
Exploring the Potential Energy Surface of Pt6 Sub-Nano Clusters Deposited over Graphene


Abstract
:1. Introduction
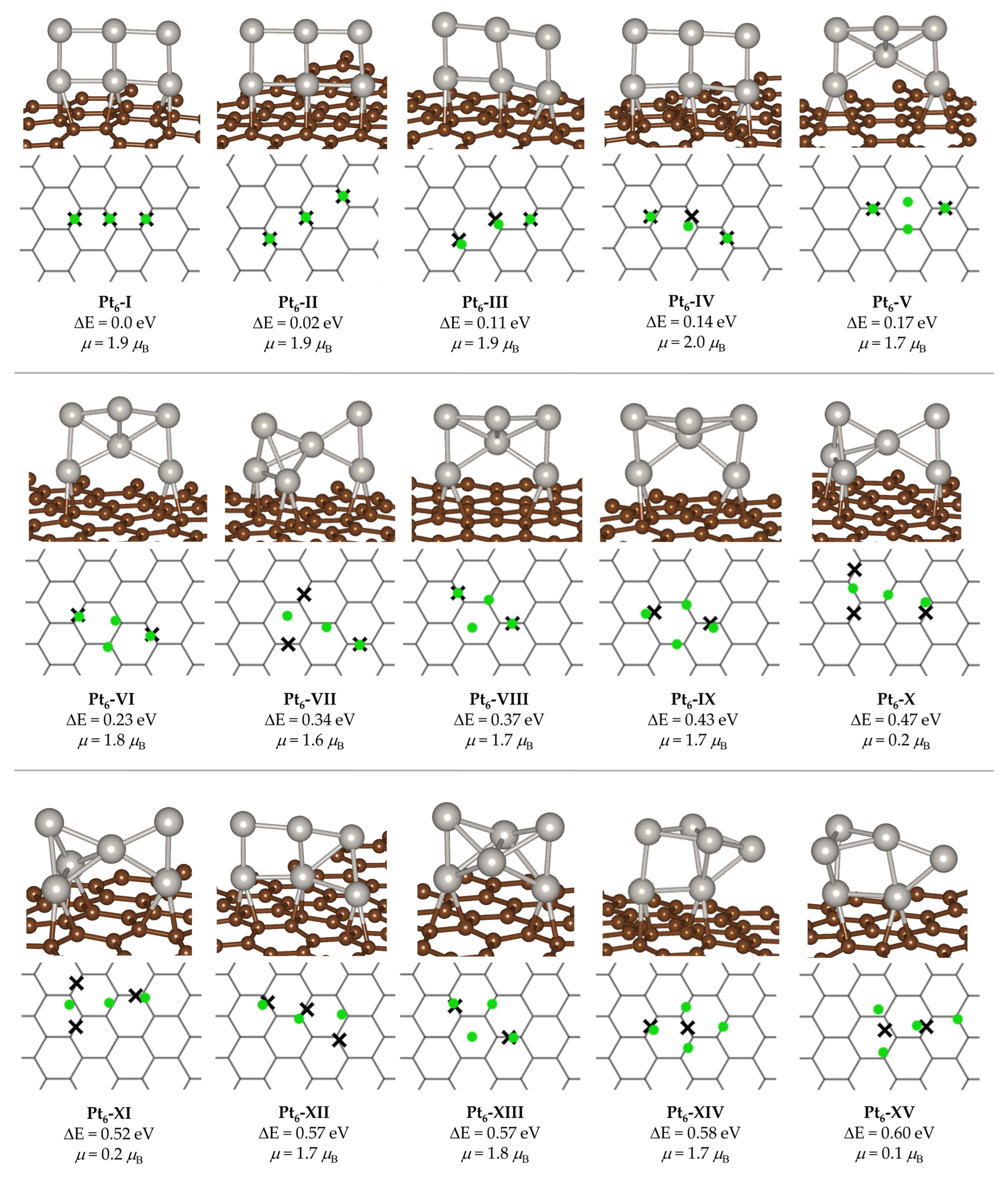
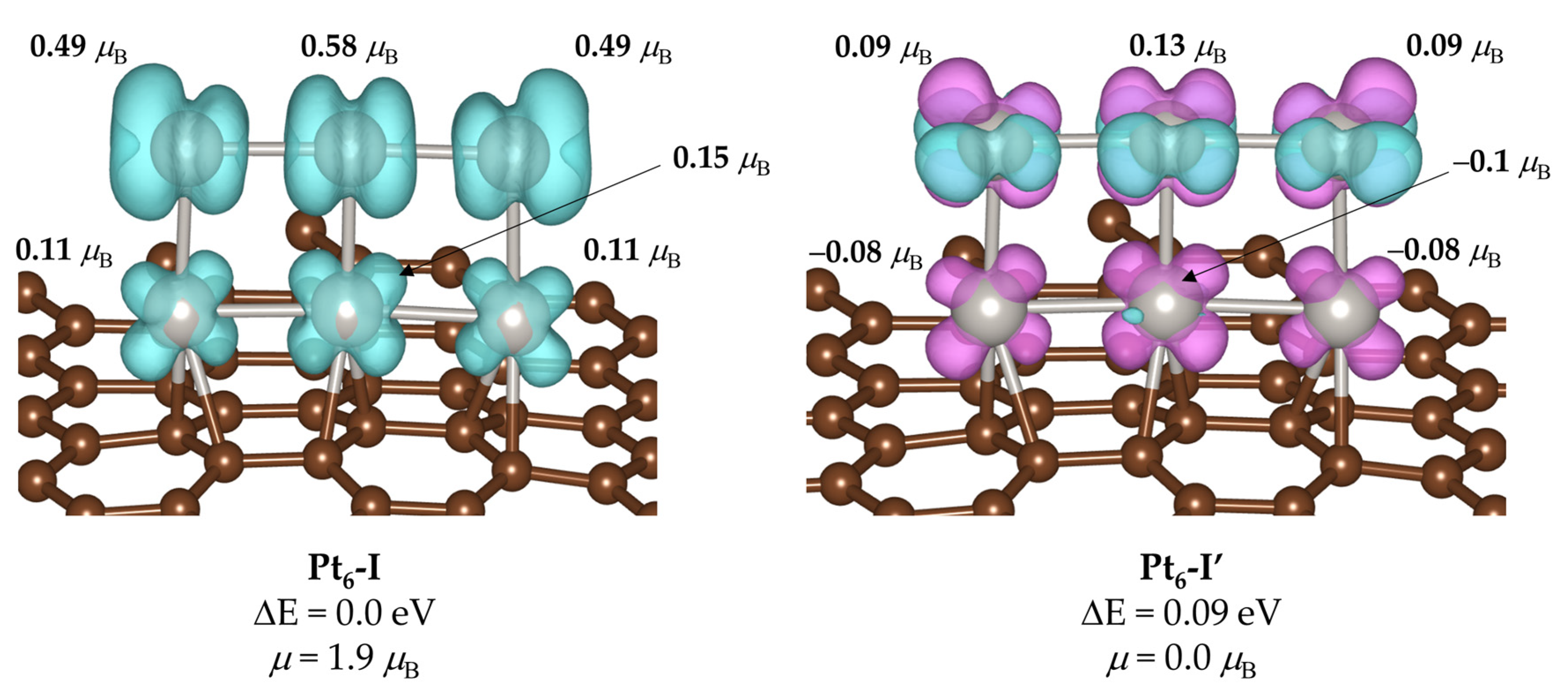
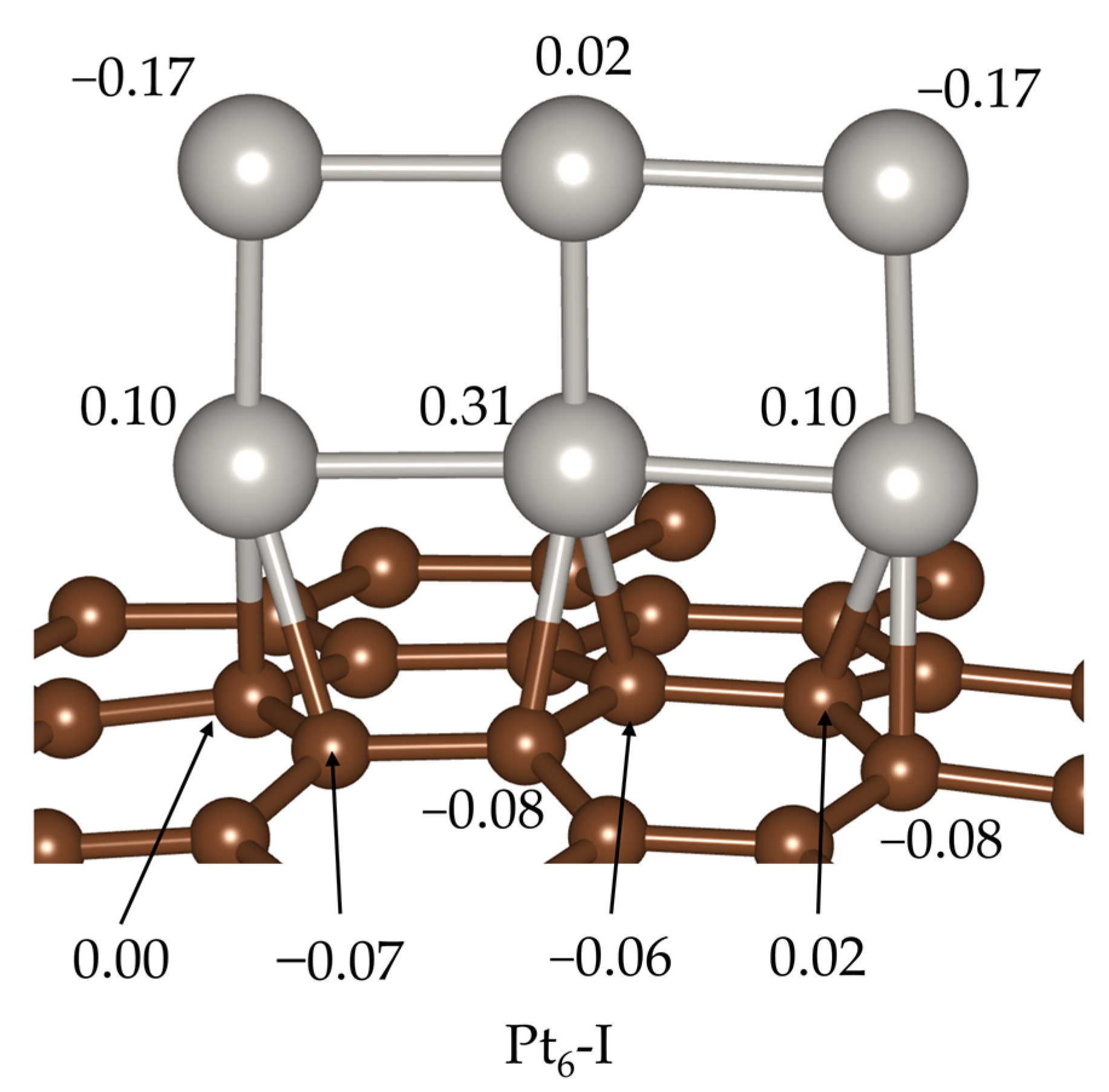
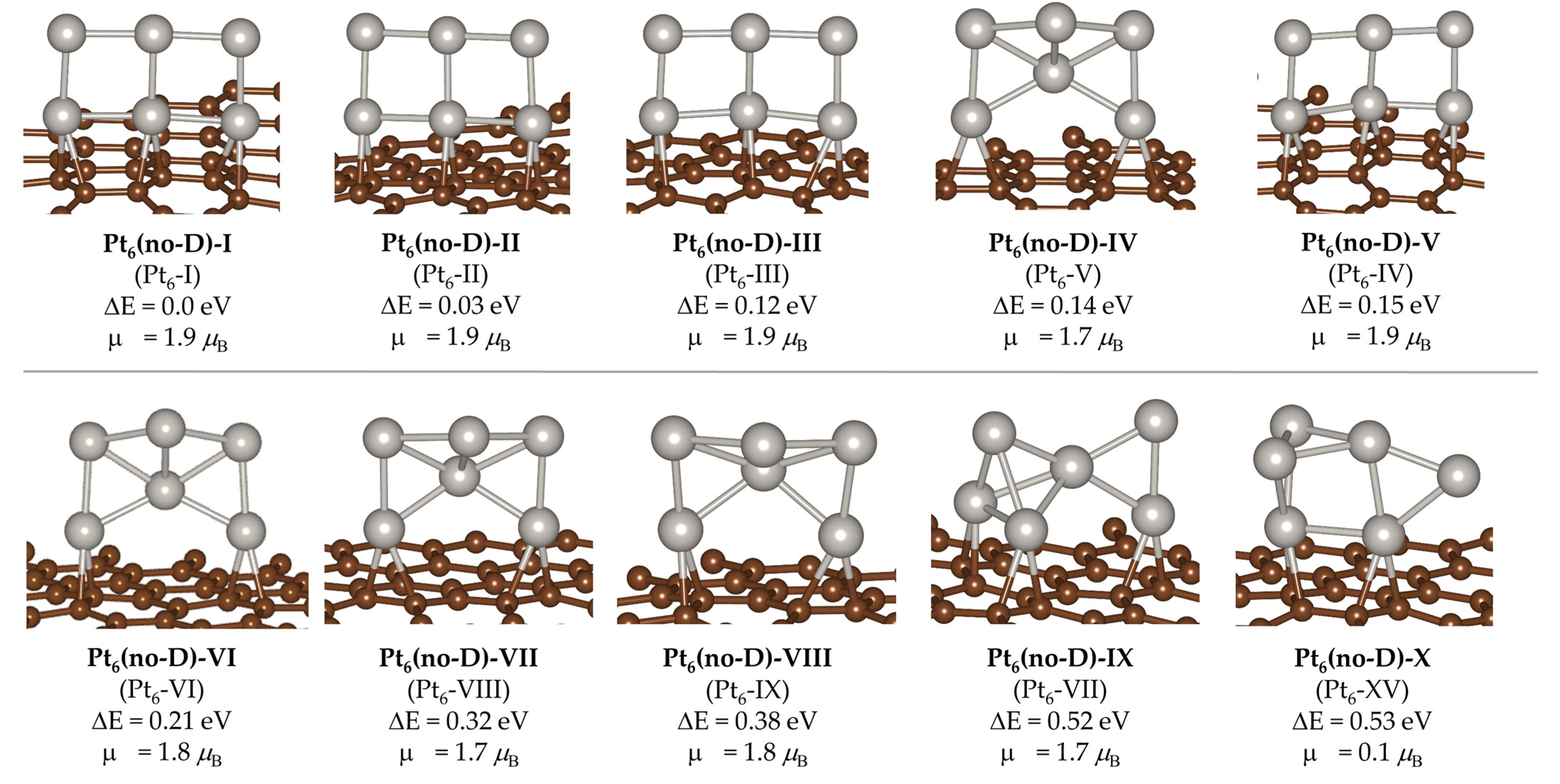
2. Results and Discussion
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tyo, E.C.; Vajda, S. Catalysis by clusters with precise numbers of atoms. Nat. Nanotechnol. 2015, 10, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, I.; Pradeep, T. Atomically Precise Clusters of Noble Metals: Emerging Link between Atoms and Nanoparticles. Chem. Rev. 2017, 117, 8208–8271. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Zeng, C.; Zhou, M.; Chen, Y. Atomically Precise Colloidal Metal Nanoclusters and Nanoparticles: Fundamentals and Opportunities. Chem. Rev. 2016, 116, 10346–10413. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A.A. Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Kong, X.; Wang, F.; Fang, R.; Li, Y. Metal Sub-nanoclusters Confined within Hierarchical Porous Carbons with High Oxidation Activity. Angew. Chem. Int. Ed. 2021, 60, 10842–10849. [Google Scholar] [CrossRef]
- Halder, A.; Curtiss, L.A.; Fortunelli, A.; Vajda, S. Perspective: Size selected clusters for catalysis and electrochemistry. J. Chem. Phys. 2018, 148, 110901. [Google Scholar] [CrossRef] [Green Version]
- Von Weber, A.; Anderson, S.L. Electrocatalysis by Mass-Selected Ptn Clusters. Acc. Chem. Res. 2016, 49, 2632–2639. [Google Scholar] [CrossRef]
- Proch, S.; Wirth, M.; White, H.S.; Anderson, S.L. Strong Effects of Cluster Size and Air Exposure on Oxygen Reduction and Carbon Oxidation Electrocatalysis by Size-Selected Ptn (n ≤ 11) on Glassy Carbon Electrodes. J. Am. Chem. Soc. 2013, 135, 3073–3086. [Google Scholar] [CrossRef]
- Ohnuma, A.; Takahashi, K.; Tsunoyama, H.; Inoue, T.; Zhao, P.; Velloth, A.; Ehara, M.; Ichikuni, N.; Tabuchi, M.; Nakajima, A. Enhanced oxygen reduction activity of size-selected platinum subnanocluster catalysts: Ptn (n = 3–9). Catal. Sci. Technol. 2022, 12, 1400–1407. [Google Scholar] [CrossRef]
- Zhai, H.; Alexandrova, A.N. Fluxionality of Catalytic Clusters: When It Matters and How to Address It. ACS. Catal. 2017, 7, 1905–1911. [Google Scholar] [CrossRef]
- Zhang, Z.; Zandkarimi, B.; Alexandrova, A.N. Ensembles of Metastable States Govern Heterogeneous Catalysis on Dynamic Interfaces. Acc. Chem. Res. 2020, 53, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Sautet, P. Metastable Structures in Cluster Catalysis from First-Principles: Structural Ensemble in Reaction Conditions and Metastability Triggered Reactivity. J. Am. Chem. Soc. 2018, 140, 2812–2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poidevin, C.; Paciok, P.; Heggen, M.; Auer, A.A. High resolution transmission electron microscopy and electronic structure theory investigation of platinum nanoparticles on carbon black. J. Chem. Phys. 2019, 150, 041705. [Google Scholar] [CrossRef] [Green Version]
- Campos-Roldaén, C.A.; Ramos-Saénchez, G.; Gonzalez-Huerta, R.G.; Vargas García, J.R.; Balbuena, P.B.; Alonso-Vante, N. Influence of sp3−sp2 Carbon Nanodomains on Metal/Support Interaction, Catalyst Durability, and Catalytic Activity for the Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2016, 8, 23260–23269. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kim, S.H.; Kwak, S.K.; Song, H.-K. Curvature-Induced Metal−Support Interaction of an Islands-by-Islands Composite of Platinum Catalyst and Carbon Nano-onion for Durable Oxygen Reduction. ACS Appl. Mater. Interfaces 2017, 9, 23302–23308. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.B.; Benedikt, U.; Auer, A.A. Interaction of Platinum Nanoparticles with Graphitic Carbon Structures: A Computational Study. ChemPhysChem 2013, 14, 2984–2989. [Google Scholar] [CrossRef] [PubMed]
- Jayabal, S.; Saranya, G.; Geng, D.; Lin, L.-Y.; Meng, X. Insight into the correlation of Pt–support interactions with electro-catalytic activity and durability in fuel cells. J. Mater. Chem. A 2020, 8, 9420–9446. [Google Scholar] [CrossRef]
- Ramos-Sanchez, G.; Balbuena, P.B. Interactions of platinum clusters with a graphite substrate. Phys. Chem. Chem. Phys. 2013, 15, 11950–11959. [Google Scholar] [CrossRef]
- Verga, L.G.; Aarons, J.; Sarwar, M.; Thompsett, D.; Russell, A.E.; Skylaris, C.-K. Effect of graphene support on large Pt nanoparticles. Phys. Chem. Chem. Phys. 2016, 18, 32713–32722. [Google Scholar] [CrossRef] [Green Version]
- Tsunoyama, H.; Ohnuma, A.; Takahashi, K.; Velloth, A.; Ehara, M.; Ichikuni, N.; Tabuchi, M.; Nakajima, A. Enhanced oxygen reduction activity of platinum subnanocluster catalysts through charge redistribution. Chem. Commun. 2019, 55, 12603–12606. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, L.; Li, X.; Liu, Y.; Wang, Y.; Yao, Q.; Xie, J.; Xue, Q.; Yan, Z.; Yuan, X.; et al. Atomic-precision Pt6 nanoclusters for enhanced hydrogen electro-oxidation. Nat. Commun. 2022, 13, 1596. [Google Scholar] [CrossRef] [PubMed]
- Rêgo, C.R.C.; Tereshchuk, P.; Oliveira, L.N.; Da Silva, J.L.F. Graphene-supported small transition-metal clusters: A density functional theory investigation within van der Waals corrections. Phys. Rev. B 2017, 95, 235422. [Google Scholar] [CrossRef]
- Lavroff, R.H.; Morgan, H.W.T.; Zhang, Z.; Poths, P.; Alexandrova, A.N. Ensemble representation of catalytic interfaces: Soloists, orchestras, and everything in-between. Chem. Sci. 2022, 13, 8003–8016. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Sundararaman, R.; Goddard, W.A., III; Arias, T.A. Grand canonical electronic density-functional theory: Algorithms and applications to electrochemistry. J. Chem. Phys. 2017, 146, 114104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchison, P.; Rice, P.S.; Warburton, R.E.; Raugei, S.; Hammes-Schiffer, S. Multilevel Computational Studies Reveal the Importance of Axial Ligand for Oxygen Reduction Reaction on Fe−N−C Materials. J. Am. Chem. Soc. 2022, 144, 16524–16534. [Google Scholar] [CrossRef]
- Melander, M.M.; Kuisma, M.J.; Christensen, T.E.K.; Honkala, K. Grand-canonical approach to density functional theory of electrocatalytic systems: Thermodynamics of solid-liquid interfaces at constant ion and electrode potentials. J. Chem. Phys. 2019, 150, 041706. [Google Scholar] [CrossRef] [Green Version]
- Basdogan, Y.; Maldonado, A.M.; Keith, J.A. Advances and challenges in modeling solvated reaction mechanisms for re-newable fuels and chemicals. WIREs Comput. Mol. Sci. 2019, 10, e1446. [Google Scholar]
- Abidi, N.; Lim, K.R.G.; Seh, Z.W.; Steinmann, S.N. Atomistic modeling of electrocatalysis: Are we there yet? WIREs Comput. Mol. Sci. 2021, 11, e1499. [Google Scholar] [CrossRef]
- Gauthier, J.A.; Ringe, S.; Dickens, C.F.; Garza, A.J.; Bell, A.T.; Head-Gordon, M.; Nørskov, J.K.; Chan, K. Challenges in Modeling Electrochemical Reaction Energetics with Polarizable Continuum Models. ACS Catal. 2019, 9, 920–931. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zandkarimi, B.; Munarriz, J.; Dickerson, C.E.; Alexandrova, A.N. Fluxionality of Subnano Clusters Reshapes the Activity Volcano of Electrocatalysis. ChemCatChem 2022, 14, e202200345. [Google Scholar] [CrossRef]
- Munarriz, J.; Zhang, Z.; Sautet, P.; Alexandrova, A.N. Graphite-supported Ptn Cluster Electrocatalysts: Major Change of Active Sites as a Function of the Applied Potential. ACS Catal. 2022, 12, 14517–14526. [Google Scholar] [CrossRef]
- Duan, Z.; Henkelman, G. Atomic-Scale Mechanisms of Electrochemical Pt Dissolution. ACS Catal. 2021, 11, 14439–14447. [Google Scholar] [CrossRef]
- Hu, X.; Chen, S.; Chen, L.; Tian, Y.; Yao, S.; Lu, Z.; Zhang, X.; Zhou, Z. What is the Real Origin of the Activity of Fe−N−C Electrocatalysts in the O2 Reduction Reaction? Critical Roles of Coordinating Pyrrolic N and Axially Adsorbing Species. J. Am. Chem. Soc. 2022, 144, 18144–18152. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gracia, J.; Sharpe, R.; Munarriz, J. Principles determining the activity of magnetic oxides for electron transfer reactions. J. Catal. 2018, 361, 331–338. [Google Scholar] [CrossRef]
- Biz, C.; Fianchini, M.; Gracia, J. Strongly Correlated Electrons in Catalysis: Focus on Quantum Exchange. ACS Catal. 2021, 11, 14249–14261. [Google Scholar] [CrossRef]
- Biz, C.; Fianchini, M.; Polo, V.; Gracia, J. Magnetism and Heterogeneous Catalysis: In Depth on the Quantum Spin-Exchange Interactions in Pt3M (M = V, Cr, Mn, Fe, Co, Ni, and Y)(111) Alloys. ACS Appl. Mater. Interfaces 2020, 12, 50484–50494. [Google Scholar] [CrossRef]
- Li, J.; Ma, J.; Ma, Z.; Zhao, E.; Du, K.; Guo, J.; Ling, T. Spin Effect on Oxygen Electrocatalysis. Adv. Energy Sustain. Res. 2021, 2, 2100034. [Google Scholar] [CrossRef]
- Sun, Y.; Ren, X.; Sun, S.; Liu, Z.; Xi, S.; Xu, Z.J. Engineering High-Spin State Cobalt Cations in Spinel Zinc Cobalt Oxide for Spin Channel Propagation and Active Site Enhancement in Water Oxidation. Angew. Chem. Int. Ed. 2021, 60, 14536–14544. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Ai, M.; Huang, C.; Yin, L.; Liu, X.; Zhang, R.; Wang, S.; Jiang, Z.; Zhang, X.; Zou, J.-J.; et al. Manipulating spin polarization of titanium dioxide for efficient photocatalysis. Nat. Commun. 2020, 11, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Cheng, Z.; Wang, X. Understanding the Mechanism of the Oxygen Evolution Reaction with Consideration of Spin. Electrochem. Energy Rev. 2021, 4, 136–145. [Google Scholar] [CrossRef]
- Groves, M.N.; Malardier-Jugroot, C.; Jugroot, M. Improving Platinum Catalyst Durability with a Doped Graphene Support. J. Phys. Chem. C 2012, 116, 10548–10556. [Google Scholar] [CrossRef]
- Li, R.; Odunlami, M.; Carbonnière, P. Low-lying Ptn cluster structures (n = 6–10) from global optimizations based on DFT potential energy surfaces: Sensitivity of the chemical ordering with the functional. Comput. Theor. Chem. 2017, 1107, 136–141. [Google Scholar] [CrossRef]
- Chaves, A.S.; Rondina, G.G.; Piotrowski, M.J.; Tereshchuk, P.; Da Silva, J.L.F. The Role of Charge States in the Atomic Structure of Cun and Ptn (n = 2−14 atoms) Clusters: A DFT Investigation. J. Phys. Chem. A 2004, 118, 10813–10821. [Google Scholar] [CrossRef]
- Xiao, L.; Wang, L. Structures of Platinum Clusters: Planar or Spherical? J. Phys. Chem. A 2004, 108, 8605–8614. [Google Scholar] [CrossRef]
- Kumar, V.; Kawazoe, Y. Evolution of atomic and electronic structure of Pt clusters: Planar, layered, pyramidal, cage, cubic, and octahedral growth. Phys. Rev. B 2008, 77, 205418. [Google Scholar] [CrossRef]
- Zandkarimi, B.; Alexandrova, A.N. Dynamics of Subnanometer Pt Clusters Can Break the Scaling Relationships in Catalysis. J. Phys. Chem. Lett. 2019, 10, 460–467. [Google Scholar] [CrossRef] [Green Version]
- Ignatov, S.K.; Razuvaev, A.G.; Loginova, A.S.; Masunov, A.E. Global Structure Optimization of Pt Clusters Based on the Modified Empirical Potentials, Calibrated using Density Functional Theory. J. Phys. Chem. C 2019, 123, 29024–29036. [Google Scholar] [CrossRef]
- Munarriz, J.; Polo, V.; Gracia, J. On the Role of Ferromagnetic Interactions in Highly Active Mo-Based Catalysts for Ammonia Synthesis. ChemPhysChem 2018, 19, 2843–2847. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B Condens. Matter Mater. Phys. 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Zhai, H.; Alexandrova, A.N. Ensemble-average representation of Pt clusters in conditions of catalysis accessed through GPU accelerated deep neural network fitting global optimization. J. Chem. Theory Comput. 2016, 12, 6213–6226. [Google Scholar] [CrossRef] [Green Version]
- Zhai, H.; Alexandrova, A.N. Local Fluxionality of Surface-Deposited Cluster Catalysts: The Case of Pt7 on Al2O3. J. Phys. Chem. Lett. 2018, 9, 1696–1702. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pt6(no-D)-# | Pt6-#’ | ∆E(Pt6(no-D)-#) | ∆E(Pt6-#’) | ∆(∆E) | Edisp |
|---|---|---|---|---|---|
| I | I | 0.00 | 0.00 | 0.00 | −7.920 |
| II | II | 0.03 | 0.02 | 0.01 | −7.930 |
| III | III | 0.12 | 0.11 | 0.01 | −7.928 |
| IV | V | 0.14 | 0.17 | −0.03 | −7.884 |
| V | IV | 0.15 | 0.14 | 0.01 | −7.936 |
| VI | VI | 0.21 | 0.23 | −0.02 | −7.900 |
| VII | VIII | 0.32 | 0.37 | −0.05 | −7.877 |
| VIII | IX | 0.38 | 0.43 | −0.05 | −7.872 |
| IX | VII | 0.52 | 0.34 | 0.18 | −8.115 |
| X | XV | 0.53 | 0.60 | −0.07 | −7.858 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrena-Espés, D.; Boneta, S.; Polo, V.; Munárriz, J. Exploring the Potential Energy Surface of Pt6 Sub-Nano Clusters Deposited over Graphene. Int. J. Mol. Sci. 2023, 24, 870. https://doi.org/10.3390/ijms24010870
Barrena-Espés D, Boneta S, Polo V, Munárriz J. Exploring the Potential Energy Surface of Pt6 Sub-Nano Clusters Deposited over Graphene. International Journal of Molecular Sciences. 2023; 24(1):870. https://doi.org/10.3390/ijms24010870
Chicago/Turabian StyleBarrena-Espés, Daniel, Sergio Boneta, Victor Polo, and Julen Munárriz. 2023. "Exploring the Potential Energy Surface of Pt6 Sub-Nano Clusters Deposited over Graphene" International Journal of Molecular Sciences 24, no. 1: 870. https://doi.org/10.3390/ijms24010870
APA StyleBarrena-Espés, D., Boneta, S., Polo, V., & Munárriz, J. (2023). Exploring the Potential Energy Surface of Pt6 Sub-Nano Clusters Deposited over Graphene. International Journal of Molecular Sciences, 24(1), 870. https://doi.org/10.3390/ijms24010870