S.; Anas, N.; Sender, L. Clinical trial of continuous infusion verapamil, bolus vinblastine, and continuous infusion VP-16 in drug-resistant pediatric tumors. Cancer Res. 1989, 49, 1063–1066. [Google Scholar]
- Cowie, F.J.; Pinkerton, C.R.; Phillips, M.; Dick, G.; Judson, I.; McCarthy, P.T.; Flanagan, R.J. Continuous-infusion verapamil with etoposide in relapsed or resistant paediatric cancers. Br. J. Cancer 1995, 71, 877–881. [Google Scholar] [CrossRef] [Green Version]
- Gatzemeier, U.; Schneider, A.; von Pawel, J. Randomised trial of vindesine and etoposide +/- dexverapamil in advanced non-small cell lung cancer: First results. J. Cancer Res. Clin. Oncol. 1995, 121 (Suppl. 3), R17-20. [Google Scholar] [CrossRef]
- Hendrick, A.M.; Harris, A.L.; Cantwell, B.M. Verapamil with mitoxantrone for advanced ovarian cancer: A negative phase II trial. Ann. Oncol. 1991, 2, 71–72. [Google Scholar] [CrossRef]
- Millward, M.J.; Cantwell, B.M.; Munro, N.C.; Robinson, A.; Corris, P.A.; Harris, A.L. Oral verapamil with chemotherapy for advanced non-small cell lung cancer: A randomised study. Br. J. Cancer 1993, 67, 1031–1035. [Google Scholar] [CrossRef] [Green Version]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Lombard, A.P.; Liu, C.; Armstrong, C.M.; Cucchiara, V.; Gu, X.; Lou, W.; Evans, C.P.; Gao, A.C. ABCB1 mediates cabazitaxel-docetaxel cross-resistance in advanced prostate cancer. Mol. Cancer 2017, 16, 2257–2266. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, A.J.; Prencipe, M.; Dowling, C.; Fan, Y.; Mulrane, L.; Gallagher, W.M.; O’Connor, D.; O’Connor, R.; Devery, A.; Corcoran, C.; et al. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol. Cancer 2011, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Lombard, A.P.; Lou, W.; Armstrong, C.M.; D’Abronzo, L.S.; Ning, S.; Evans, C.P.; Gao, A.C. Activation of the ABCB1 amplicon in docetaxel- and cabazitaxel-resistant prostate cancer cells. Mol. Cancer 2021, 20, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.; Widemann, B.C.; Pastakia, D.; Chen, C.C.; Yang, S.X.; Cole, D.; Balis, F.M. Pharmacokinetic and pharmacodynamic study of tariquidar (XR9576), a P-glycoprotein inhibitor, in combination with doxorubicin, vinorelbine, or docetaxel in children and adolescents with refractory solid tumors. Cancer Chemother. Pharm. 2015, 76, 1273–1283. [Google Scholar] [CrossRef]
- Kelly, R.J.; Draper, D.; Chen, C.C.; Robey, R.W.; Figg, W.D.; Piekarz, R.L.; Chen, X.; Gardner, E.R.; Balis, F.M.; Venkatesan, A.M.; et al. A pharmacodynamic study of docetaxel in combination with the P-glycoprotein antagonist tariquidar (XR9576) in patients with lung, ovarian, and cervical cancer. Clin. Cancer Res. 2011, 17, 569–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, J.; Edgerly, M.; Wilson, R.; Chen, C.; Rutt, A.; Bakke, S.; Robey, R.; Dwyer, A.; Goldspiel, B.; Balis, F.; et al. A phase I study of the P-glycoprotein antagonist tariquidar in combination with vinorelbine. Clin. Cancer Res. 2009, 15, 3574–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malingré, M.M.; Beijnen, J.H.; Rosing, H.; Koopman, F.J.; Jewell, R.C.; Paul, E.M.; Ten Bokkel Huinink, W.W.; Schellens, J.H. Co-administration of GF120918 significantly increases the systemic exposure to oral paclitaxel in cancer patients. Br. J. Cancer 2001, 84, 42–47. [Google Scholar] [CrossRef] [Green Version]
- Planting, A.S.; Sonneveld, P.; van der Gaast, A.; Sparreboom, A.; van der Burg, M.E.; Luyten, G.P.; de Leeuw, K.; de Boer-Dennert, M.; Wissel, P.S.; Jewell, R.C.; et al. A phase I and pharmacologic study of the MDR converter GF120918 in combination with doxorubicin in patients with advanced solid tumors. Cancer Chemother. Pharm. 2005, 55, 91–99. [Google Scholar] [CrossRef]
- Pusztai, L.; Wagner, P.; Ibrahim, N.; Rivera, E.; Theriault, R.; Booser, D.; Symmans, F.W.; Wong, F.; Blumenschein, G.; Fleming, D.R.; et al. Phase II study of tariquidar, a selective P-glycoprotein inhibitor, in patients with chemotherapy-resistant, advanced breast carcinoma. Cancer 2005, 104, 682–691. [Google Scholar] [CrossRef]
- Stewart, A.; Steiner, J.; Mellows, G.; Laguda, B.; Norris, D.; Bevan, P. Phase I trial of XR9576 in healthy volunteers demonstrates modulation of P-glycoprotein in CD56+ lymphocytes after oral and intravenous administration. Clin. Cancer Res. 2000, 6, 4186–4191. [Google Scholar] [PubMed]
- Schinkel, A.H.; Kemp, S.; Dolle, M.; Rudenko, G.; Wagenaar, E. N-glycosylation and deletion mutants of the human MDR1 P-glycoprotein. J. Biol. Chem. 1993, 268, 7474–7481. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chin, J.E.; Ueda, K.; Clark, D.P.; Pastan, I.; Gottesman, M.M.; Roninson, I.B. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 1986, 47, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Stanley, P. Golgi glycosylation. Cold Spring Harb. Perspect. Biol. 2011, 3, a005199. [Google Scholar] [CrossRef]
- Beheshti Zavareh, R.; Sukhai, M.A.; Hurren, R.; Gronda, M.; Wang, X.; Simpson, C.D.; Maclean, N.; Zih, F.; Ketela, T.; Swallow, C.J.; et al. Suppression of cancer progression by MGAT1 shRNA knockdown. PLoS ONE 2012, 7, e43721. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.W.; Clarke, D.M. Prolonged association of temperature-sensitive mutants of human P-glycoprotein with calnexin during biogenesis. J. Biol. Chem. 1994, 269, 28683–28689. [Google Scholar] [CrossRef]
- Forster, S.; Thumser, A.E.; Hood, S.R.; Plant, N. Characterization of rhodamine-123 as a tracer dye for use in in vitro drug transport assays. PLoS ONE 2012, 7, e33253. [Google Scholar] [CrossRef] [Green Version]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Loo, T.W.; Clarke, D.M. Quality control by proteases in the endoplasmic reticulum. Removal of a protease-sensitive site enhances expression of human P-glycoprotein. J. Biol. Chem. 1998, 273, 32373–32376. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Wakabayashi-Nakao, K.; Tamura, A.; Toyoda, Y.; Koshiba, S.; Ishikawa, T. Disruption of N-linked glycosylation enhances ubiquitin-mediated proteasomal degradation of the human ATP-binding cassette transporter ABCG2. FEBS J. 2009, 276, 7237–7252. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, C.; Armstrong, C.; Lou, W.; Sandher, A.; Gao, A.C. Antiandrogens inhibit ABCB1 efflux and ATPase activity and reverse docetaxel resistance in advanced prostate cancer. Clin. Cancer Res. 2015, 21, 4133–4142. [Google Scholar] [CrossRef] [PubMed]
- Lima, T.S.; Iglesias-Gato, D.; Souza, L.D.O.; Stenvang, J.; Lima, D.S.; Roder, M.A.; Brasso, K.; Moreira, J.M.A. Molecular profiling of docetaxel-resistant prostate cancer cells identifies multiple mechanisms of therapeutic resistance. Cancers 2021, 13, 1290. [Google Scholar] [CrossRef] [PubMed]
- Kuntner, C.; Bankstahl, J.P.; Bankstahl, M.; Stanek, J.; Wanek, T.; Stundner, G.; Karch, R.; Brauner, R.; Meier, M.; Ding, X.; et al. Dose-response assessment of tariquidar and elacridar and regional quantification of P-glycoprotein inhibition at the rat blood-brain barrier using (R)-[(11)C]verapamil PET. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 942–953. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, Y.; Tamada, S.; Kato, M.; Hirayama, Y.; Takeyama, Y.; Iguchi, T.; Sadar, M.D.; Nakatani, T. Androgen receptor splice variant 7 drives the growth of castration resistant prostate cancer without being involved in the efficacy of taxane chemotherapy. J. Clin. Med. 2018, 7, 444. [Google Scholar] [CrossRef] [Green Version]
- Engle, K.; Kumar, G. Cancer multidrug-resistance reversal by ABCB1 inhibition: A recent update. Eur. J. Med. Chem. 2022, 239, 114542. [Google Scholar] [CrossRef]
- Richert, N.D.; Aldwin, L.; Nitecki, D.; Gottesman, M.M.; Pastan, I. Stability and covalent modification of P-glycoprotein in multidrug-resistant KB cells. Biochemistry 1988, 27, 7607–7613. [Google Scholar] [CrossRef] [PubMed]
- Gribar, J.J.; Ramachandra, M.; Hrycyna, C.A.; Dey, S.; Ambudkar, S.V. Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system. J. Membr. Biol. 2000, 173, 203–214. [Google Scholar] [CrossRef]
- Kramer, R.; Weber, T.K.; Arceci, R.; Ramchurren, N.; Kastrinakis, W.V.; Steele, G., Jr.; Summerhayes, I.C. Inhibition of N-linked glycosylation of P-glycoprotein by tunicamycin results in a reduced multidrug resistance phenotype. Br. J. Cancer 1995, 71, 670–675. [Google Scholar] [CrossRef] [Green Version]
- Wojtowicz, K.; Januchowski, R.; Nowicki, M.; Zabel, M. Inhibition of protein glycosylation reverses the MDR phenotype of cancer cell lines. Biomed. Pharm. 2015, 74, 49–56. [Google Scholar] [CrossRef]
- Seres, M.; Cholujova, D.; Bubencikova, T.; Breier, A.; Sulova, Z. Tunicamycin depresses P-glycoprotein glycosylation without an effect on its membrane localization and drug efflux activity in L1210 cells. Int. J. Mol. Sci. 2011, 12, 7772–7784. [Google Scholar] [CrossRef] [Green Version]
- Carroll, A.G.; Voeller, H.J.; Sugars, L.; Gelmann, E.P. p53 oncogene mutations in three human prostate cancer cell lines. Prostate 1993, 23, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, F.; Brandt, D.T.; Schneikert, J.; Fuchs, J.; Grikscheit, K.; Wanzel, M.; Pavlakis, E.; Charles, J.P.; Timofeev, O.; Nist, A.; et al. Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum UDPase ENTPD5. Proc. Natl. Acad. Sci. USA 2016, 113, E8433–E8442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabi, I.R.; Shankar, J.; Dennis, J.W. The galectin lattice at a glance. J. Cell Sci. 2015, 128, 2213–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannes, L.; Billet, A. Glycosylation and raft endocytosis in cancer. Cancer Metastasis Rev. 2020, 39, 375–396. [Google Scholar] [CrossRef]
- Lee, Y.K.; Lin, T.H.; Chang, C.F.; Lo, Y.L. Galectin-3 silencing inhibits epirubicin-induced ATP binding cassette transporters and activates the mitochondrial apoptosis pathway via beta-catenin/GSK-3beta modulation in colorectal carcinoma. PLoS ONE 2013, 8, e82478. [Google Scholar] [CrossRef] [Green Version]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex Network: A systematic exploration of the human interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody against | Source | Catalog No., Clone | Dilution |
|---|---|---|---|
| ABCB1 | Cell Signaling Technology (Leiden, The Netherlands) | 12683, D3H1Q | 1:1000 |
| ABCG2 | Cell Signaling Technology | 42078, D5V2K | 1:1000 |
| GAPDH | OriGene Technologies (Rockville, MD, USA) | 5G4-6C5, 6C5 | 1:5000 |
| α-tubulin | Cell Signaling Technology | 3873, DM1A | 1:3000 |
| Reagent | Inhibition of | Applied Dose |
|---|---|---|
| Elacridar | ABCB1 & ABCG2 protein | 50 nM |
| Tariquidar | ABCB1 & ABCG2 protein | 50 nM |
| siRNA (siR-ABCB1) | ABCB1 mRNA | 20 nM |
| Tunicamycin | Glycosylation in the ER | 100 ng/mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linke, D.; Donix, L.; Peitzsch, C.; Erb, H.H.H.; Dubrovska, A.; Pfeifer, M.; Thomas, C.; Fuessel, S.; Erdmann, K. Comprehensive Evaluation of Multiple Approaches Targeting ABCB1 to Resensitize Docetaxel-Resistant Prostate Cancer Cell Lines. Int. J. Mol. Sci. 2023, 24, 666. https://doi.org/10.3390/ijms24010666
Linke D, Donix L, Peitzsch C, Erb HHH, Dubrovska A, Pfeifer M, Thomas C, Fuessel S, Erdmann K. Comprehensive Evaluation of Multiple Approaches Targeting ABCB1 to Resensitize Docetaxel-Resistant Prostate Cancer Cell Lines. International Journal of Molecular Sciences. 2023; 24(1):666. https://doi.org/10.3390/ijms24010666
Chicago/Turabian StyleLinke, Dinah, Lukas Donix, Claudia Peitzsch, Holger H. H. Erb, Anna Dubrovska, Manuel Pfeifer, Christian Thomas, Susanne Fuessel, and Kati Erdmann. 2023. "Comprehensive Evaluation of Multiple Approaches Targeting ABCB1 to Resensitize Docetaxel-Resistant Prostate Cancer Cell Lines" International Journal of Molecular Sciences 24, no. 1: 666. https://doi.org/10.3390/ijms24010666
APA StyleLinke, D., Donix, L., Peitzsch, C., Erb, H. H. H., Dubrovska, A., Pfeifer, M., Thomas, C., Fuessel, S., & Erdmann, K. (2023). Comprehensive Evaluation of Multiple Approaches Targeting ABCB1 to Resensitize Docetaxel-Resistant Prostate Cancer Cell Lines. International Journal of Molecular Sciences, 24(1), 666. https://doi.org/10.3390/ijms24010666