

Atractylodin Ameliorates Colitis via PPARα Agonism
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
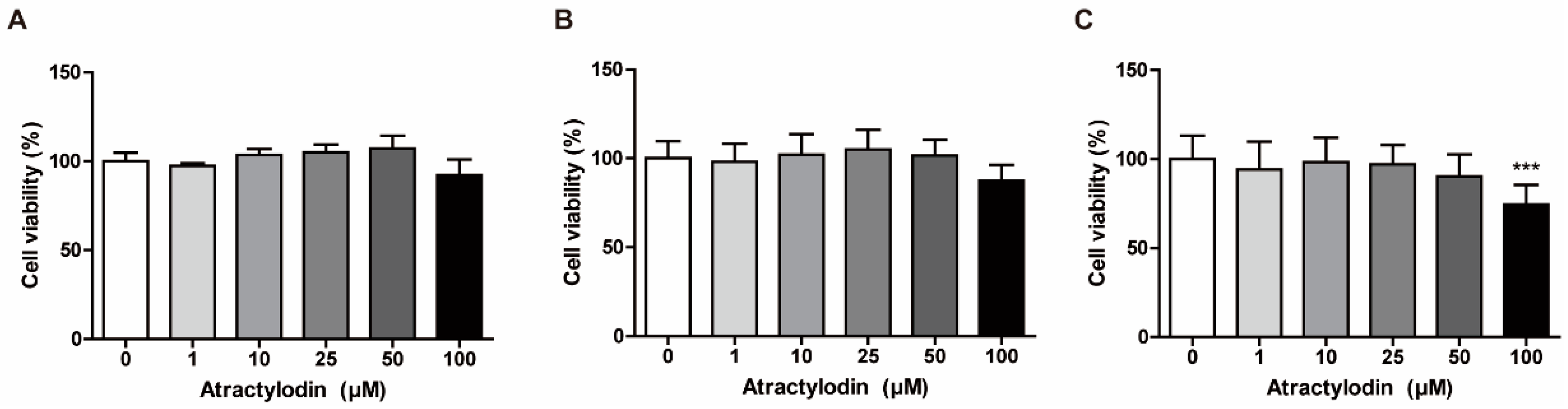
2.1. Effects of Atractylodin on the Viability of Colonic Epithelial Cells
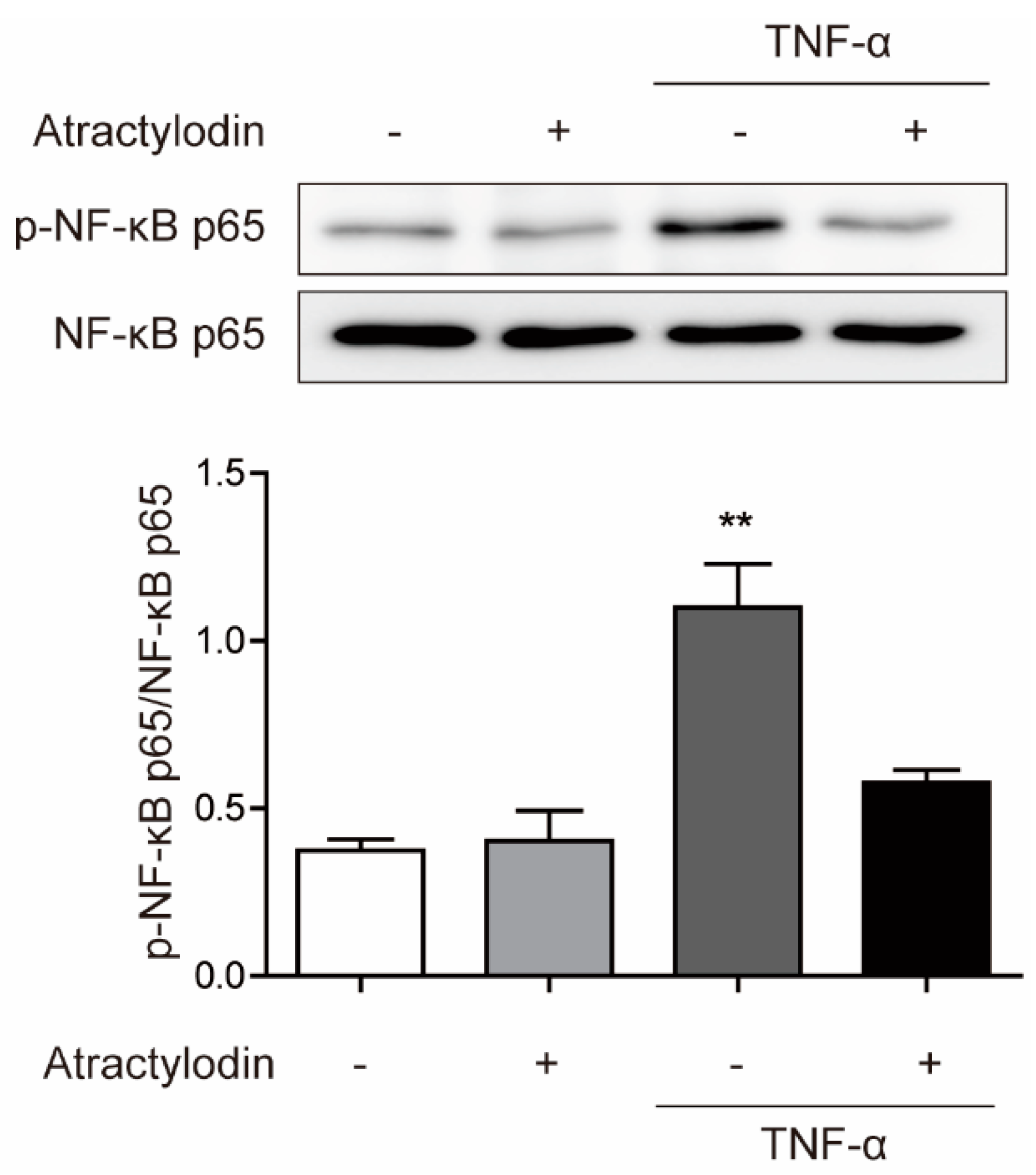
2.2. Anti-inflammatory Effects of Atractylodin in HCT116 Cells
2.3. Molecular Docking Studies for Atractylodin and PPARs
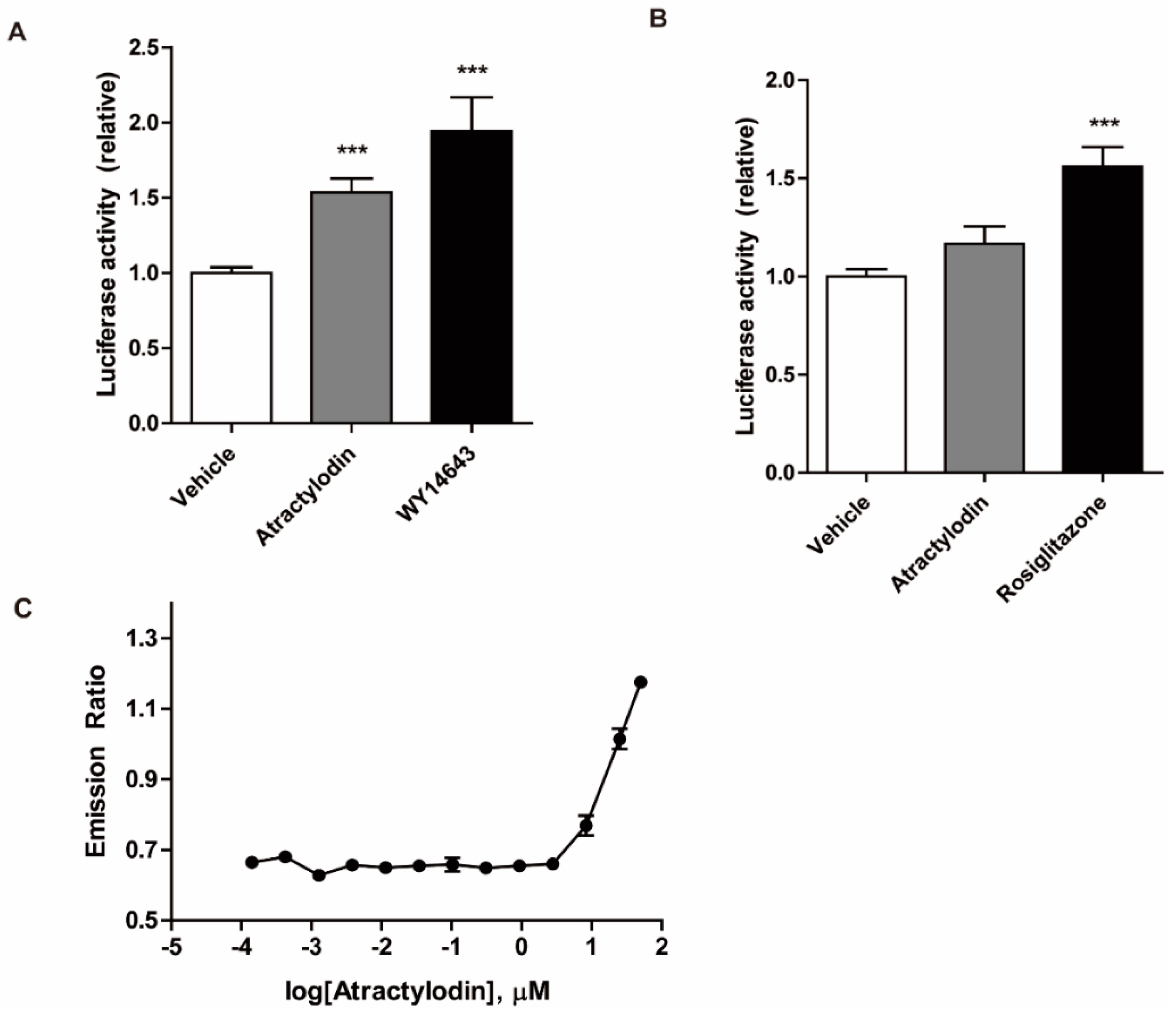
2.4. PPARα Agonism of Atractylodin
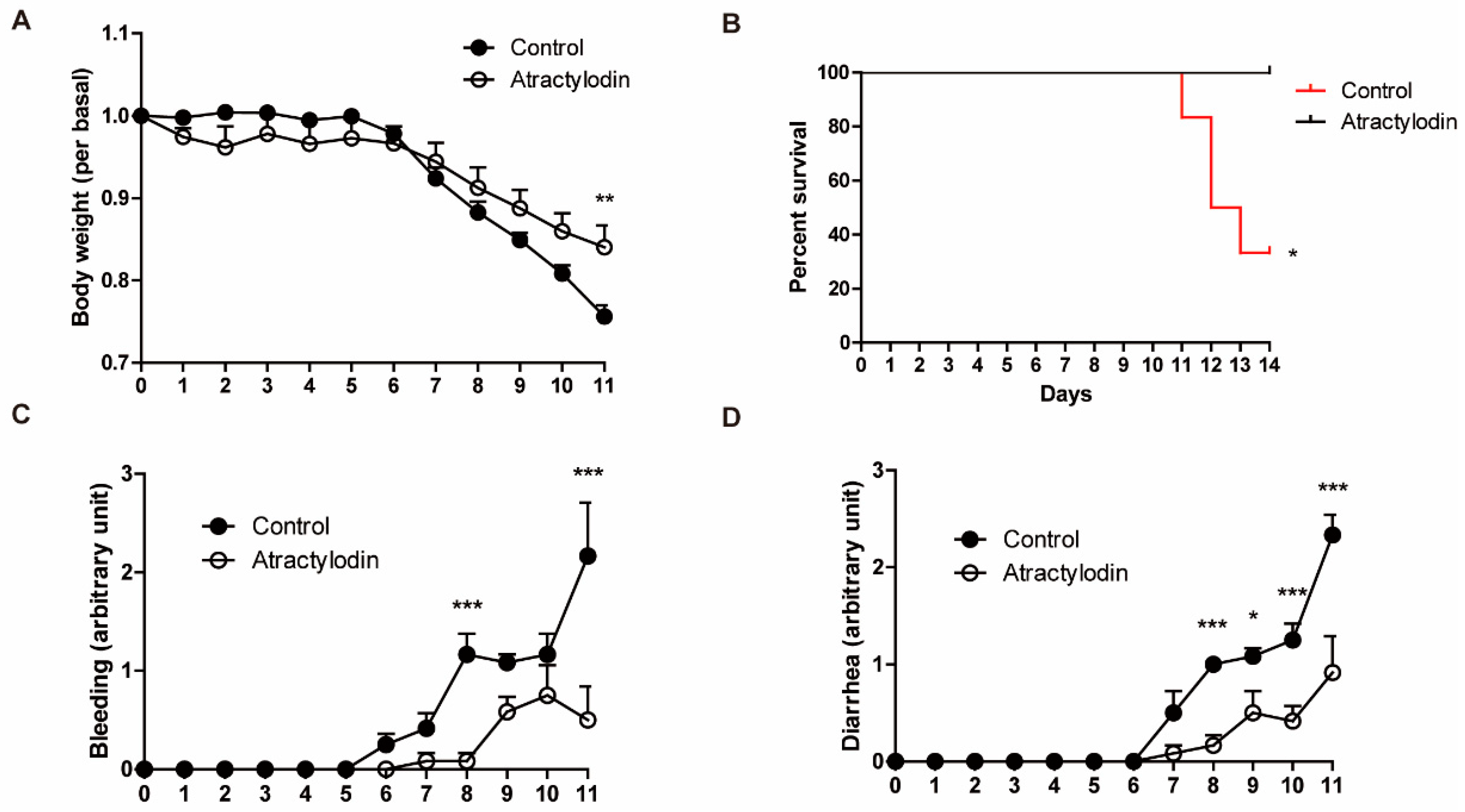
2.5. Effects of Atractylodin in a Colitis Mouse Model
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability Assay
4.3. Immunoblotting Analysis
4.4. Molecular Modeling
4.5. Luciferase Assay
4.6. Animal Model
4.7. TR-FRET-Based PPARα Binding Assay
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G.; Ng, S.C. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology 2017, 152, 313–321.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.C.; Itzkowitz, S.H. Colorectal Cancer in Inflammatory Bowel Disease: Mechanisms and Management. Gastroenterology 2022, 162, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Koonrungsesomboon, N.; Na-Bangchang, K.; Karbwang, J. Therapeutic potential and pharmacological activities of Atractylodes lancea (Thunb.) DC. Asian Pac. J. Trop. Med. 2014, 7, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Jun, X.; Fu, P.; Lei, Y.; Cheng, P. Pharmacological effects of medicinal components of Atractylodes lancea (Thunb.) DC. Chin. Med. 2018, 13, 59. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Fan, K.; Wang, K.; Bian, C. Atractylodin attenuates lipopolysaccharide-induced acute lung injury by inhibiting NLRP3 inflammasome and TLR4 pathways. J. Pharmacol. Sci. 2018, 136, 203–211. [Google Scholar] [CrossRef]
- Lyu, Z.; Ji, X.; Chen, G.; An, B. Atractylodin ameliorates lipopolysaccharide and d-galactosamine-induced acute liver failure via the suppression of inflammation and oxidative stress. Int. Immunopharmacol. 2019, 72, 348–357. [Google Scholar] [CrossRef]
- Chuang, C.H.; Cheng, Y.C.; Lin, S.C.; Lehman, C.W.; Wang, S.P.; Chen, D.Y.; Tsai, S.-W.; Lin, C.-C. Atractylodin Suppresses Dendritic Cell Maturation and Ameliorates Collagen-Induced Arthritis in a Mouse Model. J. Agric. Food Chem. 2019, 67, 6773–6784. [Google Scholar] [CrossRef]
- Yu, C.; Xiong, Y.; Chen, D.; Li, Y.; Xu, B.; Lin, Y.; Tang, Z.; Jiang, C.; Wang, L. Ameliorative effects of atractylodin on intestinal inflammation and co-occurring dysmotility in both constipation and diarrhea prominent rats. Korean J. Physiol. Pharmacol. 2017, 21, 1–9. [Google Scholar] [CrossRef]
- Qu, L.; Lin, X.; Liu, C.; Ke, C.; Zhou, Z.; Xu, K.; Cao, G.; Liu, Y. Atractylodin Attenuates Dextran Sulfate Sodium-Induced Colitis by Alleviating Gut Microbiota Dysbiosis and Inhibiting Inflammatory Response Through the MAPK Pathway. Front. Pharmacol. 2021, 12, 665376. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Behnk, A.; Morty, R.E.; Sommer, N.; Vadasz, I.; Herold, S.; Seeger, W.; Mayer, K. PPAR-alpha activation reduced LPS-induced inflammation in alveolar epithelial cells. Exp. Lung Res. 2015, 41, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Park, J.Y.; Lee, H.J.; Kim, D.H.; Chung, K.W.; Park, D.; Jeong, H.O.; Kim, H.R.; Park, C.H.; Kim, S.R.; et al. The novel PPAR alpha/gamma dual agonist MHY 966 modulates UVB-induced skin inflammation by inhibiting NF-kappaB activity. PLoS ONE 2013, 8, e76820. [Google Scholar]
- Kim, S.M.; Lee, B.; An, H.J.; Kim, D.H.; Park, K.C.; Noh, S.G.; Chung, K.W.; Lee, E.K.; Kim, K.M.; Kim, D.H.; et al. Novel PPARalpha agonist MHY553 alleviates hepatic steatosis by increasing fatty acid oxidation and decreasing inflammation during aging. Oncotarget 2017, 8, 46273–46285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, Y.T.; Nishiyama, K.; Matsuo, Y.; Kuwamura, M.; Morioka, A.; Nakajima, H.; Takeuchi, T. PPARalpha contributes to colonic protection in mice with DSS-induced colitis. Int. Immunopharmacol. 2010, 10, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Mathema, V.B.; Chaijaroenkul, W.; Na-Bangchang, K. Cytotoxic activity and molecular targets of atractylodin in cholangiocarcinoma cells. J. Pharm. Pharmacol. 2019, 71, 185–195. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Bernardes, A.; Souza, P.C.; Muniz, J.R.; Ricci, C.G.; Ayers, S.D.; Parekh, N.M.; Godoy, A.S.; Trivella, D.B.B.; Reinach, P.; Webb, P.; et al. Molecular mechanism of peroxisome proliferator-activated receptor alpha activation by WY14643: A new mode of ligand recognition and receptor stabilization. J. Mol. Biol. 2013, 425, 2878–2893. [Google Scholar] [CrossRef] [Green Version]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar] [CrossRef]
- Bruning, J.B.; Chalmers, M.J.; Prasad, S.; Busby, S.A.; Kamenecka, T.M.; He, Y.; Nettles, K.W.; Griffin, P.R. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure 2007, 15, 1258–1271. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.S.; Kim, Y.M.; Chin, Y.W. Atractylodin Inhibits Interleukin-6 by Blocking NPM-ALK Activation and MAPKs in HMC-1. Molecules 2016, 21, 1169. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Lombardi, A.; Silvestri, E.; Senese, R.; Cioffi, F.; Goglia, F.; Lanni, A.; de Lange, P. PPARs: Nuclear Receptors Controlled by, and Controlling, Nutrient Handling through Nuclear and Cytosolic Signaling. PPAR Res. 2010, 2010, 435689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decara, J.; Rivera, P.; Lopez-Gambero, A.J.; Serrano, A.; Pavon, F.J.; Baixeras, E.; de Fonseca, F.R.; Suárez, J. Peroxisome Proliferator-Activated Receptors: Experimental Targeting for the Treatment of Inflammatory Bowel Diseases. Front. Pharmacol. 2020, 11, 730. [Google Scholar] [CrossRef] [PubMed]
- Seethala, R.; Golla, R.; Ma, Z.; Zhang, H.; O’Malley, K.; Lippy, J.; Cheng, L.; Mookhtiar, K.; Farrelly, D.; Zhang, L.; et al. A rapid, homogeneous, fluorescence polarization binding assay for peroxisome proliferator-activated receptors alpha and gamma using a fluorescein-tagged dual PPARalpha/gamma activator. Anal. Biochem. 2007, 363, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, M.; Kambe, A.; Yamamoto, Y.; Arulmozhiraja, S.; Ito, S.; Nakagawa, Y.; Tokiwa, H.; Nakano, S.; Shimano, H. Elucidation of Molecular Mechanism of a Selective PPARalpha Modulator, Pemafibrate, through Combinational Approaches of X-ray Crystallography, Thermodynamic Analysis, and First-Principle Calculations. Int. J. Mol. Sci. 2020, 21, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, G.; Kang, D.; Park, C.; Kim, S.J.; Choo, J.; Lee, Y.; Yoo, J.-W.; Jung, Y.; Lee, J.; Kim, N.D.; et al. Pro-apoptotic effect of the novel benzylidene derivative MHY695 in human colon cancer cells. Oncol. Lett. 2019, 18, 3256–3264. [Google Scholar] [CrossRef] [Green Version]
- Choo, J.; Lee, Y.; Yan, X.J.; Noh, T.H.; Kim, S.J.; Son, S.; Pothoulakis, C.; Moon, H.R.; Jung, J.H.; Im, E. A Novel Peroxisome Proliferator-activated Receptor (PPAR)gamma Agonist 2-Hydroxyethyl 5-chloro-4,5-didehydrojasmonate Exerts Anti-Inflammatory Effects in Colitis. J. Biol. Chem. 2015, 290, 25609–25619. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, G.; Kim, Y.; Kim, E.-L.; Park, S.; Rhee, S.H.; Jung, J.H.; Im, E. Atractylodin Ameliorates Colitis via PPARα Agonism. Int. J. Mol. Sci. 2023, 24, 802. https://doi.org/10.3390/ijms24010802
Heo G, Kim Y, Kim E-L, Park S, Rhee SH, Jung JH, Im E. Atractylodin Ameliorates Colitis via PPARα Agonism. International Journal of Molecular Sciences. 2023; 24(1):802. https://doi.org/10.3390/ijms24010802
Chicago/Turabian StyleHeo, Gwangbeom, Yuju Kim, Eun-La Kim, Soyeong Park, Sang Hoon Rhee, Jee H. Jung, and Eunok Im. 2023. "Atractylodin Ameliorates Colitis via PPARα Agonism" International Journal of Molecular Sciences 24, no. 1: 802. https://doi.org/10.3390/ijms24010802
APA StyleHeo, G., Kim, Y., Kim, E. -L., Park, S., Rhee, S. H., Jung, J. H., & Im, E. (2023). Atractylodin Ameliorates Colitis via PPARα Agonism. International Journal of Molecular Sciences, 24(1), 802. https://doi.org/10.3390/ijms24010802