
A Dominant-Negative Mutant of ANXA7 Impairs Calcium Signaling and Enhances the Proliferation of Prostate Cancer Cells by Downregulating the IP3 Receptor and the PI3K/mTOR Pathway
 , ,
, ,
Abstract
:1. Introduction
2. Results
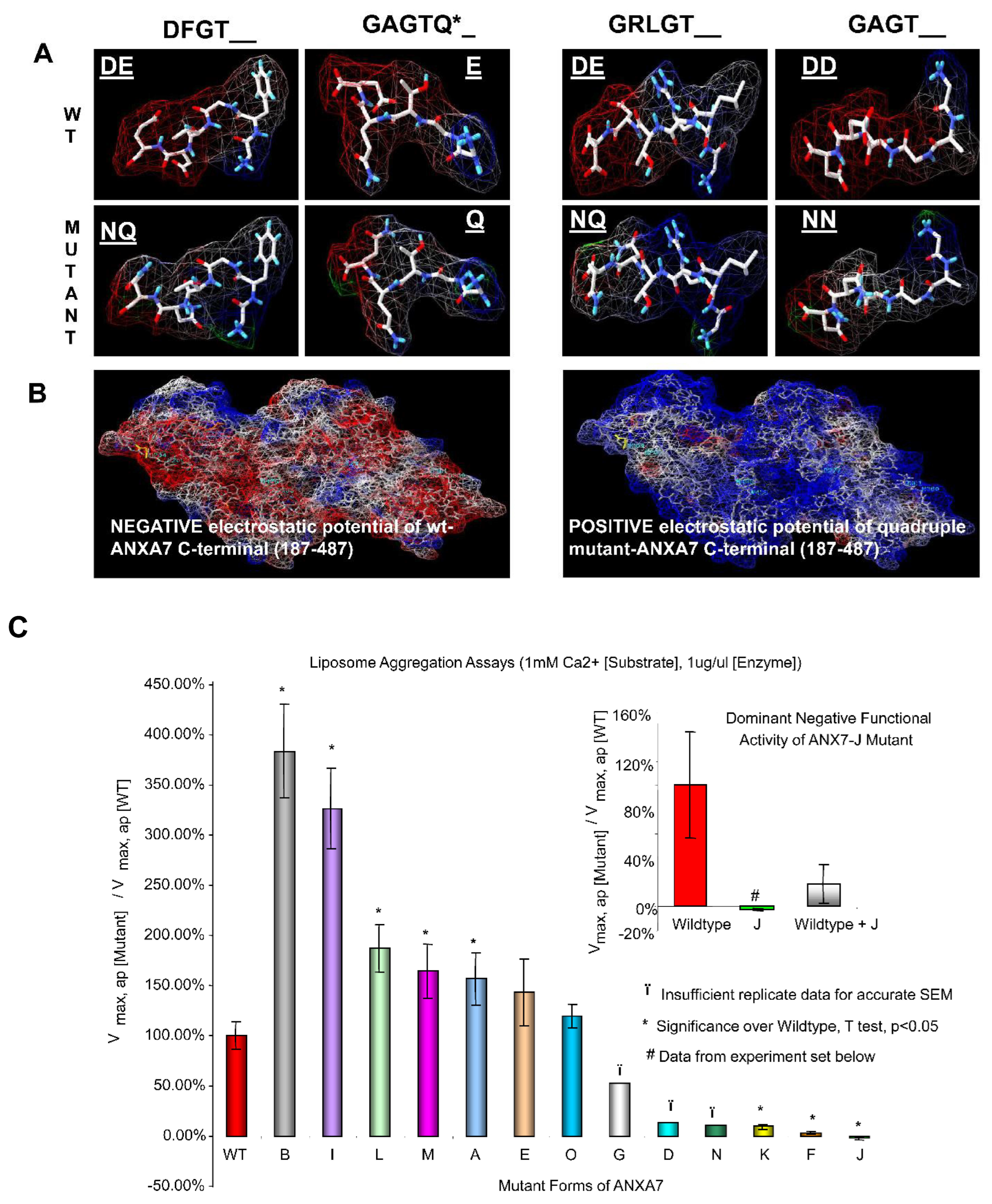
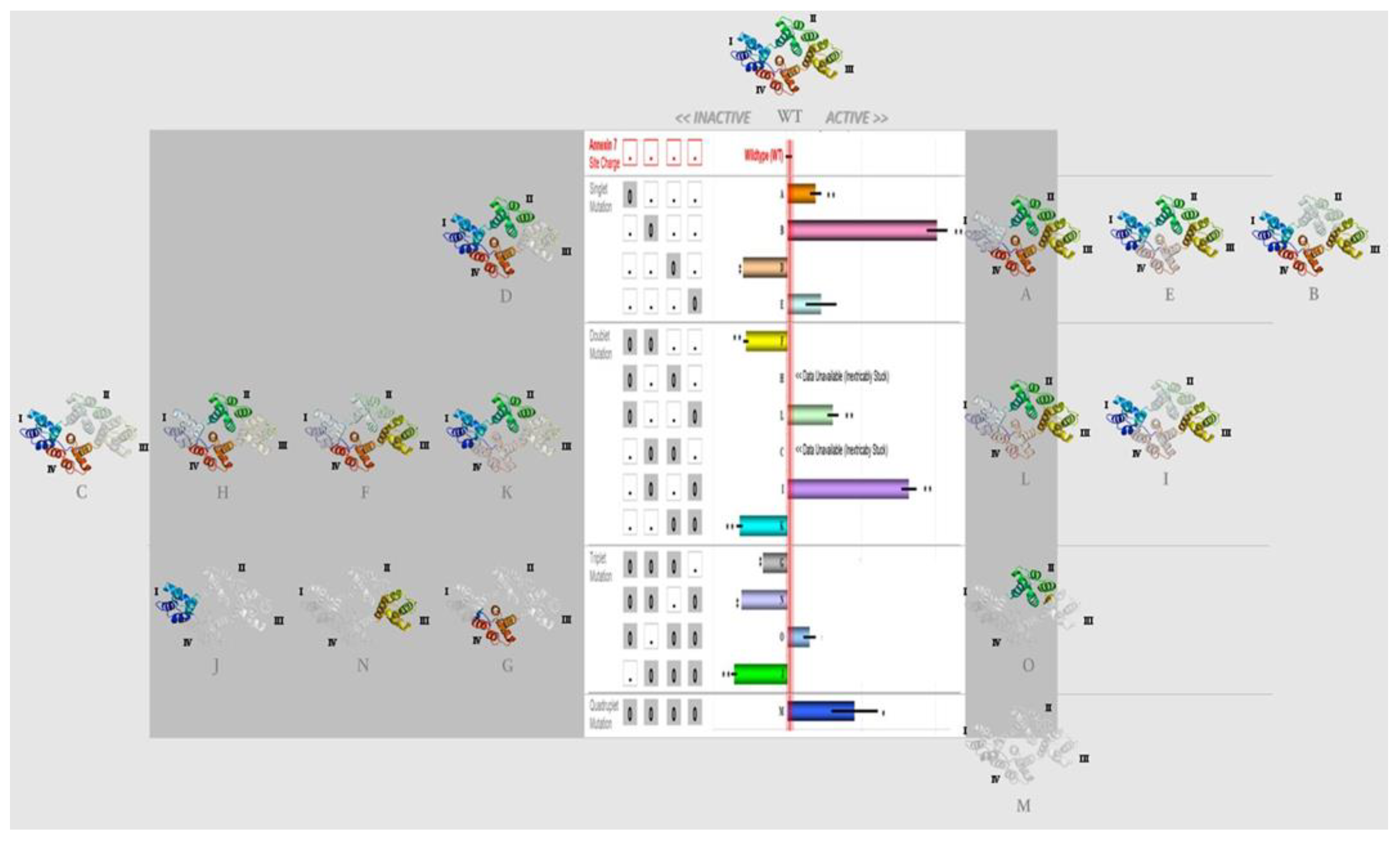
2.1. Amino Acid Residues Important for PS Liposome Aggregation
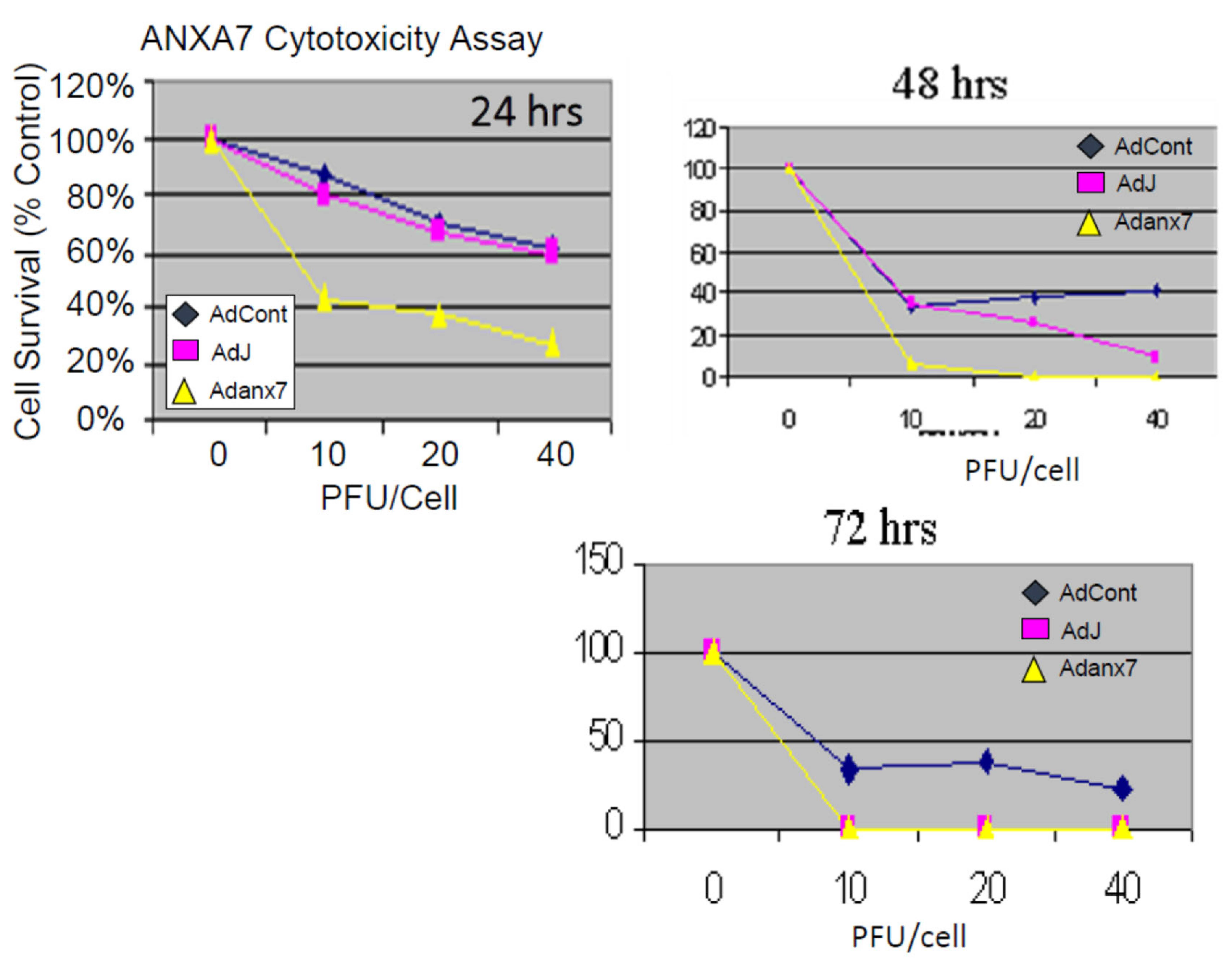
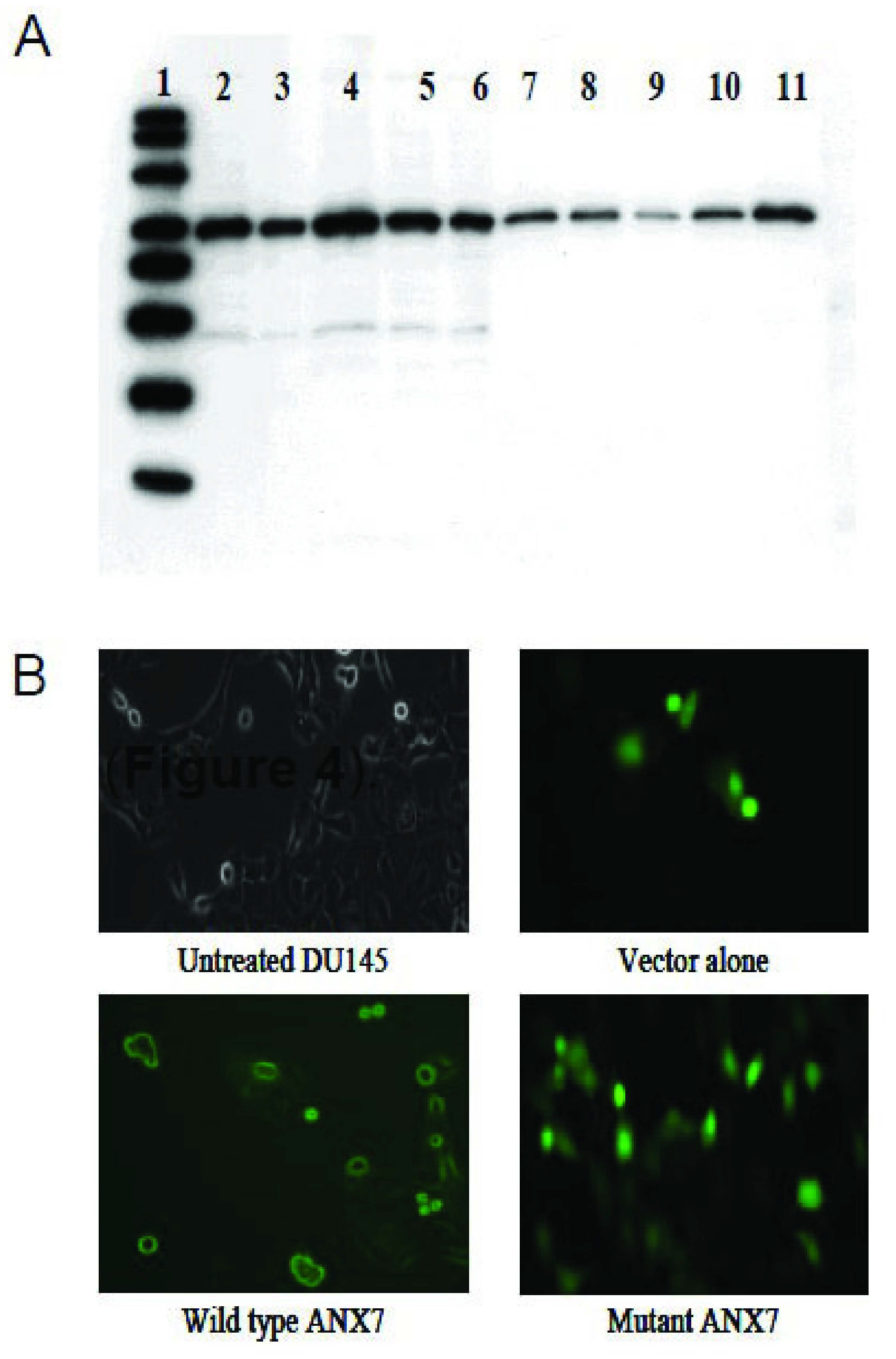
2.2. Dominant-Negative ANXA7J Does Not Kill Prostate Cancer Cells
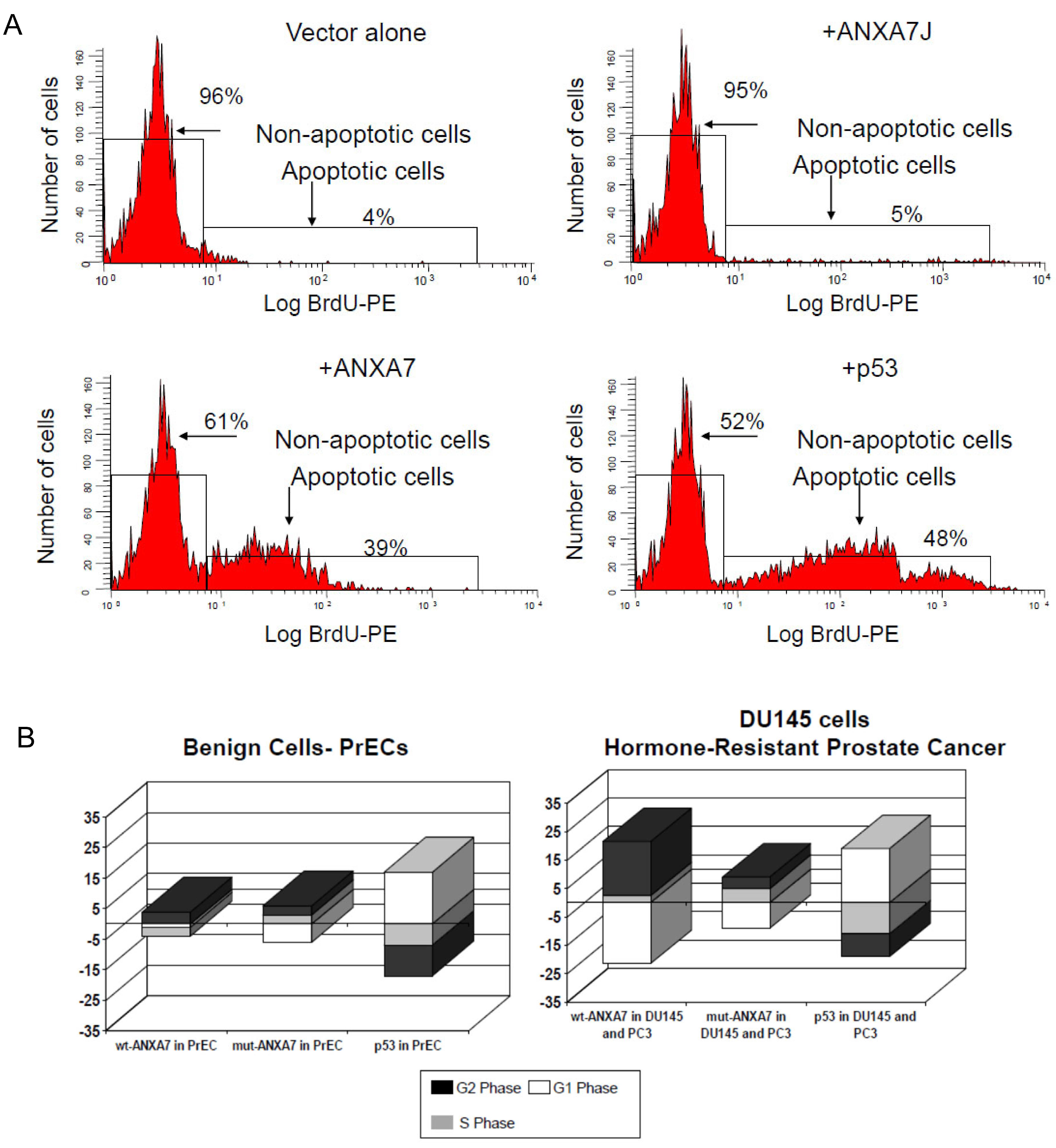
2.3. Impact of DN-ANXA7J on Apoptosis, Cell Cycle, and Cell Morphology
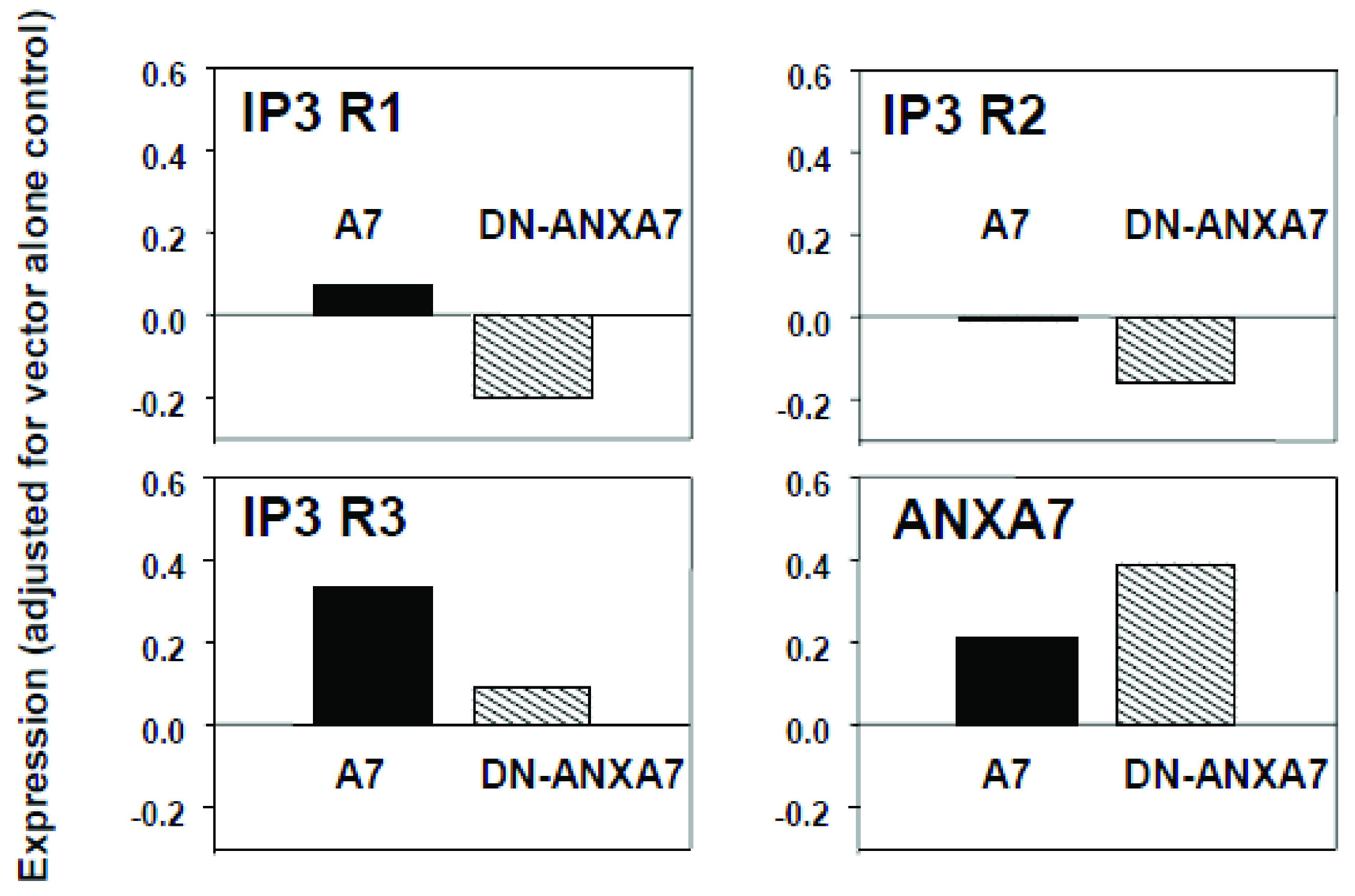
2.4. Effect of DN-ANXA7J on IP3 Receptors
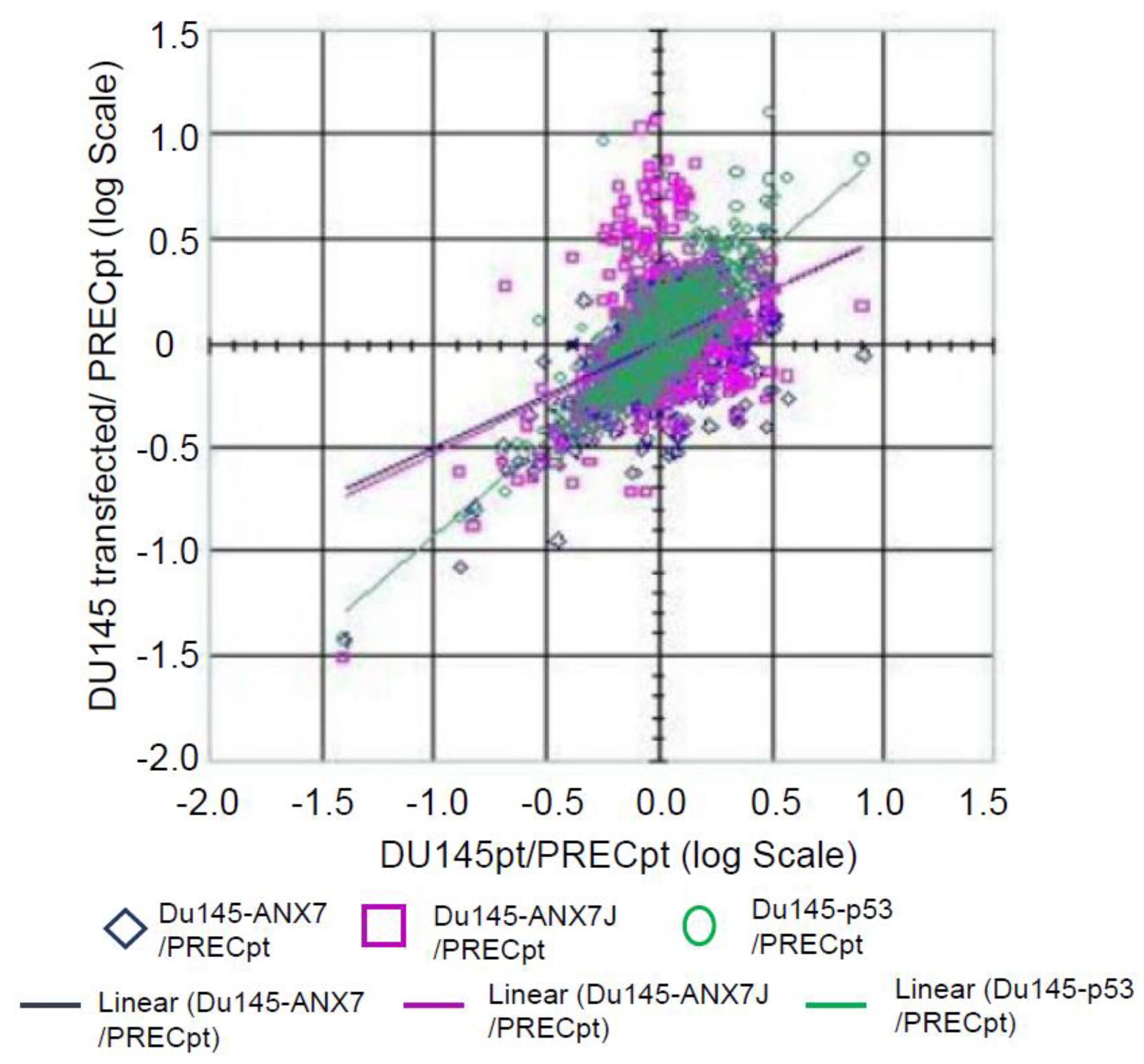
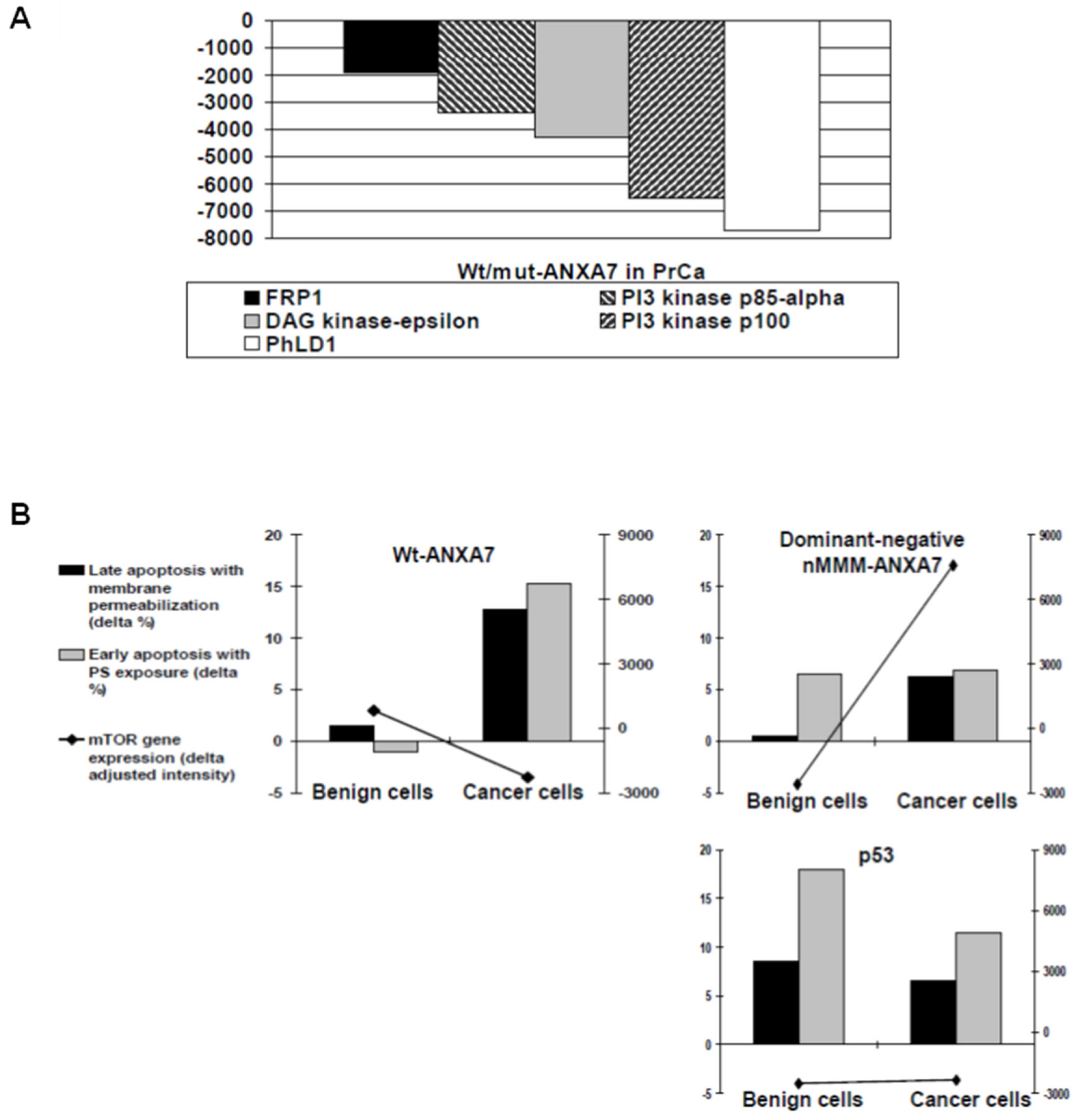
2.5. Identification of Downstream Targets of ANXA7 That Constitute the Calcium Signaling Pathway Involved in Tumorigenesis
3. Discussion
3.1. Endonexin-Fold Motif Mutations and Inhibition of Aggregation and Membrane Fusion
3.2. Inhibition of Proliferation of Prostate Cancer Cells
3.3. Sensitization of Prostate Cancer Cells to Cell Death
3.4. Identification of Signaling Pathways That May Be Responsible for the Tumor Suppressor Function
3.5. Calcium, IP3 Receptor, and Cancer
4. Materials and Methods
4.1. Site-Directed Mutagenesis
4.2. Preparation and Purification of ANXA7 Proteins In Vitro
4.3. Preparation of Phosphatidylserine-Containing Liposomes for Aggregation Assay In Vitro
4.4. Lipid Vesicle Fusion Mediated by ANXA7
4.5. Protein Modeling
4.6. Bioinformatics and Statistics
4.7. Cell Culturing and Treatment
4.8. Programmed Cell Death (PCD) and Cell Cycling
4.9. RNA Extraction and PCR
4.10. cDNA Microarray Experiment and Data Mining Using Gene Pattern and Ingenuity Pathway Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Raynal, P.; Pollard, H.B. Annexins: The problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. Biochim. Biophys. Acta. 1994, 1197, 63–93. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Torosyan, Y.; Raffeld, M.; Eidelman, O.; Pollard, H.B.; Bubendorf, L. ANXA7 expression represents hormone-relevant tumor suppression in different cancers. Int. J. Cancer 2007, 121, 2628–2636. [Google Scholar] [CrossRef]
- Srivastava, M.; Pollard, H.B. Molecular dissection of nucleolin’s role in growth and cell proliferation: New insights. FASEB J. 1999, 13, 1911–1922. [Google Scholar] [CrossRef]
- Creutz, C.E.; Pazoles, C.J.; Pollard, H.B. Identification and purification of an adrenal medullary protein (synexin) that causes calcium-dependent aggregation of isolated chromaffin granules. J. Biol. Chem. 1978, 253, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Caohuy, H.; Srivastava, M.; Pollard, H.B. Membrane fusion protein synexin (annexin VII) as a Ca2+/GTP sensor in exocytotic secretion. Proc. Natl. Acad. Sci. USA 1996, 93, 10797–10802. [Google Scholar] [CrossRef]
- Magendzo, K.; Shirvan, A.; Cultraro, C.; Srivastava, M.; Pollard, H.B.; Burns, A.L. Alternative splicing of human synexin mRNA in brain, cardiac, and skeletal muscle alters the unique N-terminal domain. J. Biol. Chem. 1991, 266, 3228–3232. [Google Scholar] [CrossRef]
- Clemen, C.S.; Herr, C.; Lie, A.A.; Noegel, A.A.; Schröder, R. Annexin VII: An astroglial protein exhibiting a Ca2+-dependent subcellular distribution. Neuroreport 2001, 12, 1139–1144. [Google Scholar] [CrossRef]
- Dowling, L.G.; Creutz, C.E. Comparison of synexin isotypes in secretory and non-secretory tissues. Biochem. Biophys. Res. Commun. 1985, 132, 382–389. [Google Scholar] [CrossRef]
- Glezer, I.I.; Leranth, C.; Morgane, P.J.; Hof, P.R. Calcium-binding protein-containing neuronal populations in mammalian visual cortex: A comparative study in whales, insectivores, bats, rodents, and primates. Cereb. Cortex 1993, 3, 249–272. [Google Scholar] [CrossRef]
- Srivastava, M.; Bubendorf, L.; Srikantan, V.; Fossom, L.; Nolan, L.; Glasman, M.; Leighton, X.; Fehrle, W.; Pittaluga, S.; Raffeld, M.; et al. ANX7, a candidate tumor suppressor gene for prostate cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 4575–4580. [Google Scholar] [CrossRef] [PubMed]
- Eidelman, O.; Zhang, J.; Srivastava, M.; Pollard, H.B. Cystic fibrosis and the use of pharmacogenomics to determine surrogate endpoints for drug discovery. Am. J. Pharm. 2001, 1, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Kumar, P.; Leighton, X.; Glasman, M.; Goping, G.; Eidelman, O.; Pollard, H.B. Influence of the Anx7 (+/−) knockout mutation and fasting stress on the genomics of the mouse adrenal gland. Ann. N. Y. Acad. Sci. 2002, 971, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Montagna, C.; Leighton, X.; Glasman, M.; Naga, S.; Eidelman, O.; Ried, T.; Pollard, H.B. Haploinsufficiency of Anx7 tumor suppressor gene and consequent genomic instability promotes tumorigenesis in the Anx7(+/−) mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 14287–14292. [Google Scholar] [CrossRef]
- Watson, W.; Srivastava, M.; Leighton, X.; Glasman, M.; Faraday, M.; Fossam, L.; Pollard, H.; Verma, A. Annexin 7 mobilizes calcium from endoplasmic reticulum stores in brain. Biochim. Biophys. Acta. 2004, 1742, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Torosyan, Y.; Dobi, A.; Naga, S.; Mezhevaya, K.; Glasman, M.; Norris, C.; Jiang, G.; Mueller, G.; Pollard, H.; Srivastava, M. Distinct effects of annexin A7 and p53 on arachidonate lipoxygenation in prostate cancer cells involve 5-lipoxygenase transcription. Cancer Res. 2006, 66, 9609–9616. [Google Scholar] [CrossRef]
- Leighton, X.; Bera, A.; Eidelman, O.; Bubendorf, L.; Zellweger, T.; Banerjee, J.; Gelmann, E.P.; Pollard, H.B.; Srivastava, M. Tissue microarray analysis delineate potential prognostic role of Annexin A7 in prostate cancer progression. PLoS ONE 2018, 13, e0205837. [Google Scholar] [CrossRef]
- Arrang, J.M.; Garbarg, M.; Lancelot, J.C.; LeComte, J.M.; Pollard, H.; Robba, M.; Schunack, W.; Schwartz, J.C. Potential interest in powerful and specific ligands for the histamine H3 receptor. Allerg. Immunol. 1988, 20, 327–329. [Google Scholar]
- Arrang, J.M.; Garbarg, M.; Lancelot, J.C.; Lecomte, J.M.; Pollard, H.; Robba, M.; Schunack, W.; Schwartz, J.C. Highly potent and selective ligands for a new class H3 of histamine receptor. Investig. Radiol. 1988, 23, S130–S132. [Google Scholar] [CrossRef]
- Brocklehurst, K.W.; Pollard, H.B. Pertussis toxin stimulates delayed-onset, Ca2+-dependent catecholamine release and the ADP-ribosylation of a 40 kDa protein in bovine adrenal chromaffin cells. FEBS Lett. 1988, 234, 439–445. [Google Scholar] [CrossRef]
- Forsberg, E.; Pollard, H. Ba2+-induced ATP release from adrenal medullary chromaffin cells is mediated by Ba2+ entry through both voltage- and receptor-gated Ca2+ channels. Neuroscience 1988, 27, 711–715. [Google Scholar] [CrossRef]
- Nassar-Gentina, V.; Pollard, H.B.; Rojas, E. Electrical activity in chromaffin cells of intact mouse adrenal gland. Am. J. Physiol. Physiol. 1988, 254, C675–C683. [Google Scholar] [CrossRef]
- Pollard, H.B.; Burns, A.L.; Rojas, E. A molecular basis for synexin-driven, calcium-dependent membrane fusion. J. Exp. Biol. 1988, 139, 267–286. [Google Scholar] [CrossRef] [PubMed]
- Pollard, H.B.; Rojas, E. Ca2+-activated synexin forms highly selective, voltage-gated Ca2+ channels in phosphatidylserine bilayer membranes. Proc. Natl. Acad. Sci. USA 1988, 85, 2974–2978. [Google Scholar] [CrossRef] [PubMed]
- Ramu, N.; Ramu, A.; Cole, D.E.; Balis, F.; Poplack, D.G.; Pollard, H.B. Mechanism of acquired resistance to methotrexate in P388 murine leukemia cells and in their doxorubicin-resistant subline. Isr. J. Med. Sci. 1988, 24, 477–482. [Google Scholar]
- Ronco, P.; Pollard, H.; Galceran, M.; Delauche, M.; Schwartz, J.C.; Verroust, P. Distribution of enkephalinase (membrane metalloendopeptidase, E.C. 3.4.24.11) in rat organs. Detection using a monoclonal antibody. Lab Investig. 1988, 58, 210–217. [Google Scholar] [PubMed]
- Burns, A.L.; Magendzo, K.; Shirvan, A.; Srivastava, M.; Rojas, E.; Alijani, M.R.; Pollard, H.B. Calcium channel activity of purified human synexin and structure of the human synexin gene. Proc. Natl. Acad. Sci. USA 1989, 86, 3798–3802. [Google Scholar] [CrossRef]
- Ling, F.; Zhang, H.; Sun, Y.; Meng, J.; Sanches, J.G.P.; Huang, H.; Zhang, Q.; Yu, X.; Wang, B.; Hou, L.; et al. AnnexinA7 promotes epithelial-mesenchymal transition by interacting with Sorcin and contributes to aggressiveness in hepatocellular carcinoma. Cell Death Dis. 2021, 12, 1018. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Guo, D.; Sha, Y.; Zhang, C.; Jiang, Y.; Hong, L.; Zhang, J.; Jiang, Y.; Lu, L.; Huang, H. ANXA7 promotes the cell cycle, proliferation and cell adhesion-mediated drug resistance of multiple myeloma cells by up-regulating CDC5L. Aging 2020, 12, 11100–11115. [Google Scholar] [CrossRef]
- Bera, A.; Biring, S. A quantitative characterization of interaction between prion protein with nucleic acids. Biochem. Biophys. Rep. 2018, 14, 114–124. [Google Scholar] [CrossRef]
- Leighton, X.; Bera, A.; Eidelman, O.; Eklund, M.; Puthillathu, N.; Pollard, H.B.; Srivastava, M. High ANXA7 Potentiates Eucalyptol Toxicity in Hormone-refractory Prostate Cancer. Anticancer Res. 2018, 38, 3831–3842. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Atwater, I.; Glasman, M.; Leighton, X.; Goping, G.; Caohuy, H.; Miller, G.; Pichel, J.; Westphal, H.; Mears, D.; et al. Defects in inositol 1,4,5-trisphosphate receptor expression, Ca2+ signaling, and insulin secretion in the anx7(+/−) knockout mouse. Proc. Natl. Acad. Sci. USA 1999, 96, 13783–13788. [Google Scholar] [CrossRef]
- Srivastava, M.; Pollard, H.B. Low in vivo levels of human anx7 (annexin vii) gene expression are due to endogenous inhibitory promoter sequences. Cell Biol. Int. 2000, 24, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Sohma, H.; Creutz, C.E.; Gasa, S.; Ohkawa, H.; Akino, T.; Kuroki, Y. Differential lipid specificities of the repeated domains of annexin IV. Biochim. Biophys. Acta 2001, 1546, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Montaville, P.; Neumann, J.-M.; Russo-Marie, F.; Ochsenbein, F.; Sanson, A. A new consensus sequence for phosphatidylserine recognition by annexins. J. Biol. Chem. 2002, 277, 24684–24693. [Google Scholar] [CrossRef] [PubMed]
- Szalai, G.; Krishnamurthy, R.; Hajnóczky, G. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J. 1999, 18, 6349–6361. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Grewal, T.; Wason, S.J.; Enrich, C.; Rentero, C. Annexins—Insights from knockout mice. Biol. Chem. 2016, 397, 1031–1053. [Google Scholar] [CrossRef]
- Heitmann, A.S.B.; Zanjani, A.A.H.; Klenow, M.B.; Mularski, A.; Sønder, S.L.; Lund, F.W.; Boye, T.L.; Dias, C.; Bendix, P.M.; Simonsen, A.C.; et al. Phenothiazines alter plasma membrane properties and sensitize cancer cells to injury by inhibiting annexin-mediated repair. J. Biol. Chem. 2021, 297, 101012. [Google Scholar] [CrossRef]
- Srivastava, M.; Bubendorf, L.; Nolan, L.; Glasman, M.; Leighton, X.; Miller, G.; Fehrle, W.; Raffeld, M.; Eidelman, O.; Kallioniemi, O.P.; et al. ANX7 as a bio-marker in prostate and breast cancer progression. Dis. Markers 2001, 17, 115–120. [Google Scholar] [CrossRef]
- Rosengarth, A.; Luecke, H. A calcium-driven conformational switch of the N-terminal and core domains of annexin A1. J. Mol. Biol. 2003, 326, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Paweletz, C.P.; Ornstein, D.K.; Roth, M.J.; Bichsel, V.E.; Gillespie, J.W.; Calvert, V.S.; Vocke, C.D.; Hewitt, S.; Duray, P.H.; Herring, J.; et al. Loss of annexin 1 correlates with early onset of tumorigenesis in esophageal and prostate carcinoma. Cancer Res. 2000, 60, 6293–6297. [Google Scholar] [PubMed]
- Rodrigo, J.P.; Garcia-Pedrero, J.M.; Fernandez, M.P.; Morgan, R.O.; Suárez, C.; Herrero, A. Annexin A1 expression in nasopharyngeal carcinoma correlates with squamous differentiation. Am. J. Rhinol. 2005, 19, 483–487. [Google Scholar] [CrossRef]
- Shen, D.; Chang, H.R.; Chen, Z.; He, J.; Lonsberry, V.; Elshimali, Y.; Chia, D.; Seligson, D.; Goodglick, L.; Nelson, S.F.; et al. Loss of annexin A1 expression in human breast cancer detected by multiple high-throughput analyses. Biochem. Biophys. Res. Commun. 2005, 326, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Petrella, A.; Festa, M.; Ercolino, S.; Zerilli, M.; Stassi, G.; Solito, E.; Parente, L. Annexin-1 downregulation in thyroid cancer correlates to the degree of tumor differentiation. Cancer Biol. Ther. 2006, 5, 643–647. [Google Scholar] [CrossRef]
- Croxtall, J.D.; Wu, H.-L.; Yang, H.-Y.; Smith, B.; Sutton, C.; Chang, B.-I.; Shi, G.-Y.; Flower, R. Lipocortin 1 co-associates with cytokeratins 8 and 18 in A549 cells via the N-terminal domain. Biochim. Biophys. Acta 1998, 1401, 39–51. [Google Scholar] [CrossRef]
- Skryma, R.; Mariot, P.; Bourhis, X.; Coppenolle, F.; Shuba, Y.; Abeele, F.V.; Legrand, G.; Humez, S.; Boilly, B.; Prevarskaya, N. Store depletion and store-operated Ca2+ current in human prostate cancer LNCaP cells: Involvement in apoptosis. J. Physiol. 2000, 527, 71–83. [Google Scholar] [CrossRef]
- Legrand, G.; Humez, S.; Slomianny, C.; Dewailly, E.; Abeele, F.V.; Mariot, P.; Wuytack, F.; Prevarskaya, N. Ca2+ pools and cell growth. Evidence for sarcoendoplasmic Ca2+-ATPases 2B involvement in human prostate cancer cell growth control. J. Biol. Chem. 2001, 276, 47608–47614. [Google Scholar] [CrossRef]
- McConkey, D.J.; Orrenius, S. The Role of Calcium in the Regulation of Apoptosis. Biochem. Biophys. Res. Commun. 1997, 239, 357–366. [Google Scholar] [CrossRef]
- Lam, M.; Dubyak, G.; Chen, L.; Nuñez, G.; Miesfeld, R.L.; Distelhorst, C.W. Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc. Natl. Acad. Sci. USA 1994, 91, 6569–6573. [Google Scholar] [CrossRef]
- Bian, X.; Hughes, F.M.; Huang, Y.; Cidlowski, J.A.; Putney, J.W., Jr. Roles of cytoplasmic Ca2+ and intracellular Ca2+ stores in induction and suppression of apoptosis in S49 cells. Am. J. Physiol. 1997, 272, C1241–C1249. [Google Scholar] [CrossRef]
- Nakamura, K.; Bossy-Wetzel, E.; Burns, K.; Fadel, M.P.; Lozyk, M.; Goping, I.S.; Opas, M.; Bleackley, R.C.; Green, D.; Michalak, M. Changes in endoplasmic reticulum luminal environment affect cell sensitivity to apoptosis. J. Cell Biol. 2000, 150, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Ferrari, D.; Magalhães, P.; Schulze-Osthoff, K.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J. Cell Biol. 2000, 148, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Foyouzi-Youssefi, R.; Arnaudeau, S.; Borner, C.; Kelley, W.L.; Tschopp, J.; Lew, D.P.; Demaurex, N.; Krause, K.-H. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2000, 97, 5723–5728. [Google Scholar] [CrossRef] [PubMed]
- Pollard, H.B.; Caohuy, H.; Minton, A.P.; Srivastava, M. Synexin (annexin VII) hypothesis for Ca2+/GTP-regulated exocytosis. Adv. Pharmacol. 1998, 42, 81–87. [Google Scholar]
- Srivastava, M.; Pollard, H.B.; Fleming, P.J. Mouse cytochrome b561: cDNA cloning and expression in rat brain, mouse embryos, and human glioma cell lines. DNA Cell Biol. 1998, 17, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, S.; Petranka, J.G.; Cheran, S.K.; Biermann, J.A.; Barrett, J.C.; Murphy, E. Reduced Capacitative Calcium Entry Correlates with Vesicle Accumulation and Apoptosis. J. Biol. Chem. 1999, 274, 8261–8268. [Google Scholar] [CrossRef]
- Danilczyk, U.G.; Cohen-Doyle, M.F.; Williams, D.B. Functional Relationship between Calreticulin, Calnexin, and the Endoplasmic Reticulum Luminal Domain of Calnexin. J. Biol. Chem. 2000, 275, 13089–13097. [Google Scholar] [CrossRef]
- Torosyan, Y.; Simakova, O.; Naga, S.; Mezhevaya, K.; Leighton, X.; Diaz, J.; Huang, W.; Pollard, H.; Srivastava, M. Annexin-A7 protects normal prostate cells and induces distinct patterns of RB-associated cytotoxicity in androgen-sensitive and -resistant prostate cancer cells. Int. J. Cancer 2009, 125, 2528–2539. [Google Scholar] [CrossRef]
- Jorgensen, P.; Tyers, M. How Cells Coordinate Growth and Division. Curr. Biol. 2004, 14, R1014–R1027. [Google Scholar] [CrossRef] [PubMed]
- Corradetti, M.N.; Guan, K.-L. Upstream of the mammalian target of rapamycin: Do all roads pass through mTOR? Oncogene 2006, 25, 6347–6360. [Google Scholar] [CrossRef]
- Hisamoto, K.; Bender, J.R. Vascular cell signaling by membrane estrogen receptors. Steroids 2005, 70, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Yang, L.; Feldman, R.I.; Sun, X.-M.; Bhalla, K.N.; Jove, R.; Nicosia, S.V.; Cheng, J.Q. Activation of phosphatidylinositol 3-kinase/Akt pathway by androgen through interaction of p85alpha, androgen receptor, and Src. J. Biol. Chem. 2003, 278, 42992–43000. [Google Scholar] [CrossRef]
- Lefai, E.; Roques, M.; Vega, N.; Laville, M.; Vidal, H. Expression of the splice variants of the p85alpha regulatory subunit of phosphoinositide 3-kinase in muscle and adipose tissue of healthy subjects and type 2 diabetic patients. Biochem. J. 2001, 360, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Bunting, M.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Molecular Cloning of a Novel Human Diacylglycerol Kinase Highly Selective for Arachidonate-containing Substrates. J. Biol. Chem. 1996, 271, 10237–10241. [Google Scholar] [CrossRef]
- Klein, J. Functions and pathophysiological roles of phospholipase D in the brain. J. Neurochem. 2005, 94, 1473–1487. [Google Scholar] [CrossRef]
- Nakashima, S.; Nozawa, Y. Possible role of phospholipase D in cellular differentiation and apoptosis. Chem. Phys. Lipids 1999, 98, 153–164. [Google Scholar] [CrossRef]
- Xu, L.; Shen, Y.; Joseph, T.; Bryant, A.; Luo, J.-Q.; Frankel, P.; Rotunda, T.; Foster, D.A. Mitogenic phospholipase D activity is restricted to caveolin-enriched membrane microdomains. Biochem. Biophys. Res. Commun. 2000, 273, 77–83. [Google Scholar] [CrossRef]
- Foster, D.A. Regulation of mTOR by phosphatidic acid? Cancer Res. 2007, 67, 1–4. [Google Scholar] [CrossRef]
- Levy, B.D.; Fokin, V.V.; Clark, J.M.; Wakelam, M.J.O.; Petasis, N.A.; Serhan, C.N. Polyisoprenyl phosphate (PIPP) signaling regulates phospholipase D activity: A ‘stop’ signaling switch for aspirin-triggered lipoxin A4. FASEB J. 1999, 13, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Furuya, Y.; Ohta, S.; Shimazaki, J. Induction of apoptosis in an androgen-independent mouse cell line by transforming growth factor-beta 1. J. Exp. Ther. Oncol. 1996, 1, 377–384. [Google Scholar] [PubMed]
- Srivastava, M.; Bubendorf, L.; Raffeld, M.; Bucher, C.; Torhorst, J.; Sauter, G.; Olsen, C.; Kallioniemi, O.P.; Eidelman, O.; Pollard, H.B. Prognostic impact of ANX7-GTPase in metastatic and HER2-negative breast cancer patients. Clin. Cancer Res. 2004, 10, 2344–2350. [Google Scholar] [CrossRef]
- Kuijpers, G.A.J.; Lee, G.; Pollard, H.B. Immunolocalization of synexin (annexin VII) in adrenal chromaffin granules and chromaffin cells: Evidence for a dynamic role in the secretory process. Cell Tissue Res. 1992, 269, 323–330. [Google Scholar] [CrossRef]
- Srivastava, M.; Eidelman, O.; Leighton, X.; Glasman, M.; Goping, G.; Pollard, H.B. Anx7 is required for nutritional control of gene expression in mouse pancreatic islets of Langerhans. Mol. Med. 2002, 8, 781–797. [Google Scholar] [CrossRef]
- Baines, L.S.; Jindal, R.M. Comment: Kidney Exchange to Overcome Financial Barriers to Kidney Transplantation. Am. J. Transplant. 2017, 17, 2742. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Mattson, M.P.; Chan, S.L. Calcium orchestrates apoptosis. Nat. Cell Biol. 2003, 5, 1041–1043. [Google Scholar] [CrossRef]
- Demaurex, N.; Distelhorst, C. Apoptosis—The calcium connection. Science 2003, 300, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.L.; Magendzo, K.; Srivastava, M.; Rojas, E.; Parra, C.; De La Fuente, M.; Cultraro, C.; Shirvan, A.; Vogel, T.; Heldman, J.; et al. Human synexin (annexin VII) polymorphisms: Tissue specificity and expression in Escherichia coli. Biochem. Soc. Trans. 1990, 18, 1118–1121. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.P.; Dowben, R.M. Water permeability of phospholipid vesicles. J. Membr. Biol. 1970, 3, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Caohuy, H.; Pollard, H.B. Activation of Annexin 7 by Protein Kinase C in Vitroand in Vivo. J. Biol. Chem. 2001, 276, 12813–12821. [Google Scholar] [CrossRef] [PubMed]
- Raffaniello, R.; Raufman, J. Guanine nucleotides activate multiple signaling pathways in permeabilized gastric chief cells. Evidence for GTP gamma S-induced calcium-independent pepsinogen secretion. J. Biol. Chem. 1993, 268, 8491–8496. [Google Scholar] [CrossRef]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deléage, G. NPS@: Network Protein Sequence Analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar] [CrossRef]
- Quinodoz, M.; Royer-Bertrand, B.; Cisarova, K.; Di Gioia, S.A.; Superti-Furga, A.; Rivolta, C. DOMINO: Using Machine Learning to Predict Genes Associated with Dominant Disorders. Am. J. Hum. Genet. 2017, 101, 623–629. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Singlet (Mutation Site) | Doublet (Mutation Sites) | Triplet (Mutation Sites) | Quadruplet (Mutation Sites) |
|---|---|---|---|
| A (1) | C (2, 3) | G (1, 2, 3) | M (1, 2, 3, 4) |
| B (2) | F (1, 2) | J (2, 3, 4) | |
| D (3) | H (1, 3) | N (1, 2, 4) | |
| E (4) | I (2, 4) | O (1, 3, 4) | |
| K (3, 4) | |||
| L (1, 4) |
| Corrective Effect of: | |||
|---|---|---|---|
| Gene Name | ANXA7 | ANX7AJ | P53 |
| ribosomal protein S6 kinase II alpha 3 (S6KII-alpha 3); ribosomal S6 kinase 2 (RSK2); insulin-stimulated protein kinase 1 (ISPK1) | 3.5 | 1.6 | −0.1 |
| bcl2 homologous antagonist/killer (BAK) | −0.2 | 0.2 | 3.3 |
| MHC class II HLA-DR-beta (DR2-DQW1/DR4 DQW3) precursor | 0.1 | 0.1 | 3.1 |
| B4-2 protein | 2.8 | 1.5 | 0.5 |
| integrin alpha E precursor (ITGAE); mucosal lymphocyte-1 antigen; hml-1 antigen; CD103 antigen | 2.7 | 0.0 | −0.1 |
| integrin alpha 8 (ITGA8) | 2.5 | 0.7 | −0.1 |
| DNA fragmentation factor 45 (DFF45) | 2.5 | 0.4 | 0.9 |
| hyaluronan receptor (RHAMM) | 2.5 | 0.0 | −0.2 |
| CDC25C; M-phase inducer phosphatase 3 | 2.4 | −0.1 | −0.2 |
| cadherin 6 precursor (CDH6); kidney cadherin (K-cadherin) | 2.4 | 0.4 | 0.1 |
| cation-independent mannose-6-phosphate receptor precursor (CI man-6-P receptor; CI-MPR); insulin-like growth factor II receptor (IGFR II) | 0.9 | −0.1 | 2.4 |
| clone PO2ST9 (brain striatum) | −0.2 | −0.3 | 2.3 |
| sonic hedgehog (SHH) | 2.1 | 0.0 | 0.1 |
| transforming growth factor beta2 precursor (TGF-beta2; TGFB2); glioblastoma-derived T-cell suppressor factor (G-TSF); bsc-1 cell growth inhibitor; polyergin; cetermin | 2.1 | 0.3 | 0.2 |
| c-myc oncogene | 0.0 | 0.0 | 2.0 |
| HLA-DR antigen-associated invariant subunit | 0.1 | 2.0 | −0.2 |
| heparin-binding growth factor 2 precursor (HBGF2); prostatropin; basic fibroblast growth factor (BFGF; FGFB; FGF2) | 0.9 | −0.7 | 2.0 |
| ras-like protein TC10 | 2.0 | 0.5 | −0.1 |
| interferon-induced guanylate-binding protein 1; guanine nucleotide-binding protein 1 | −0.5 | 1.9 | −0.1 |
| skeletal muscle phosphorylase B kinase gamma catalytic subunit | 1.9 | −0.1 | −0.2 |
| interleukin-7 (IL-7) | 0.1 | 1.9 | −0.2 |
| cyclin-dependent kinase regulatory subunit (CKS2) | 1.9 | 0.5 | 1.1 |
| eukaryotic translation initiation factor 3 beta subunit (EIF-3 beta); EIF3 P116 | −0.1 | −0.2 | 1.9 |
| semaphorin; CD100 | 1.9 | 0.3 | −0.3 |
| ephrin type-B receptor 4 precursor; tyrosine-protein kinase receptor HTK | 0.9 | 1.9 | 0.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, M.; Bera, A.; Eidelman, O.; Tran, M.B.; Jozwik, C.; Glasman, M.; Leighton, X.; Caohuy, H.; Pollard, H.B. A Dominant-Negative Mutant of ANXA7 Impairs Calcium Signaling and Enhances the Proliferation of Prostate Cancer Cells by Downregulating the IP3 Receptor and the PI3K/mTOR Pathway. Int. J. Mol. Sci. 2023, 24, 8818. https://doi.org/10.3390/ijms24108818
Srivastava M, Bera A, Eidelman O, Tran MB, Jozwik C, Glasman M, Leighton X, Caohuy H, Pollard HB. A Dominant-Negative Mutant of ANXA7 Impairs Calcium Signaling and Enhances the Proliferation of Prostate Cancer Cells by Downregulating the IP3 Receptor and the PI3K/mTOR Pathway. International Journal of Molecular Sciences. 2023; 24(10):8818. https://doi.org/10.3390/ijms24108818
Chicago/Turabian StyleSrivastava, Meera, Alakesh Bera, Ofer Eidelman, Minh B. Tran, Catherine Jozwik, Mirta Glasman, Ximena Leighton, Hung Caohuy, and Harvey B. Pollard. 2023. "A Dominant-Negative Mutant of ANXA7 Impairs Calcium Signaling and Enhances the Proliferation of Prostate Cancer Cells by Downregulating the IP3 Receptor and the PI3K/mTOR Pathway" International Journal of Molecular Sciences 24, no. 10: 8818. https://doi.org/10.3390/ijms24108818
APA StyleSrivastava, M., Bera, A., Eidelman, O., Tran, M. B., Jozwik, C., Glasman, M., Leighton, X., Caohuy, H., & Pollard, H. B. (2023). A Dominant-Negative Mutant of ANXA7 Impairs Calcium Signaling and Enhances the Proliferation of Prostate Cancer Cells by Downregulating the IP3 Receptor and the PI3K/mTOR Pathway. International Journal of Molecular Sciences, 24(10), 8818. https://doi.org/10.3390/ijms24108818