

The Potential Role of Regulated Cell Death in Dry Eye Diseases and Ocular Surface Dysfunction
 , , and
, , and
Abstract
:1. Introduction
2. Oxidative Stress and Inflammation in DED
3. Regulated Cell Death (RCD): An Emerging Field
4. Targeting RCD as an Innovative Strategy in DED
4.1. Ferroptosis
Possible Implication of Ferroptosis in Dry Eye and Ocular Surface Dysfunction
4.2. Necroptosis
Possible Implication in Dry Eye and Ocular Surface Dysfunction
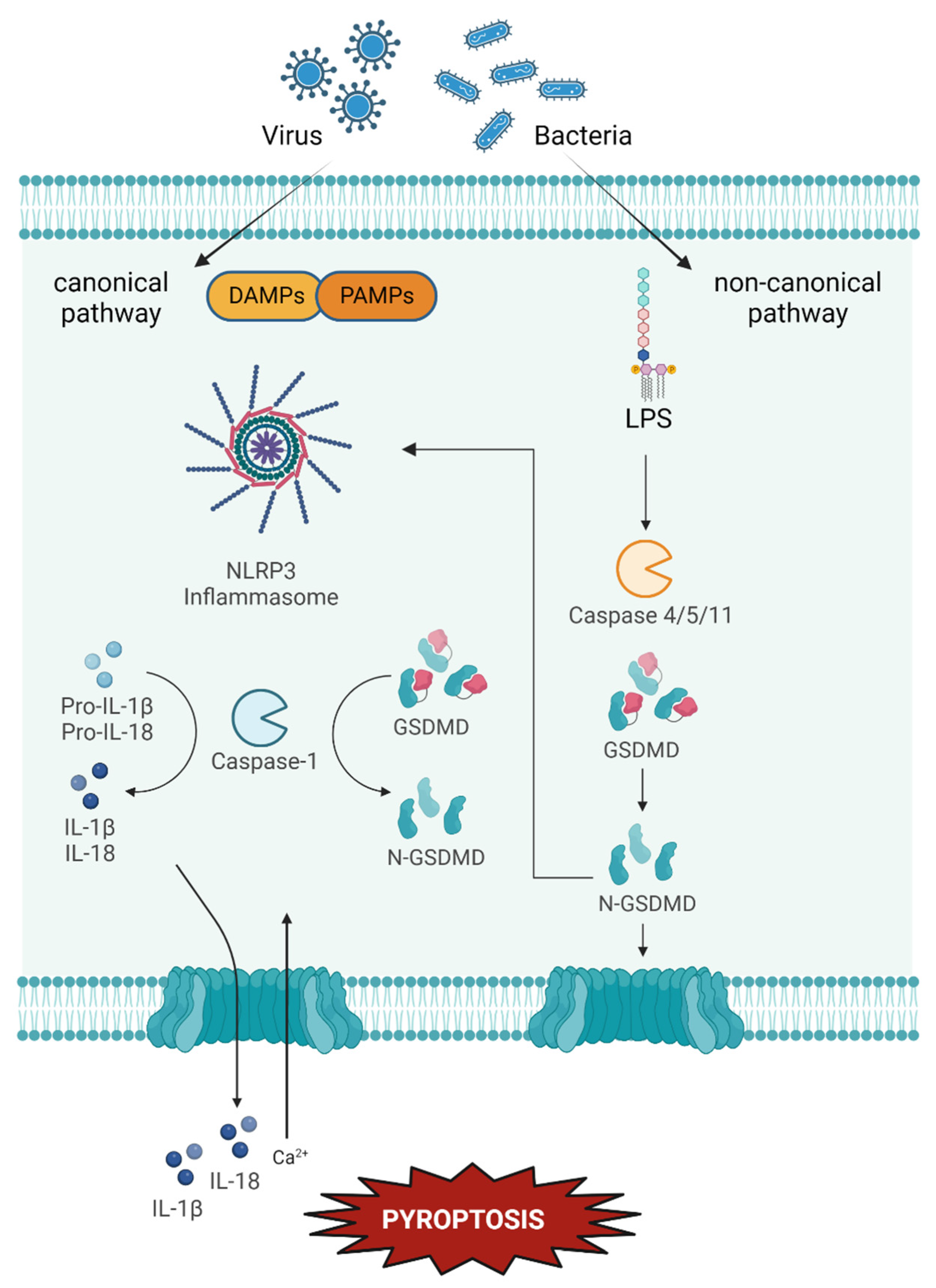
4.3. Pyroptosis
Possible Implication in Dry Eye and Ocular Surface Dysfunction
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-HNE | 4-Hydroxynonenal |
| ACD | Accidental Cell Death |
| ADDE | Aqueous Deficient Dry Eye |
| BAK | Benzalkonium Chloride |
| CASP1 | Caspase 1 |
| DAMP | Damage-associated Molecular Patterns |
| DED | Dry Eye Disease |
| EDE | Evaporative dry eye |
| FDA | Food and Drug Administration |
| GPX4 | Glutathione Peroxidase 4 |
| GSDMD | Gasdermin D |
| GSH | Glutathione |
| HCE | Human corneal epithelial cells |
| ICAM-1 | Intracellular adhesion molecule-1 |
| IL | Interleukine |
| IF | Interferon |
| IFNAR1 | Interferon Receptor 1 |
| JAK | Janus Kinases |
| JNK | c-Jun N-Terminal Kinase |
| LIP | Labile Iron Pool |
| LOX | Lipoxygenases |
| MAPK | Mitogen-Activated Protein Kinase |
| MDA | Malondialdehyde |
| MGD | Meibomian gland dysfunction |
| MLKL | Mixed Lineage Kinase Domain Like Pseudokinase |
| MMP | Matric Metalloprotease |
| NET | Neutrophil extracellular trap |
| NFκB | Nuclear factor Kappa Beta |
| NSSDE | Non-Sjögren Syndrome Dry Eye |
| OSDI | Ocular surface disease index |
| PAMP | Pathogen-associated Molecular Patterns |
| PCD | Programmed cell death |
| PM | Particulate Matter |
| PUFA | Polyunsaturated Fatty Acids |
| RAS | Rat Sarcoma virus protein |
| RCD | Regulated Cell Death |
| RIPK1 | Receptor-Interacting serine/threonine-Protein Kinase 1 |
| RIPK3 | Receptor-Interacting serine/threonine-Protein Kinase 3 |
| ROS | Radical Oxygen Species |
| SOD | Superoxide Dismutase |
| SSDE | Sjögren Syndrome Dry Eye |
| TLR4 | Toll-like Receptor 4 |
| TNF | Tumour Necrosis Factor |
| ZBP1 | Z-DNA binding protein 1 |
References
- Stapleton, F.; Alves, M.; Bunya, V.Y.; Jalbert, I.; Lekhanont, K.; Malet, F.; Na, K.S.; Schaumberg, D.; Uchino, M.; Vehof, J.; et al. TFOS DEWS II Epidemiology Report. Ocul. Surf. 2017, 15, 334–365. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.P.; Nelson, J.D.; Azar, D.T.; Belmonte, C.; Bron, A.J.; Chauhan, S.K.; de Paiva, C.S.; Gomes, J.A.P.; Hammitt, K.M.; Jones, L.; et al. TFOS DEWS II Report Executive Summary. Ocul. Surf. 2017, 15, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.W.; Zhu, X.P.; Pan, S.Y.; Yang, H.; Xiao, X.H. Prevalence and Risk Factors of Dry Eye Disease in Young and Middle-Aged Office Employee: A Xi’an Study. Int. J. Ophthalmol. 2021, 14, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.K.; Mohan, R.; Gokhale, N.; Matalia, H.; Mehta, P. Inflammation and Dry Eye Disease-Where Are We? Int. J. Ophthalmol. 2022, 15, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Perrigan, D.; Morgan, A.; Quintero, S.; Perrigin, J.; Brown, S.; Bergmanson, J. Comparison of Osmolarity Values of Selected Ocular Lubricants. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3901. [Google Scholar]
- Sullivan, D.A.; Rocha, E.M.; Aragona, P.; Clayton, J.A.; Ding, J.; Golebiowski, B.; Hampel, U.; McDermott, A.M.; Schaumberg, D.A.; Srinivasan, S.; et al. TFOS DEWS II Sex, Gender, and Hormones Report. Ocul. Surf. 2017, 15, 284–333. [Google Scholar] [CrossRef]
- Truong, S.; Cole, N.; Stapleton, F.; Golebiowski, B. Sex Hormones and the Dry Eye. Clin. Exp. Optom. 2014, 97, 324–336. [Google Scholar] [CrossRef]
- Matossian, C.; McDonald, M.; Donaldson, K.E.; Nichols, K.K.; Maciver, S.; Gupta, P.K. Dry Eye Disease: Consideration for Women’s Health. J. Women’s Health 2019, 28, 502–514. [Google Scholar] [CrossRef] [Green Version]
- Gagliano, C.; Caruso, S.; Napolitano, G.; Malaguarnera, G.; Cicinelli, M.V.; Amato, R.; Reibaldi, M.; Incarbone, G.; Bucolo, C.; Drago, F.; et al. Low Levels of 17-β-Oestradiol, Oestrone and Testosterone Correlate with Severe Evaporative Dysfunctional Tear Syndrome in Postmenopausal Women: A Case-Control Study. Br. J. Ophthalmol. 2014, 98, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Ablamowicz, A.F.; Nichols, J.J.; Nichols, K.K. Association between Serum Levels of Testosterone and Estradiol with Meibomian Gland Assessments in Postmenopausal Women. Investig. Ophthalmol. Vis. Sci. 2016, 57, 295–300. [Google Scholar] [CrossRef]
- Sullivan, D.A.; Sullivan, B.D.; Evans, J.E.; Schirra, F.; Yamagami, H.; Liu, M.; Richards, S.M.; Suzuki, T.; Schaumberg, D.A.; Sullivan, R.M.; et al. Androgen Deficiency, Meibomian Gland Dysfunction, and Evaporative Dry Eye. Ann. N. Y. Acad. Sci. 2002, 966, 211–222. [Google Scholar] [CrossRef]
- Sriprasert, I.; Warren, D.W.; Mircheff, A.K.; Stanczyk, F.Z. Dry Eye in Postmenopausal Women: A Hormonal Disorder. Menopause 2016, 23, 343–351. [Google Scholar] [CrossRef]
- Nuzzi, R.; Caselgrandi, P. Sex Hormones and Their Effects on Ocular Disorders and Pathophysiology: Current Aspects and Our Experience. Int. J. Mol. Sci. 2022, 23, 3269. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.A.P.; Azar, D.T.; Baudouin, C.; Efron, N.; Hirayama, M.; Horwath-Winter, J.; Kim, T.; Mehta, J.S.; Messmer, E.M.; Pepose, J.S.; et al. TFOS DEWS II Iatrogenic Report. Ocul. Surf. 2017, 15, 511–538. [Google Scholar] [CrossRef] [PubMed]
- Baudouin, C.; Labbé, A.; Liang, H.; Pauly, A. Progress in Retinal and Eye Research Preservatives in Eyedrops: The Good, the Bad and the Ugly Q. Prog. Retin. Eye Res. 2010, 29, 312–334. [Google Scholar] [CrossRef] [PubMed]
- Fraunfelder, F.T.; Sciubba, J.J.; Mathers, W.D. The Role of Medications in Causing Dry Eye. J. Ophthalmol. 2012, 2012, 285851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, J.P.; Nichols, K.K.; Akpek, E.K.; Caffery, B.; Dua, H.S.; Joo, C.K.; Liu, Z.; Nelson, J.D.; Nichols, J.J.; Tsubota, K.; et al. TFOS DEWS II Definition and Classification Report. Ocul. Surf. 2017, 15, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Jarad, N. Treatment of Dry Eye Disease. Table of Artificial Tears Solutions. Chron. Respir. Dis. 2016, 13, 173–188. [Google Scholar] [CrossRef] [Green Version]
- Akpek, E.K.; Bunya, V.Y.; Saldanha, I.J. Sjögren’s Syndrome: More Than Just Dry Eye. Cornea 2019, 38, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, T.H.; Dogru, M.; Matsumoto, Y.; Kojima, T.; Kaido, M.; Ibrahim, O.M.A.; Sato, E.A.; Igarashi, A.; Ichihashi, Y.; Satake, Y.; et al. Evaluation of Lipid Oxidative Stress Status in Sjögren Syndrome Patients. Investig. Ophthalmol. Vis. Sci. 2013, 54, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Chhadva, P.; Raquel, G.; Galor, A. Meibomian Gland Disease: The Role of Gland Dysfunction in Dry Eye Disease. Physiol. Behav. 2016, 176, 139–148. [Google Scholar] [CrossRef]
- Foulks, G.N. Introduction to the Report of the International Dry Eye WorkShop; Ethis Communications: New York, NY, USA, 2007; ISBN 9786468600. [Google Scholar]
- Woodward, A.M.; Senchyna, M.; Argüeso, P. Differential Contribution of Hypertonic Electrolytes to Corneal Epithelial Dysfunction. Exp. Eye Res. 2012, 100, 98–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudouin, C.; Aragona, P.; Messmer, E.M.; Tomlinson, A.; Calonge, M.; Boboridis, K.G.; Akova, Y.A.; Geerling, G.; Labetoulle, M.; Rolando, M. Role of Hyperosmolarity in the Pathogenesis and Management of Dry Eye Disease: Proceedings of the Ocean Group Meeting. Ocul. Surf. 2013, 11, 246–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, S.; Shibuya, M.; Nakashima, H.; Hisamura, R.; Masuda, N.; Imagawa, T.; Uehara, M.; Tsubota, K. Involvement of Oxidative Stress on Corneal Epithelial Alterations in a Blink-Suppressed Dry Eye. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1552–1558. [Google Scholar] [CrossRef]
- Seen, S.; Tong, L. Dry Eye Disease and Oxidative Stress. Acta Ophthalmol. 2018, 96, e412–e420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joossen, C.; Baan, A.; Moreno-Cinos, C.; Joossens, J.; Cools, N.; Moons, L.; Lemmens, K.; Lambeir, A.; Fransen, E.; Delputte, P.; et al. A Novel Serine Protease Inhibitor as Potential Treatment for Dry Eye Syndrome and Ocular Inflammation. Sci. Rep. 2020, 10, 17268. [Google Scholar] [CrossRef] [PubMed]
- Clayton, J.A. Dry Eye. N. Engl. J. Med. 2018, 378, 2212–2223. [Google Scholar] [CrossRef]
- Hampel, U.; Garreis, F. The Human Meibomian Gland Epithelial Cell Line as a Model to Study Meibomian Gland Dysfunction. Exp. Eye Res. 2017, 163, 46–52. [Google Scholar] [CrossRef]
- Balasopoulou, A.; Κokkinos, P.; Pagoulatos, D.; Plotas, P.; Makri, O.E.; Georgakopoulos, C.D.; Vantarakis, A.; Li, Y.; Liu, J.J.; Qi, P.; et al. Anatomy of Cornea and Ocular Surface. BMC Ophthalmol. 2017, 17, 1. [Google Scholar] [CrossRef]
- Kumagai, N.; Fukuda, K.; Fujitsu, Y.; Yamamoto, K.; Nishida, T. Role of Structural Cells of the Cornea and Conjunctiva in the Pathogenesis of Vernal Keratoconjunctivitis. Prog. Retin. Eye Res. 2006, 25, 165–187. [Google Scholar] [CrossRef]
- Buckner, C.A.; Lafrenie, R.M.; Dénommée, J.A.; Caswell, J.M.; Want, D.A.; Gan, G.G.; Leong, Y.C.; Bee, P.C.; Chin, E.; Teh, A.K.H.; et al. The Physiology of Tear Film. Intech 2021, 11, 13. [Google Scholar]
- Cwiklik, L. Tear Film Lipid Layer: A Molecular Level View. Biochim. Biophys. Acta - Biomembr. 2016, 1858, 2421–2430. [Google Scholar] [CrossRef] [PubMed]
- Conrady, C.D.; Joos, Z.P.; Patel, B.C.K. Review: The Lacrimal Gland and Its Role in Dry Eye. J. Ophthalmol. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bron, A.J.; Tiffany, J.M.; Gouveia, S.M.; Yokoi, N.; Voon, L.W. Functional Aspects of the Tear Film Lipid Layer. Exp. Eye Res. 2004, 78, 347–360. [Google Scholar] [CrossRef]
- Zhang, X.; Vimalin Jeyalatha, M.; Qu, Y.; He, X.; Ou, S.; Bu, J.; Jia, C.; Wang, J.; Wu, H.; Liu, Z.; et al. Dry Eye Management: Targeting the Ocular Surface Microenvironment. Int. J. Mol. Sci. 2017, 18, 1398. [Google Scholar] [CrossRef] [Green Version]
- Hori, Y. Secreted Mucins on the Ocular Surface. Investig. Ophthalmol. Vis. Sci. 2018, 59, DES151–DES156. [Google Scholar] [CrossRef] [Green Version]
- Shimazaki, J. Definition and Diagnostic Criteria of Dry Eye Disease: Historical Overview and Future Directions. Investig. Ophthalmol. Vis. Sci. 2018, 59, DES7–DES12. [Google Scholar] [CrossRef] [Green Version]
- Arif, S.A.; Khan, M.I.; Abid, M.S.; Babar, A.; Arif, M.A.; Jahanzaib, H.M.; Khan, I. Frequency and Impact of Individual Symptoms on Quality of Life in Dry Eye Disease in Patients Presenting to a Tertiary Care Hospital. J. Pak. Med. Assoc. 2021, 71, 1063–1068. [Google Scholar] [CrossRef]
- Nichols, K.K. Patient-Reported Symptoms in Dry Dye Disease. Ocul. Surf. 2006, 4, 137–145. [Google Scholar] [CrossRef]
- Khanal, S.; Tomlinson, A.; McFadyen, A.; Diaper, C.; Ramaesh, K. Dry Eye Diagnosis. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1407–1414. [Google Scholar] [CrossRef]
- Wolffsohn, J.S.; Arita, R.; Chalmers, R.; Djalilian, A.; Dogru, M.; Dumbleton, K.; Gupta, P.K.; Karpecki, P.; Lazreg, S.; Pult, H.; et al. TFOS DEWS II Diagnostic Methodology Report. Ocul. Surf. 2017, 15, 539–574. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Chan, B.C.L.; Tsang, M.S.M.; Gao, X.; Leung, P.C.; Lam, C.W.K.; Hu, J.M.; Wong, C.K. Current Advances in Mechanisms and Treatment of Dry Eye Disease: Toward Anti-Inflammatory and Immunomodulatory Therapy and Traditional Chinese Medicine. Front. Med. 2022, 8, 815075. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Rupenthal, I.D. Modern Approaches to the Ocular Delivery of Cyclosporine A. Drug Discov. Today 2016, 21, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Gote, V.; Pal, D.; Ogundele, A.; Mitra, A.K. Ocular Pharmacokinetics of a Topical Ophthalmic Nanomicellar Solution of Cyclosporine (Cequa®) for Dry Eye Disease. Pharm. Res. 2019, 36. [Google Scholar] [CrossRef] [PubMed]
- Othman, T.M.; Mousa, A.; Gikandi, P.W.; AbdelMabod, M.; Abdelrahman, A.M. Efficacy and Safety of Using Topical Cyclosporine A for Treatment of Moderate to Severe Dry Eye Disease. Saudi J. Ophthalmol. 2018, 32, 217–221. [Google Scholar] [CrossRef]
- Soiza, R.L.; Donaldson, A.I.C.; Myint, P.K. Vaccine against Arteriosclerosis: An Update. Ther. Adv. Vaccines 2018, 9, 259–261. [Google Scholar] [CrossRef] [Green Version]
- Beckman, K.; Katz, J.; Majmudar, P.; Rostov, A. Loteprednol Etabonate for the Treatment of Dry Eye Disease. J. Ocul. Pharmacol. Ther. 2020, 36, 497–511. [Google Scholar] [CrossRef]
- Katroscik, J.T. FDA Approves New Prescription Medication for Dry Eye Disease. Pharm. Today 2022, 28, 18. [Google Scholar] [CrossRef]
- Tauber, J.; Wirta, D.L.; Sall, K.; Majmudar, P.A.; Willen, D.; Krösser, S. A Randomized Clinical Study (SEECASE) to Assess Efficacy, Safety, and Tolerability of NOV03 for Treatment of Dry Eye Disease. Cornea 2021, 40, 1132–1140. [Google Scholar] [CrossRef]
- Walsh, K.; Jones, L. The Use of Preservatives in Dry Eye Drops. Clin. Ophthalmol. 2019, 13, 1409–1425. [Google Scholar] [CrossRef] [Green Version]
- Tong, L.; Lan, W.; Lim, R.R.; Chaurasia, S.S. S100A Proteins as Molecular Targets in the Ocular Surface Inflammatory Diseases. Ocul. Surf. 2014, 12, 23–31. [Google Scholar] [CrossRef]
- Mohamed, H.B.; Abd El-Hamid, B.N.; Fathalla, D.; Fouad, E.A. Current Trends in Pharmaceutical Treatment of Dry Eye Disease: A Review. Eur. J. Pharm. Sci. 2022, 175, 106206. [Google Scholar] [CrossRef]
- Talens-Estarelles, C.; García-Marqués, J.V.; Cerviño, A.; García-Lázaro, S. Digital Display Use and Contact Lens Wear: Effects on Dry Eye Signs and Symptoms. Ophthalmic Physiol. Opt. 2022, 797–806. [Google Scholar] [CrossRef]
- Al-Mohtaseb, Z.; Schachter, S.; Lee, B.S.; Garlich, J.; Trattler, W. The Relationship between Dry Eye Disease and Digital Screen Use. Clin. Ophthalmol. 2021, 15, 3811–3820. [Google Scholar] [CrossRef]
- Behar-Cohen, F.; Baillet, G.; de Ayguavives, T.; Garcia, P.O.; Krutmann, J.; Peña-García, P.; Reme, C.; Wolffsohn, J.S. Ultraviolet Damage to the Eye Revisited: Eye-Sun Protection Factor (E-SPF®), a New Ultraviolet Protection Label for Eyewear. Clin. Ophthalmol. 2014, 8, 87–104. [Google Scholar] [CrossRef] [Green Version]
- Deng, R.; Hua, X.; Li, J.; Chi, W.; Zhang, Z.; Lu, F.; Zhang, L.; Pflugfelder, S.C.; Li, D.Q. Oxidative Stress Markers Induced by Hyperosmolarity in Primary Human Corneal Epithelial Cells. PLoS ONE 2015, 10, e0126561. [Google Scholar] [CrossRef] [Green Version]
- Macri, A.; Scanarotti, C.; Bassi, A.M.; Giuffrida, S.; Sangalli, G.; Traverso, C.E.; Iester, M. Evaluation of Oxidative Stress Levels in the Conjunctival Epithelium of Patients with or without Dry Eye, and Dry Eye Patients Treated with Preservative-Free Hyaluronic Acid 0.15 % and Vitamin B12 Eye Drops. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 425–430. [Google Scholar] [CrossRef]
- Wakamatsu, T.H.; Dogru, M.; Ayako, I.; Takano, Y.; Matsumoto, Y.; Ibrahim, O.M.A.; Okada, N.; Satake, Y.; Fukagawa, K.; Shimazaki, J.; et al. Evaluation of Lipid Oxidative Stress Status and Inflammation in Atopic Ocular Surface Disease. Mol. Vis. 2010, 16, 2465–2475. [Google Scholar]
- Gaschier, M.M.; Stockwell, B.R. Lipid Peroxidation in Cell Death. Physiol. Behav. 2018, 176, 139–148. [Google Scholar] [CrossRef]
- Choi, W.; Lian, C.; Ying, L.; Kim, G.E.; You, I.C.; Park, S.H.; Yoon, K.C. Expression of Lipid Peroxidation Markers in the Tear Film and Ocular Surface of Patients with Non-Sjogren Syndrome: Potential Biomarkers for Dry Eye Disease. Curr. Eye Res. 2016, 41, 1143–1149. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, T.; Li, J.; Xia, M.; Li, Y.; Wang, X.; Liu, C.; Zheng, T.; Chen, R.; Kan, D.; et al. Oxidative Stress and 4-Hydroxy-2-Nonenal (4-HNE): Implications in the Pathogenesis and Treatment of Aging-Related Diseases. J. Immunol. Res. 2022, 2022. [Google Scholar] [CrossRef]
- Hessen, M.; Akpek, E.K. Dry Eye: An Inflammatory Ocular Disease. J. Ophthalmic Vis. Res. 2014, 9, 240–250. [Google Scholar]
- Yamaguchi, T. Inflammatory Response in Dry Eye. Investig. Ophthalmol. Vis. Sci. 2018, 59, DES192–DES199. [Google Scholar] [CrossRef] [Green Version]
- Ganesalingam, K.; Ismail, S.; Sherwin, T.; Craig, J.P. Molecular Evidence for the Role of Inflammation in Dry Eye Disease. Clin. Exp. Optom. 2019, 102, 446–454 . [Google Scholar] [CrossRef]
- Liu, H.; Gambino, F.; Algenio, C.S.; Wu, C.; Gao, Y.; Bouchard, C.S.; Qiao, L.; Bu, P.; Zhao, S. Inflammation and Oxidative Stress Induced by Lipid Peroxidation Metabolite 4-Hydroxynonenal in Human Corneal Epithelial Cells. Graefe’s Arch. Clin. Exp. Ophthalmol. 2020, 258, 1717–1725. [Google Scholar] [CrossRef]
- Lan, W.; Petznick, A.; Heryati, S.; Rifada, M.; Tong, L. Nuclear Factor-ΚB: Central Regulator in Ocular Surface Inflammation and Diseases. Ocul. Surf. 2012, 10, 137–148. [Google Scholar] [CrossRef]
- Stern, M.E.; Pflugfelder, S.C. Inflammation in Dry Eye. Ocul. Surf. 2004, 2, 124–130. [Google Scholar] [CrossRef]
- Rolando, M.; Zierhut, M. The Ocular Surface and Tear Film and Their Dysfunction in Dry Eye Disease. Surv. Ophthalmol. 2001, 45, 203–210. [Google Scholar] [CrossRef]
- Yeh, S.; Song, X.J.; Farley, W.; Li, D.Q.; Stern, M.E.; Pflugfelder, S.C. Apoptosis of Ocular Surface Cells in Experimentally Induced Dry Eye. Investig. Ophthalmol. Vis. Sci. 2003, 44, 124–129. [Google Scholar] [CrossRef] [Green Version]
- Shi, K.; Yin, Q.; Tang, X.; Yu, X.; Zheng, S.; Shentu, X. Necroptosis Contributes to Airborne Particulate Matter-Induced Ocular Surface Injury. Toxicology 2022, 470, 153140. [Google Scholar] [CrossRef]
- Chen, M.; Rong, R.; Xia, X. Spotlight on Pyroptosis: Role in Pathogenesis and Therapeutic Potential of Ocular Diseases. J. Neuroinflamm. 2022, 19, 183. [Google Scholar] [CrossRef] [PubMed]
- Hantera, M. Tear Film Biomarkers in Dry Eye Disease. US Ophthalmic Rev. 2020, 13, 68. [Google Scholar] [CrossRef]
- Ahmad, S.S. Update on the Role of Impression Cytology in Ocular Surface Disease. Taiwan J. Ophthalmol. 2017, 8, 53–55. [Google Scholar] [CrossRef]
- Chiva, A. Tear Biomarkers in Dry Eye Disease. Eur. Ophthalmic Rev. 2019, 13, 21. [Google Scholar] [CrossRef]
- Navel, V.; Sapin, V.; Henrioux, F.; Blanchon, L.; Labbé, A.; Chiambaretta, F.; Baudouin, C.; Dutheil, F. Oxidative and Antioxidative Stress Markers in Dry Eye Disease: A Systematic Review and Meta-Analysis. Acta Ophthalmol. 2022, 100, 45–57. [Google Scholar] [CrossRef]
- Akkurt Arslan, M.; Kolman, I.; Pionneau, C.; Chardonnet, S.; Magny, R.; Baudouin, C.; Brignole-Baudouin, F.; Kessal, K. Proteomic Analysis of Tears and Conjunctival Cells Collected with Schirmer Strips Using Timstof pro: Preanalytical Considerations. Metabolites 2022, 12, 2. [Google Scholar] [CrossRef]
- Long, J.S.; Ryan, K.M. New Frontiers in Promoting Tumour Cell Death: Targeting Apoptosis, Necroptosis and Autophagy. Oncogene 2012, 31, 5045–5060. [Google Scholar] [CrossRef] [Green Version]
- Vogt, C. ALYTES OBSTETRICANS. Biol. Cent. 1842, 2, 1–114. [Google Scholar]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Lockshin, A.; Williams, M. Programmed Cell Death. Endocrine Potentiation of the Breakdown of the Intersegmental Muscles of Silkmoths. J. Insect Physiol. 1964, 10, 643–649. [Google Scholar] [CrossRef]
- Lockshin, R.A.; Williams, C.M. Programmed Cell Death-I. Cytology of Degeneration in the Intersegmental Muscles of the Pernyi Silkmoth. J. Insect Physiol. 1965, 11, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Ameisen, J.C. On the Origin, Evolution, and Nature of Programmed Cell Death: A Timeline of Four Billion Years. Cell Death Differ. 2002, 9, 367–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadeel, B.; Orrenius, S. Apoptosis: A Basic Biological Phenomenon with Wide-Ranging Implications in Human Disease. J. Intern. Med. 2005, 258, 479–517. [Google Scholar] [CrossRef] [PubMed]
- Armenta, D.A.; Dixon, S.J. Investigating Nonapoptotic Cell Death Using Chemical Biology Approaches. Cell Chem. Biol. 2020, 27, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus Accessory Aspects of Cell Death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; El-Deiry, W.S.; Golstein, P.; Peter, M.E.; Vaux, D.; Vandenabeele, P.; Zhivotovsky, B.; Blagosklonny, M.V.; Malorni, W.; Knight, R.A.; et al. Classification of Cell Death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005, 12, 1463–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The Molecular Machinery of Regulated Cell Death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated Necrosis: The Expanding Network of Non-Apoptotic Cell Death Pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Linkermann, A.; Stockwell, B.R.; Krautwald, S.; Anders, H.J. Regulated Cell Death and Inflammation: An Auto-Amplification Loop Causes Organ Failure. Nat. Rev. Immunol. 2014, 14, 759–767. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A Regulated Inflammatory Mode of Cell Death. J. Neuroinflamm. 2018, 15, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and Its Role in Inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Brignole, F.; De Saint-Jean, M.; Goldschild, M.; Becquet, F.; Goguel, A.; Baudouin, C. Expression of Fas-Fas Ligand Antigens and Apoptotic Marker APO2.7 by the Human Conjunctival Epithelium. Positive Correlation with Class II HLA DR Expression in Inflammatory Ocular Surface Disorders. Exp. Eye Res. 1998, 67, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, H.; Zeng, W.; Wei, J. Edaravone Protects against Hyperosmolarity-Induced Oxidative Stress and Apoptosis in Primary Human Corneal Epithelial Cells. PLoS ONE 2017, 12, e0174437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitoux, M.A.; Kessal, K.; Melik Parsadaniantz, S.; Claret, M.; Guerin, C.; Baudouin, C.; Brignole-Baudouin, F.; Réaux-Le Goazigo, A. Benzalkonium Chloride-Induced Direct and Indirect Toxicity on Corneal Epithelial and Trigeminal Neuronal Cells: Proinflammatory and Apoptotic Responses in Vitro. Toxicol. Lett. 2020, 319, 74–84. [Google Scholar] [CrossRef]
- Bonneau, N.; Brignole-Baudouin, F.; Guerin, C.; Melik-Parsadaniantz, S.; Baudouin, C.; Réaux-Le Goazigo, A. Effects of CX3CR1 deficiency on trigeminal cell death in an in vitro Toxicity-Induced Dry Eye model. Investig. Ophthalmol. Vis. Sci. 2022, 63, 684-F0138. [Google Scholar]
- Baudouin, C.; Messmer, E.M.; Aragona, P.; Geerling, G.; Akova, Y.A.; Benítez-Del-Castillo, J.; Boboridis, K.G.; Merayo-Lloves, J.; Rolando, M.; Labetoulle, M. Revisiting the Vicious Circle of Dry Eye Disease: A Focus on the Pathophysiology of Meibomian Gland Dysfunction. Br. J. Ophthalmol. 2016, 100, 300–306. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Maiorino, M. Lipid Peroxidation and Ferroptosis: The Role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Bayır, H.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Patel, S.J.; Amoscato, A.A.; Lamade, A.M.; Yang, Q.; Vladimirov, G.K.; Philpott, C.C.; Kagan, V.E. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem. Biol. 2020, 27, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Pratt, D.A. The Chemical Basis of Ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Proneth, B. Selenium: Tracing Another Essential Element of Ferroptotic Cell Death. Cell Chem. Biol. 2020, 27, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in Cancer Therapy: A Novel Approach to Reversing Drug Resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Tan, Q.; Fang, Y.; Gu, Q. Mechanisms of Modulation of Ferroptosis and Its Role in Central Nervous System Diseases. Front. Pharmacol. 2021, 12, 657033. [Google Scholar] [CrossRef]
- Ni, L.; Yuan, C.; Wu, X. Targeting Ferroptosis in Acute Kidney Injury. Cell Death Dis. 2022, 13, 182. [Google Scholar] [CrossRef]
- Van Coillie, S.; Van San, E.; Goetschalckx, I.; Wiernicki, B.; Mukhopadhyay, B.; Tonnus, W.; Choi, S.M.; Roelandt, R.; Dumitrascu, C.; Lamberts, L.; et al. Targeting Ferroptosis Protects against Experimental (Multi)Organ Dysfunction and Death. Nat. Commun. 2022, 13, 657033. [Google Scholar] [CrossRef]
- Luo, L.; Mo, G.; Huang, D. Ferroptosis in Hepatic Ischemia-Reperfusion Injury: Regulatory Mechanisms and New Methods for Therapy (Review). Mol. Med. Rep. 2021, 23, 1. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.J.; Song, W.T.; Yao, F.; Zhang, X.; Peng, J.; Luo, X.J.; Xia, X.B. Involvement of Regulated Necrosis in Blinding Diseases: Focus on Necroptosis and Ferroptosis. Exp. Eye Res. 2020, 191, 107922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sheng, S.; Wang, W.; Dai, J.; Zhong, Y. Molecular Mechanisms of Iron Mediated Programmed Cell Death and Its Roles in Eye Diseases. Front. Nutr. 2022, 9, 844757. [Google Scholar] [CrossRef] [PubMed]
- Rowsey, T.G.; Karamichos, D. The Role of Lipids in Corneal Diseases and Dystrophies: A Systematic Review. Clin. Transl. Med. 2017, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, Y.Y.; Tong, L. Barrier Function in the Ocular Surface: From Conventional Paradigms to New Opportunities. Ocul. Surf. 2015, 13, 103–109. [Google Scholar] [CrossRef] [PubMed]
- de Paiva, C.S.; St. Leger, A.J.; Caspi, R.R. Mucosal Immunology of the Ocular Surface. Mucosal Immunol. 2022, 15, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.J. The Environment and the Eye. Eye 2004, 18, 1235–1250. [Google Scholar] [CrossRef] [Green Version]
- Cejková, J.; Cejka, C. The Role of Oxidative Stress in Corneal Diseases and Injuries. Histol. Histopathol. 2015, 30, 893–900. [Google Scholar] [CrossRef]
- Sakai, O.; Uchida, T.; Imai, H.; Ueta, T. Glutathione Peroxidase 4 Plays an Important Role in Oxidative Homeostasis and Wound Repair in Corneal Epithelial Cells. FEBS Open Bio 2016, 6, 1238–1247. [Google Scholar] [CrossRef]
- Ogawa, Y. Glutathione Peroxidase 4, a Unique Antioxidant Enzyme, Plays a Role in Protecting Ocular Surface Mucosal Epithelia. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1657. [Google Scholar] [CrossRef] [Green Version]
- Katsinas, N.; Rodríguez-Rojo, S.; Enríquez-De-salamanca, A. Olive Pomace Phenolic Compounds and Extracts Can Inhibit Inflammatory-and Oxidative-Related Diseases of Human Ocular Surface Epithelium. Antioxidants 2021, 10, 1150. [Google Scholar] [CrossRef] [PubMed]
- Lovatt, M.; Adnan, K.; Kocaba, V.; Dirisamer, M.; Peh, G.S.L.; Mehta, J.S. Peroxiredoxin-1 Regulates Lipid Peroxidation in Corneal Endothelial Cells. Redox Biol. 2020, 30, 101417. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Zeng, H.; Wang, B.; Yang, X.; He, D.; Wang, L.; Ouyang, H.; Yuan, J. AKR1C1 Protects Corneal Epithelial Cells Against Oxidative Stress-Mediated Ferroptosis in Dry Eye. Invest. Ophthalmol. Vis. Sci. 2022, 63, 3. [Google Scholar] [CrossRef] [PubMed]
- Bucolo, C.; Fidilio, A.; Platania, C.B.M.; Geraci, F.; Lazzara, F.; Drago, F. Antioxidant and Osmoprotecting Activity of Taurine in Dry Eye Models. J. Ocul. Pharmacol. Ther. 2018, 34, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Čejková, J.; Ardan, T.; Šimonová, Z.; Čejka, Č.; Malec, J.; Dotřelova, D.; Brůnová, B. Decreased Expression of Antioxidant Enzymes in the Conjunctival Epithelium of Dry Eye (Sjögren’s Syndrome) and Its Possible Contribution to the Development of Ocular Surface Oxidative Injuries. Histol. Histopathol. 2008, 23, 1477–1483. [Google Scholar] [CrossRef]
- Magny, R.; Kessal, K.; Regazzetti, A.; Ben Yedder, A.; Baudouin, C.; Mélik Parsadaniantz, S.; Brignole-Baudouin, F.; Laprévote, O.; Auzeil, N. Lipidomic Analysis of Epithelial Corneal Cells Following Hyperosmolarity and Benzalkonium Chloride Exposure: New Insights in Dry Eye Disease. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 2020, 1865, 158728. [Google Scholar] [CrossRef] [PubMed]
- Augustin, A.J.; Spitznas, M.; Kaviani, N.; Meller, D.; Koch, F.H.J.; Grus, F.; Göbbels, M.J. Oxidative Reactions in the Tear Fluid of Patients Suffering from Dry Eyes. Graefe’s Arch. Clin. Exp. Ophthalmol. 1995, 233, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Dogru, M.; Kojima, T.; Simsek, C.; Tsubotav, K. Potential Role of Oxidative Stress in Ocular Surface Inflammation and Dry Eye Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, DES163–DES168. [Google Scholar] [CrossRef] [Green Version]
- Chhadva, P.; Alexander, A.; McClellan, A.L.; McManus, K.T.; Seiden, B.; Galor, A. The Impact of Conjunctivochalasis on Dry Eye Symptoms and Signs. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2867–2871. [Google Scholar] [CrossRef] [Green Version]
- Ward, S.K.; Wakamatsu, T.H.; Dogru, M.; Ibrahim, O.M.A.; Kaido, M.; Ogawa, Y.; Matsumoto, Y.; Igarashi, A.; Ishida, R.; Shimazaki, J.; et al. The Role of Oxidative Stress and Inflammation in Conjunctivochalasis. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1994–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxidants Redox Signal. 2018, 29, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Nezzar, H.; Mbekeani, J.N.; Noblanc, A.; Chiambaretta, F.; Drevet, J.R.; Kocer, A. Investigation of Antioxidant Systems in Human Meibomian Gland and Conjunctival Tissues. Exp. Eye Res. 2017, 165, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Ran, Q.; Liang, H.; Ikeno, Y.; Qi, W.; Prolla, T.A.; Ii, L.J.R.; Wolf, N.; Vanremmen, H.; Richardson, A. Reduction in Glutathione Peroxidase 4 Increases Life Span Through Increased Sensitivity to Apoptosis. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 932–942. [Google Scholar] [CrossRef] [Green Version]
- Sakai, O.; Uchida, T.; Imai, H.; Ueta, T.; Amano, S. Role of Glutathione Peroxidase 4 in Conjunctival Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Telegina, D.V.; Kozhevnikova, O.S.; Kolosova, N.G. Molecular Mechanisms of Cell Death in Retina during Development of Age-Related Macular Degeneration. Adv. Gerontol. 2017, 7, 17–24. [Google Scholar] [CrossRef]
- Totsuka, K.; Ueta, T.; Uchida, T.; Roggia, M.F.; Nakagawa, S.; Vavvas, D.G.; Honjo, M.; Aihara, M. Oxidative Stress Induces Ferroptotic Cell Death in Retinal Pigment Epithelial Cells. Exp. Eye Res. 2019, 181, 316–324. [Google Scholar] [CrossRef]
- Henning, Y.; Blind, U.S.; Larafa, S.; Matschke, J.; Fandrey, J. Hypoxia Aggravates Ferroptosis in RPE Cells by Promoting the Fenton Reaction. Cell Death Dis. 2022, 13, 662. [Google Scholar] [CrossRef]
- Shu, W.; Dunaief, J.L. Potential Treatment of Retinal Diseases with Iron Chelators. Pharmaceuticals 2018, 11, 112. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione Depletion Induces Ferroptosis, Autophagy, and Premature Cell Senescence in Retinal Pigment Epithelial Cells Article. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Gao, M.; Liang, J.; Chen, Y.; Wang, Y. SLC7A11 Reduces Laser-Induced Choroidal Neovascularization by Inhibiting RPE Ferroptosis and VEGF Production. Front. Cell Dev. Biol. 2021, 9, 639851. [Google Scholar] [CrossRef]
- Sakamoto, K.; Suzuki, T.; Takahashi, K.; Koguchi, T.; Hirayama, T.; Mori, A.; Nakahara, T.; Nagasawa, H.; Ishii, K. Iron-Chelating Agents Attenuate NMDA-Induced Neuronal Injury via Reduction of Oxidative Stress in the Rat Retina. Exp. Eye Res. 2018, 171, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes (Basel) 2020, 11, 1120. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Oveson, B.C.; Jo, Y.J.; Lauer, T.W.; Usui, S.; Komeima, K.; Xie, B.; Campochiaro, P.A. Increased Expression of Glutathione Peroxidase 4 Strongly Protects Retina from Oxidative Damage. Antioxid. Redox Signal. 2009, 11, 715–724. [Google Scholar] [CrossRef]
- Raman, T.; Ramar, M.; Arumugam, M.; Nabavi, S.M.; Varsha, M.K.N.S. Cytoprotective Mechanism of Action of Curcumin against Cataract. Pharmacol. Reports 2016, 68, 561–569. [Google Scholar] [CrossRef]
- Yan, H.F.; Tuo, Q.Z.; Yin, Q.Z.; Lei, P. The Pathological Role of Ferroptosis in Ischemia/Reperfusion-Related Injury. Zool. Res. 2020, 41, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jiang, L.; Zhong, Y.; Zhang, Y.; Yin, Q.; Li, S.; Zhang, X.; Han, H.; Yao, K. Ferrostatin-1-Loaded Liposome for Treatment of Corneal Alkali Burn via Targeting Ferroptosis. Bioeng. Transl. Med. 2022, 7, e10276. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, S.; Berghe, T.V.; Vandenabeele, P. Initiation and Execution Mechanisms of Necroptosis: An Overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.; Peng, W.; Liu, Y.; Yan, C.; Maki, J.; Degterev, A.; Yuan, J.; Shi, Y. Structural Basis of RIP1 Inhibition by Necrostatins. Structure 2013, 21, 493–499. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma Membrane Translocation of Trimerized MLKL Protein Is Required for TNF-Induced Necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, K.D.; Davis, M.A.; Daniels, B.P.; Olsen, T.M.; Ralli- Jain, P.; Gale, M., Jr.; Oberst, A. MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1β Independent of Gasdermin-D. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.Q.; Chen, X.; Cai, Q.; Yang, Z.H.; Huang, D.; Wu, R.; et al. RIP1 Autophosphorylation Is Promoted by Mitochondrial ROS and Is Essential for RIP3 Recruitment into Necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearney, C.J.; Martin, S.J. An Inflammatory Perspective on Necroptosis. Mol. Cell 2017, 65, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xie, X.; Ren, Y.; Shao, Z.; Zhang, N.; Li, L.; Ding, X.; Zhang, L. The Role of Necroptosis in Disease and Treatment. MedComm 2021, 2, 730–755. [Google Scholar] [CrossRef] [PubMed]
- Baidya, R.; Crawford, D.H.G.; Gautheron, J.; Wang, H.; Bridle, K.R. Necroptosis in Hepatosteatotic Ischaemia-Reperfusion Injury. Int. J. Mol. Sci. 2020, 21, 5931. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J. Necroptosis in Acute Kidney Injury. Nephron 2018, 139, 342–348. [Google Scholar] [CrossRef]
- Coornaert, I.; Hofmans, S.; Devisscher, L.; Augustyns, K.; Van Der Veken, P.; De Meyer, G.R.Y.; Martinet, W. Novel Drug Discovery Strategies for Atherosclerosis That Target Necrosis and Necroptosis. Expert Opin. Drug Discov. 2018, 13, 477–488. [Google Scholar] [CrossRef]
- Dionísio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Molecular Mechanisms of Necroptosis and Relevance for Neurodegenerative Diseases, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 353. [Google Scholar]
- Chalkias, A.; Xanthos, T. Acute Kidney Injury. Lancet 2012, 380, 1904. [Google Scholar] [CrossRef]
- Lin, W.; Chen, M.; Cissé, Y.; Chen, X.; Bai, L. Roles and Mechanisms of Regulated Necrosis in Corneal Diseases: Progress and Perspectives. J. Ophthalmol. 2022, 2022, 2695212. [Google Scholar] [CrossRef]
- Do, Y.J.; Sul, J.W.; Jang, K.H.; Kang, N.S.; Kim, Y.H.; Kim, Y.G.; Kim, E. A Novel RIPK1 Inhibitor That Prevents Retinal Degeneration in a Rat Glaucoma Model. Exp. Cell Res. 2017, 359, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Hanus, J.; Anderson, C.; Wang, S. RPE Necroptosis in Response to Oxidative Stress and in AMD. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, M.; Huang, X.; Liang, W.; Li, G.; Lu, X.; Li, Y.; Pan, H.; Shi, L.; Zhu, H.; et al. Ubiquitination of RIPK1 Regulates Its Activation Mediated by TNFR1 and TLRs Signaling in Distinct Manners. Nat. Commun. 2020, 11, 6364. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Chen, M.; Zheng, D.; He, J.; Song, M.; Mo, L.; Feng, J.; Lan, J. A Novel Damage Mechanism: Contribution of the Interaction between Necroptosis and ROS to High Glucose-Induced Injury and Inflammation in H9c2 Cardiac Cells. Int. J. Mol. Med. 2017, 40, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Kessal, K.; Daull, P.; Cimbolini, N.; Feraille, L.; Grillo, S.; Docquier, M.; Baudouin, C.; Brignole-Baudouin, F.; Garrigue, J.S. Comparison of Two Experimental Mouse Dry Eye Models through Inflammatory Gene Set Enrichment Analysis Based on a Multiplexed Transcriptomic Approach. Int. J. Mol. Sci. 2021, 22, 10770. [Google Scholar] [CrossRef]
- Pflugfelder, S.C.; De Paiva, C.S.; Tong, L.; Luo, L.; Stern, M.E.; Li, D.Q. Stress-Activated Protein Kinase Signaling Pathways in Dry Eye and Ocular Surface Disease. Ocul. Surf. 2005, 3, S154–S157. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahiki, K.; Wang, L.; Handa, J.T. The Impact of Oxidative Stress and Inflammation on RPE Degeneration in Non-Neovascular AMD. Rev. del Col. Am. Cardiol. 2018, 72, 2964–2979. [Google Scholar] [CrossRef]
- Jang, K.H.; Do, Y.J.; Koo, T.S.; Choi, J.S.; Song, E.J.; Hwang, Y.; Bae, H.J.; Lee, J.H.; Kim, E. Protective Effect of RIPK1-Inhibitory Compound in in Vivo Models for Retinal Degenerative Disease. Exp. Eye Res. 2019, 180, 8–17. [Google Scholar] [CrossRef]
- Murakami, Y.; Matsumoto, H.; Roh, M.; Suzuki, J.; Hisatomi, T.; Ikeda, Y.; Miller, J.W. Receptor Interacting Protein Kinase Mediates Necrotic Cone but Not Rod Cell Death in a Mouse Model of Inherited Degeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 14598–14603. [Google Scholar] [CrossRef] [Green Version]
- Fink, S.L.; Cookson, B.T. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Cookson, B.T.; Brennan, M.A. Pro-Inflammatory Programmed Cell Death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, S.M.; Karki, R.; Kanneganti, T.-D. Molecular Mechanisms and Functions of Atg11 in Selective Autophagy. Immunol Rev. 2018, 277, 61–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Q.; Yi, T.; Chen, C. NF-ΚB-Gasdermin D (GSDMD) Axis Couples Oxidative Stress and NACHT, LRR and PYD Domains-Containing Protein 3 (NLRP3) Inflammasome-Mediated Cardiomyocyte Pyroptosis Following Myocardial Infarction. Med. Sci. Monit. 2018, 24, 6044–6052. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, X.; Yuan, S.; Wen, S.; Liu, X.; Wang, C.; Qu, Z.; Li, J.; Liu, H.; Sun, L.; et al. TLR4/NF-ΚB Signaling Induces GSDMD-Related Pyroptosis in Tubular Cells in Diabetic Kidney Disease. Front. Endocrinol. (Lausanne) 2019, 10, 603. [Google Scholar] [CrossRef] [Green Version]
- Sangiuliano, B.; Pérez, N.M.; Moreira, D.F.; Belizário, J.E. Cell Death-Associated Molecular-Pattern Molecules: Inflammatory Signaling and Control. Mediators Inflamm. 2014, 2014, 821043. [Google Scholar] [CrossRef]
- Gritsenko, A.; Yu, S.; Martin-Sanchez, F.; Diaz-del-Olmo, I.; Nichols, E.M.; Davis, D.M.; Brough, D.; Lopez-Castejon, G. Priming Is Dispensable for NLRP3 Inflammasome Activation in Human Monocytes In Vitro. Front. Immunol. 2020, 11, 2318. [Google Scholar] [CrossRef]
- Wang, C.; Ruan, J. Mechanistic Insights into Gasdermin Pore Formation and Regulation in Pyroptosis. J. Mol. Biol. 2022, 434, 167297. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, Pyroptosis and Apoptosis: An Intricate Game of Cell Death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Kopitar-Jerala, N. The Role of Interferons in Inflammation and Inflammasome Activation. Front. Immunol. 2017, 8, 1331. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 Is the Molecular Switch for Apoptosis, Necroptosis and Pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Ting, D.S.J.; Ho, C.S.; Deshmukh, R.; Said, D.G.; Dua, H.S. Infectious Keratitis: An Update on Epidemiology, Causative Microorganisms, Risk Factors, and Antimicrobial Resistance. Eye 2021, 35, 1084–1101. [Google Scholar] [CrossRef]
- Sharma, P.; Sharma, N.; Mishra, P.; Joseph, J.; Mishra, D.K.; Garg, P.; Roy, S. Differential Expression of Antimicrobial Peptides in Streptococcus Pneumoniae Keratitis and STAT3-Dependent Expression of LL-37 by Streptococcus Pneumoniae in Human Corneal Epithelial Cells. Pathogens 2019, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Liu, X.; Liu, X.; Shi, Y.; Jin, X.; Zhang, N.; Li, X. Wedelolactone Ameliorates Pseudomonas aeruginosa -Induced Inflammation and Corneal Injury by Suppressing Caspase-4 / 5 / 11 / GSDMD-Mediated Non-Canonical Pyroptosis. Exp. Eye Res. 2021, 211, 108750. [Google Scholar] [CrossRef]
- Qu, W.; Wang, Y.; Wu, Y.; Liu, Y.; Chen, K.; Liu, X.; Zou, Z.; Huang, X.; Wu, M. Triggering Receptors Expressed on Myeloid Cells 2 Promotes Corneal Resistance against Pseudomonas aeruginosa by Inhibiting Caspase-1-Dependent Pyroptosis. Front. Immunol. 2018, 9, 1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, F.; Xiao, Y.; Zaheer, M.; Volpe, E.A.; Pflugfelder, S.C.; Li, D.; Paiva, C.S. De Inhibition of NLRP3 Inflammasome Pathway by Butyrate Improves Corneal Wound Healing in Corneal Alkali Burn. Int. J. Mol. Sci. 2017, 18, 562. [Google Scholar] [CrossRef]
- Zheng, Q.; Ren, Y.; Reinach, P.S.; She, Y.; Xiao, B.; Hua, S.; Qu, J.; Chen, W. Reactive Oxygen Species Activated NLRP3 Inflammasomes Prime Environment-Induced Murine Dry Eye. Exp. Eye Res. 2014, 125, 1–8. [Google Scholar] [CrossRef]
- Niu, L.; Li, L.; Xing, C.; Luo, B.; Hu, C.; Song, M.; Niu, J.; Ruan, Y.; Sun, X.; Lei, Y. Airborne Particulate Matter (PM2.5) Triggers Cornea Inflammation and Pyroptosis via NLRP3 Activation. Ecotoxicol. Environ. Saf. 2021, 207, 111306. [Google Scholar] [CrossRef]
- Chen, H.; Gan, X.; Li, Y.; Gu, J.; Liu, Y.; Deng, Y.; Wang, X.; Hong, Y.; Hu, Y.; Su, L.; et al. NLRP12- and NLRC4-Mediated Corneal Epithelial Pyroptosis Is Driven by GSDMD Cleavage Accompanied by IL-33 Processing in Dry Eye. Ocul. Surf. 2020, 18, 783–794. [Google Scholar] [CrossRef]
- Wang, B.; Zeng, H.; Zuo, X.; Yang, X.; Wang, X.; He, D.; Yuan, J. TLR4-Dependent DUOX2 Activation Triggered Oxidative Stress and Promoted HMGB1 Release in Dry Eye. Front. Med. 2022, 8, 781616. [Google Scholar] [CrossRef]
- Zhang, J.; Dai, Y.; Yang, Y.; Xu, J. Calcitriol Alleviates Hyperosmotic Stress-Induced Corneal Epithelial Cell Damage via Inhibiting the NLRP3-ASC-Caspase-1-GSDMD Pyroptosis Pathway in Dry Eye Disease. J. Inflamm. Res. 2021, 14, 2955. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jin, X.; Wang, J.; Li, X.; Zhang, H. Dexamethasone Attenuates Dry Eye-Induced Pyroptosis by Regulating the Kcnq1ot1/Mir-214 Cascade. SSRN Electron. J. 2022, 186, 109073. [Google Scholar] [CrossRef]
- Vakrakou, A.G.; Svolaki, I.P.; Evangelou, K.; Gorgoulis, V.G.; Manoussakis, M.N. Cell-Autonomous Epithelial Activation of AIM2 (Absent in Melanoma-2) in Fl Ammasome by Cytoplasmic DNA Accumulations in Primary Sjögren’s Syndrome. J. Autoimmun. 2019, 2, 102381. [Google Scholar] [CrossRef]
- Ambati, J.; Atkinson, J.P.; Gelfand, B.D. Immunology of Age-Related Macular Degeneration. Nat. Rev. Immunol. 2013, 13, 438–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, S.L.; Campbell, M.; Ozaki, E.; Salomon, R.G.; Mori, A.; Kenna, P.F.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Lavelle, E.C.; et al. NLRP3 Has a Protective Role in Age-Related Macular Degeneration through the Induction of IL-18 by Drusen Components. Nat. Med. 2012, 18, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Su, S.; Jiang, S.; Sun, X.; Wang, J. Role of Amyloid β-Peptide in the Pathogenesis of Age-Related Macular Degeneration. BMJ Open Ophthalmol. 2021, 6, 1–6. [Google Scholar] [CrossRef]
- Liu, R.T.; Gao, J.; Cao, S.; Sandhu, N.; Cui, J.Z.; Chou, C.L.; Fang, E.; Matsubara, J.A. Inflammatory Mediators Induced by Amyloid-Beta in the Retina and RPE In Vivo: Implications for Inflammasome Activation in Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2225–2237. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, A.; Niskanen, H.; Suuronen, T.; Kinnunen, K.; Salminen, A.; Kaarniranta, K. Oxidative Stress Activates NLRP3 Inflammasomes in ARPE-19 Cells-Implications for Age-Related Macular Degeneration (AMD). Immunol. Lett. 2012, 147, 29–33. [Google Scholar] [CrossRef]
- Baudouin, C.; Kolko, M.; Melik-Parsadaniantz, S.; Messmer, E.M. Inflammation in Glaucoma: From the Back to the Front of the Eye, and Beyond. Prog. Retin. Eye Res. 2021, 83, 100916. [Google Scholar] [CrossRef]
- Rossi, T.; Romano, M.R.; Iannetta, D.; Romano, V.; Gualdi, L.; D’Agostino, I.; Ripandelli, G. Cataract Surgery Practice Patterns Worldwide: A Survey. BMJ Open Ophthalmol. 2021, 6, e000464. [Google Scholar] [CrossRef]
- Rosenzweig, H.L.; Woods, A.; Clowers, J.S.; Planck, S.R.; Rosenbaum, J.T. The NLRP3 Inflammasome Is Active but Not Essential in Endotoxin-Induced Uveitis. Inflamm. Res. 2012, 61, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Liu, S.; Li, P.; Yao, K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int. J. Mol. Sci. 2022, 23, 4883. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DED Biomarkers on the Ocular Surface | Biomarkers [73] | Possible Correlation with RCD |
|---|---|---|
| Inflammatory biomarkers [74] | TNF-α | ✓ Necroptosis |
| MMP-9 | ✕ (Apoptosis) | |
| IL-1β | ✓ Pyroptosis | |
| IL-6 | ✓ Necroptosis | |
| IL-17A | ✕ (Apoptosis) | |
| IL-18 | ✓ Pyroptosis | |
| Tear film biomarkers [75,76,77] | ROS | ✓ Ferroptosis, necroptosis |
| Hyperosmolarity | ✓ Ferroptosis | |
| Lipid peroxidation | ✓ Ferroptosis | |
| ↓Lactoferrin | ✕ n.d.* | |
| ↓Lysozyme | ✕ n.d.* | |
| Chemokyne/cytokines | ✓ Necroptosis, pyroptosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarpellini, C.; Ramos Llorca, A.; Lanthier, C.; Klejborowska, G.; Augustyns, K. The Potential Role of Regulated Cell Death in Dry Eye Diseases and Ocular Surface Dysfunction. Int. J. Mol. Sci. 2023, 24, 731. https://doi.org/10.3390/ijms24010731
Scarpellini C, Ramos Llorca A, Lanthier C, Klejborowska G, Augustyns K. The Potential Role of Regulated Cell Death in Dry Eye Diseases and Ocular Surface Dysfunction. International Journal of Molecular Sciences. 2023; 24(1):731. https://doi.org/10.3390/ijms24010731
Chicago/Turabian StyleScarpellini, Camilla, Alba Ramos Llorca, Caroline Lanthier, Greta Klejborowska, and Koen Augustyns. 2023. "The Potential Role of Regulated Cell Death in Dry Eye Diseases and Ocular Surface Dysfunction" International Journal of Molecular Sciences 24, no. 1: 731. https://doi.org/10.3390/ijms24010731
APA StyleScarpellini, C., Ramos Llorca, A., Lanthier, C., Klejborowska, G., & Augustyns, K. (2023). The Potential Role of Regulated Cell Death in Dry Eye Diseases and Ocular Surface Dysfunction. International Journal of Molecular Sciences, 24(1), 731. https://doi.org/10.3390/ijms24010731