
Chemerin as an Inducer of β Cell Proliferation Mediates Mitochondrial Homeostasis and Promotes β Cell Mass Expansion

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. The Circulating Chemerin Level Is Correlated with T2DM in Humans and Mice
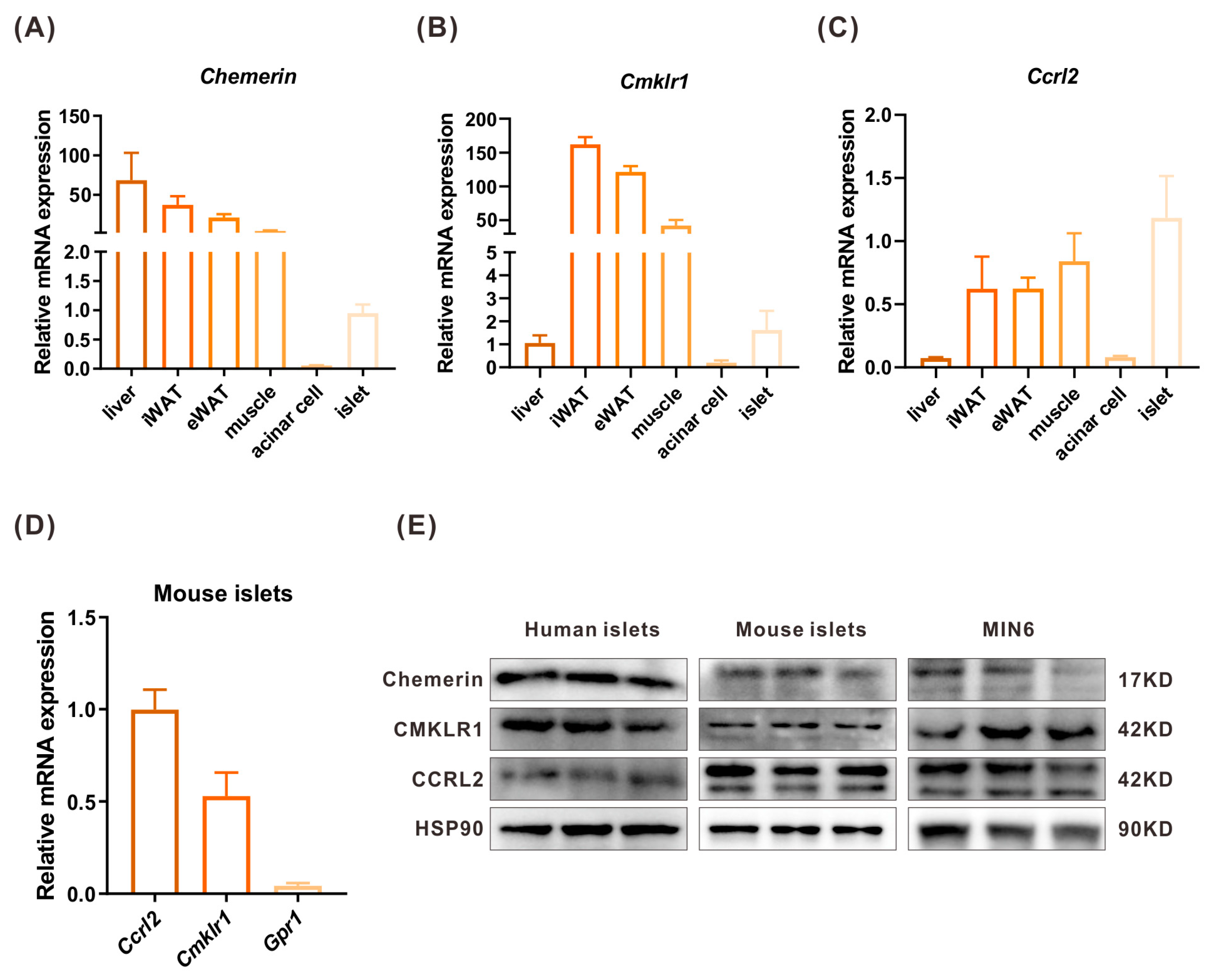
2.2. The Expressions of Chemerin, CMKLR1, and CCRL2 in Islets and Peripheral Tissues
2.3. Chemerin Can Promote the Proliferation and Inhibit the Apoptosis of MIN6 In Vitro
2.4. Islets Area and β Cell Mass Are Upregulated in Chemerin-Overexpressed Transgenic Mice
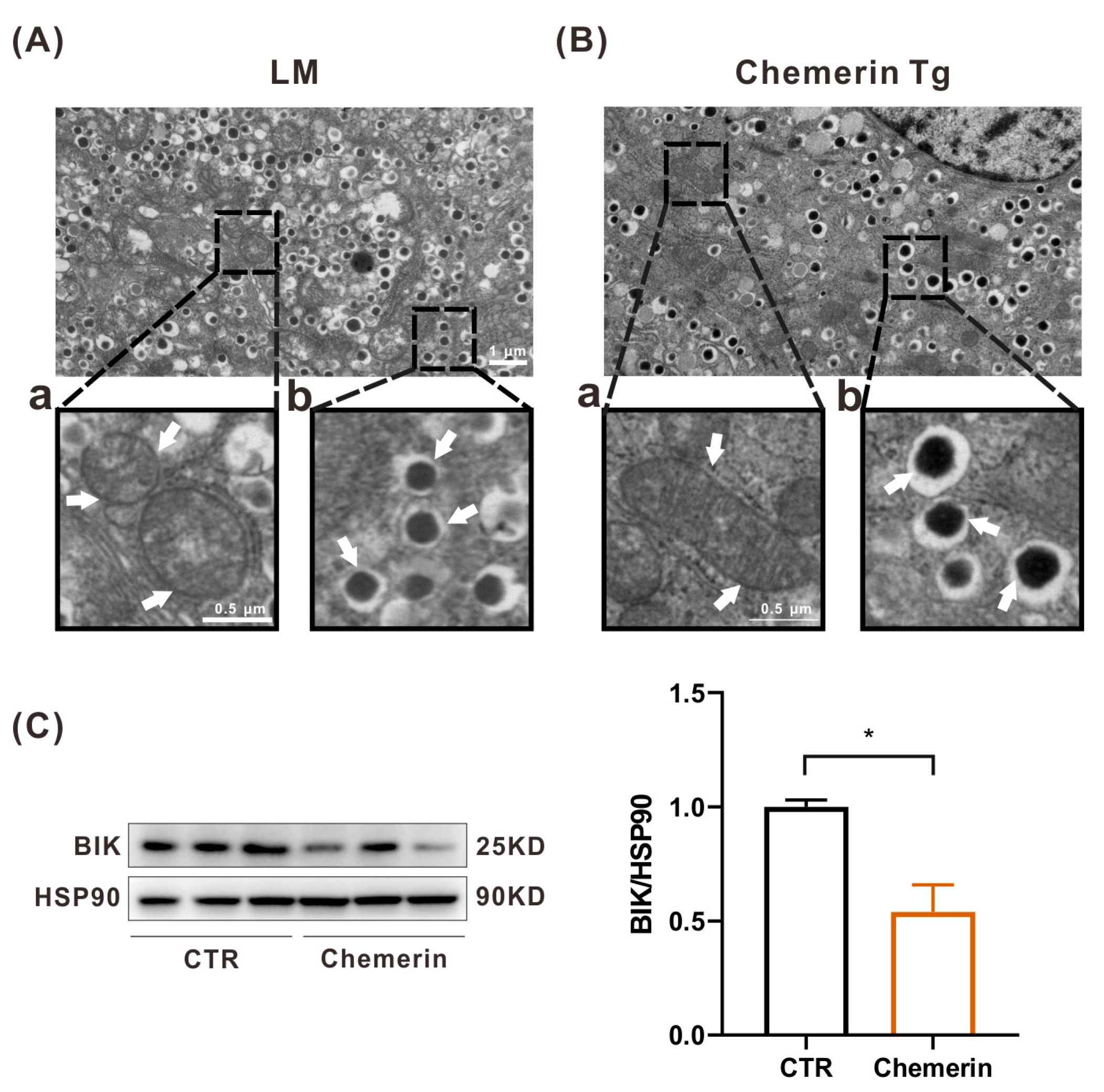
2.5. Chemerin Sustains Mitochondrial Homeostasis to Prevent Apoptosis
2.6. Chemerin Improves β Cell Function In Vitro
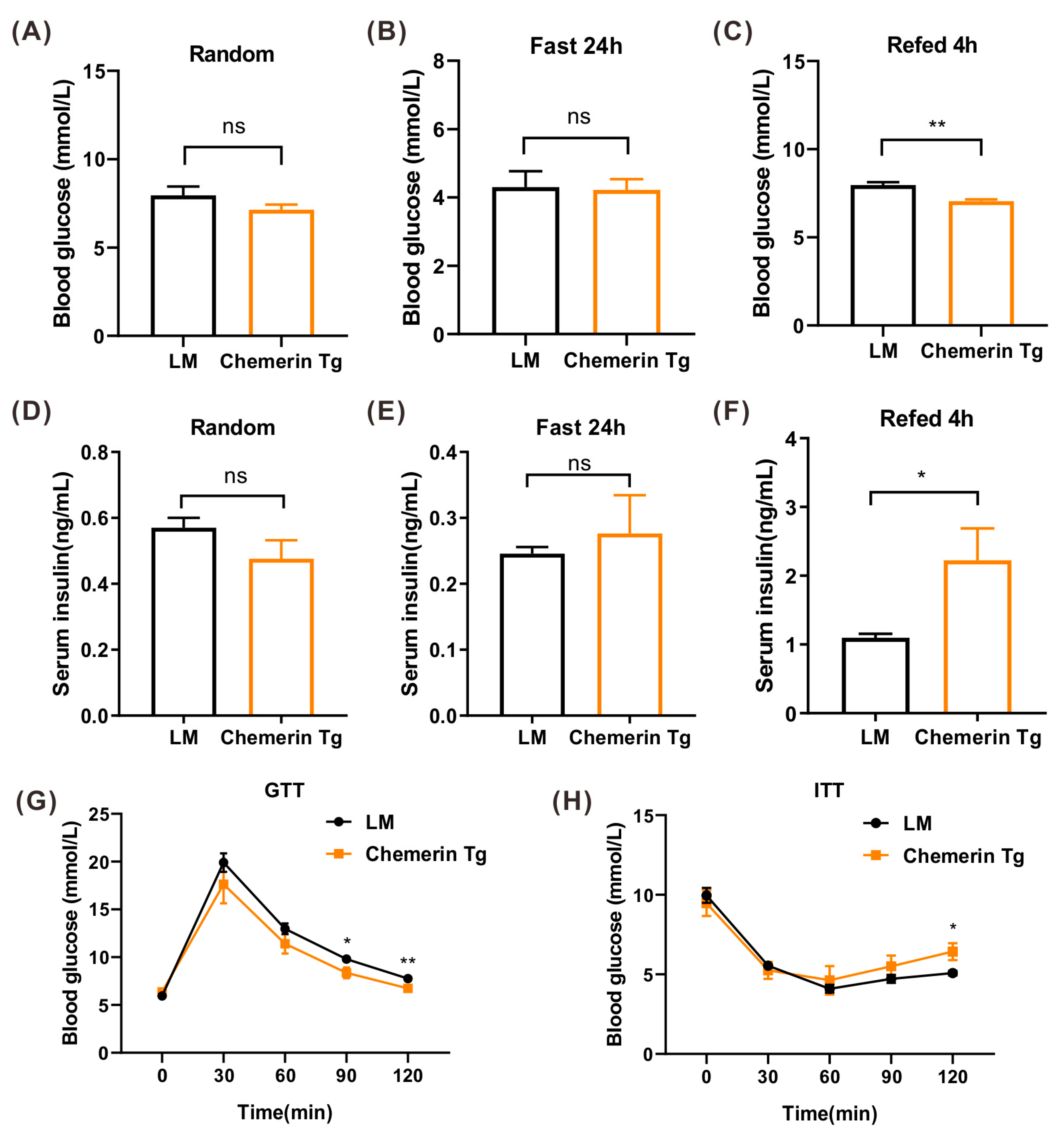
2.7. Chemerin Improves Glucose Tolerance In Vivo
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Glucose and Insulin Tolerance Test
4.3. Pancreatic Islets Isolation
4.4. Glucose-Stimulated Insulin Secretion (GSIS)
4.5. H&E Staining and Immunofluorescence Staining
4.6. Cell Culture
4.7. Apoptosis Assay
4.8. EdU Cell Proliferation Assay
4.9. Cell Counting Kit-8(CCK-8) Cell Viability Assay
4.10. Western Blot
4.11. Real-Time Quantitative PCR
4.12. Transmission Electron Microscope
4.13. [Ca2+]c and NAD(P)H Fluorescence Measurements
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klein, S.; Gastaldelli, A.; Yki-Järvinen, H.; Scherer, P.E. Why does obesity cause diabetes? Cell Metab. 2022, 34, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Gaglia, J.; Bonner-Weir, S. Inadequate β-cell mass is essential for the pathogenesis of type 2 diabetes. Lancet Diabetes Endocrinol. 2020, 8, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Dunmore, S.J.; Brown, J.E. The role of adipokines in β-cell failure of type 2 diabetes. J. Endocrinol. 2013, 216, T37–T45. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Sergeev, P.; Ehses, J.A.; Mathe, Z.; Bosco, D.; Berney, T.; Dayer, J.M.; Reinecke, M.; Halban, P.A.; Donath, M.Y. Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc. Natl. Acad. Sci. USA 2004, 101, 8138–8143. [Google Scholar] [CrossRef]
- Huang, R.; Bai, X.; Li, X.; Wang, X.; Zhao, L. Retinol-Binding Protein 4 Activates STRA6, Provoking Pancreatic β-Cell Dysfunction in Type 2 Diabetes. Diabetes 2021, 70, 449–463. [Google Scholar] [CrossRef]
- Wang, R.; Hu, W. Asprosin promotes β-cell apoptosis by inhibiting the autophagy of β-cell via AMPK-mTOR pathway. J. Cell. Physiol. 2021, 236, 215–221. [Google Scholar] [CrossRef]
- Takahashi, M.; Okimura, Y.; Iguchi, G.; Nishizawa, H.; Yamamoto, M.; Suda, K.; Kitazawa, R.; Fujimoto, W.; Takahashi, K.; Zolotaryov, F.N.; et al. Chemerin regulates β-cell function in mice. Sci. Rep. 2011, 1, 123. [Google Scholar] [CrossRef]
- Nagpal, S.; Patel, S.; Jacobe, H.; DiSepio, D.; Ghosn, C.; Malhotra, M.; Teng, M.; Duvic, M.; Chandraratna, R.A. Tazarotene-induced gene 2 (TIG2), a novel retinoid-responsive gene in skin. J. Investig. Dermatol. 1997, 109, 91–95. [Google Scholar] [CrossRef]
- Wittamer, V.; Franssen, J.D.; Vulcano, M.; Mirjolet, J.F.; Le Poul, E.; Migeotte, I.; Brézillon, S.; Tyldesley, R.; Blanpain, C.; Detheux, M.; et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 2003, 198, 977–985. [Google Scholar] [CrossRef]
- Rourke, J.L.; Dranse, H.J.; Sinal, C.J. Towards an integrative approach to understanding the role of chemerin in human health and disease. Obes. Rev. 2013, 14, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Bozaoglu, K.; Bolton, K.; McMillan, J.; Zimmet, P.; Jowett, J.; Collier, G.; Walder, K.; Segal, D. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology 2007, 148, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Goralski, K.B.; McCarthy, T.C.; Hanniman, E.A.; Zabel, B.A.; Butcher, E.C.; Parlee, S.D.; Muruganandan, S.; Sinal, C.J. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J. Biol. Chem. 2007, 282, 28175–28188. [Google Scholar] [CrossRef] [PubMed]
- Meder, W.; Wendland, M.; Busmann, A.; Kutzleb, C.; Spodsberg, N.; John, H.; Richter, R.; Schleuder, D.; Meyer, M.; Forssmann, W.G. Characterization of human circulating TIG2 as a ligand for the orphan receptor ChemR23. FEBS Lett. 2003, 555, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Zabel, B.A.; Nakae, S.; Zúñiga, L.; Kim, J.Y.; Ohyama, T.; Alt, C.; Pan, J.; Suto, H.; Soler, D.; Allen, S.J.; et al. Mast cell-expressed orphan receptor CCRL2 binds chemerin and is required for optimal induction of IgE-mediated passive cutaneous anaphylaxis. J. Exp. Med. 2008, 205, 2207–2220. [Google Scholar] [CrossRef]
- Barnea, G.; Strapps, W.; Herrada, G.; Berman, Y.; Ong, J.; Kloss, B.; Axel, R.; Lee, K.J. The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. USA 2008, 105, 64–69. [Google Scholar] [CrossRef]
- Lin, Y.; Xiao, L.; Cai, Q.; Zhu, C.; Li, S.; Li, B.; Liu, T.; Zhang, Q.; Wang, Y.; Li, Y.; et al. The chemerin-CMKLR1 axis limits thermogenesis by controlling a beige adipocyte/IL-33/type 2 innate immunity circuit. Sci. Immunol. 2021, 6, eabg9698. [Google Scholar] [CrossRef]
- Huang, C.L.; Xiao, L.L.; Xu, M.; Li, J.; Li, S.F.; Zhu, C.S.; Lin, Y.L.; He, R.; Li, X. Chemerin deficiency regulates adipogenesis is depot different through TIMP1. Genes Dis. 2021, 8, 698–708. [Google Scholar] [CrossRef]
- Li, J.; Zhang, T.; Huang, C.; Xu, M.; Xie, W.; Pei, Q.; Xie, X.; Wang, B.; Li, X. Chemerin located in bone marrow promotes osteogenic differentiation and bone formation via Akt/Gsk3β/β-catenin axis in mice. J. Cell. Physiol. 2021, 236, 6042–6054. [Google Scholar] [CrossRef]
- Takahashi, M.; Inomata, S.; Okimura, Y.; Iguchi, G.; Fukuoka, H.; Miyake, K.; Koga, D.; Akamatsu, S.; Kasuga, M.; Takahashi, Y. Decreased serum chemerin levels in male Japanese patients with type 2 diabetes: Sex dimorphism. Endocr. J. 2013, 60, 37–44. [Google Scholar] [CrossRef]
- Ferland, D.J.; Mullick, A.E.; Watts, S.W. Chemerin as a Driver of Hypertension: A Consideration. Am. J. Hypertens. 2020, 33, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Akgul Balaban, Y.; Yilmaz, N.; Kalayci, M.; Unal, M.; Turhan, T. Irisin and chemerin levels in patients with type 2 diabetes mellitus. Acta Endocrinol. (Buchar.) 2019, 15, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Mir, M.M.; Mir, R.; Alghamdi, M.A.A.; Wani, J.I.; Sabah, Z.U.; Jeelani, M.; Marakala, V.; Sohail, S.K.; O’Haj, M.; Alharthi, M.H.; et al. Differential Association of Selected Adipocytokines, Adiponectin, Leptin, Resistin, Visfatin and Chemerin, with the Pathogenesis and Progression of Type 2 Diabetes Mellitus (T2DM) in the Asir Region of Saudi Arabia: A Case Control Study. J. Pers. Med. 2022, 12, 735. [Google Scholar] [CrossRef] [PubMed]
- Balcazar, N.; Sathyamurthy, A.; Elghazi, L.; Gould, A.; Weiss, A.; Shiojima, I.; Walsh, K.; Bernal-Mizrachi, E. mTORC1 activation regulates beta-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. J. Biol. Chem. 2009, 284, 7832–7842. [Google Scholar] [CrossRef]
- Georgia, S.; Hinault, C.; Kawamori, D.; Hu, J.; Meyer, J.; Kanji, M.; Bhushan, A.; Kulkarni, R.N. Cyclin D2 is essential for the compensatory beta-cell hyperplastic response to insulin resistance in rodents. Diabetes 2010, 59, 987–996. [Google Scholar] [CrossRef]
- Desgraz, R.; Herrera, P.L. Pancreatic neurogenin 3-expressing cells are unipotent islet precursors. Development 2009, 136, 3567–3574. [Google Scholar] [CrossRef]
- Teta, M.; Long, S.Y.; Wartschow, L.M.; Rankin, M.M.; Kushner, J.A. Very slow turnover of beta-cells in aged adult mice. Diabetes 2005, 54, 2557–2567. [Google Scholar] [CrossRef]
- Stewart, A.F.; Hussain, M.A.; García-Ocaña, A.; Vasavada, R.C.; Bhushan, A.; Bernal-Mizrachi, E.; Kulkarni, R.N. Human β-cell proliferation and intracellular signaling: Part 3. Diabetes 2015, 64, 1872–1885. [Google Scholar] [CrossRef]
- MacKenzie, S.H.; Clark, A.C. Targeting cell death in tumors by activating caspases. Curr. Cancer Drug Targets 2008, 8, 98–109. [Google Scholar] [CrossRef]
- Repický, A.; Jantová, S.; Milata, V. [Signal pathways of cell proliferation and death as targets of potential chemotherapeutics]. Ceska Slov. Farm. 2008, 57, 4–10. [Google Scholar]
- Germain, M.; Mathai, J.P.; McBride, H.M.; Shore, G.C. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005, 24, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Guest, P.C. Biogenesis of the Insulin Secretory Granule in Health and Disease. Adv. Exp. Med. Biol. 2019, 1134, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, K.; Ravier, M.A.; Schraenen, A.; Creemers, J.W.; Van de Plas, R.; Granvik, M.; Van Lommel, L.; Waelkens, E.; Chimienti, F.; Rutter, G.A.; et al. Insulin crystallization depends on zinc transporter ZnT8 expression, but is not required for normal glucose homeostasis in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14872–14877. [Google Scholar] [CrossRef] [PubMed]
- Henquin, J.C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 2000, 49, 1751–1760. [Google Scholar] [CrossRef]
- Quoix, N.; Cheng-Xue, R.; Mattart, L.; Zeinoun, Z.; Guiot, Y.; Beauvois, M.C.; Henquin, J.C.; Gilon, P. Glucose and pharmacological modulators of ATP-sensitive K+ channels control [Ca2+]c by different mechanisms in isolated mouse alpha-cells. Diabetes 2009, 58, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Shimabukuro, M.; Koyama, K.; Chen, G.; Wang, M.Y.; Trieu, F.; Lee, Y.; Newgard, C.B.; Unger, R.H. Direct antidiabetic effect of leptin through triglyceride depletion of tissues. Proc. Natl. Acad. Sci. USA 1997, 94, 4637–4641. [Google Scholar] [CrossRef]
- Pick, A.; Clark, J.; Kubstrup, C.; Levisetti, M.; Pugh, W.; Bonner-Weir, S.; Polonsky, K.S. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes 1998, 47, 358–364. [Google Scholar] [CrossRef]
- Tanabe, K.; Okuya, S.; Tanizawa, Y.; Matsutani, A.; Oka, Y. Leptin induces proliferation of pancreatic beta cell line MIN6 through activation of mitogen-activated protein kinase. Biochem. Biophys. Res. Commun. 1997, 241, 765–768. [Google Scholar] [CrossRef]
- Uchida, T.; Nakamura, T.; Hashimoto, N.; Matsuda, T.; Kotani, K.; Sakaue, H.; Kido, Y.; Hayashi, Y.; Nakayama, K.I.; White, M.F.; et al. Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nat. Med. 2005, 11, 175–182. [Google Scholar] [CrossRef]
- Scherer, P.E.; Williams, S.; Fogliano, M.; Baldini, G.; Lodish, H.F. A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 1995, 270, 26746–26749. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Ohara-Imaizumi, M.; Kubota, N.; Hashimoto, S.; Eto, K.; Kanno, T.; Kubota, T.; Wakui, M.; Nagai, R.; Noda, M.; et al. Adiponectin induces insulin secretion in vitro and in vivo at a low glucose concentration. Diabetologia 2008, 51, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Li, X.; Liu, C.; Yang, J.; Ye, L.; Tang, J.; Gu, Y.; Yang, Y.; Hong, J.; Zhang, Y.; et al. Globular adiponectin augments insulin secretion from pancreatic islet beta cells at high glucose concentrations. Endocrine 2006, 30, 217–221. [Google Scholar] [CrossRef]
- Wijesekara, N.; Krishnamurthy, M.; Bhattacharjee, A.; Suhail, A.; Sweeney, G.; Wheeler, M.B. Adiponectin-induced ERK and Akt phosphorylation protects against pancreatic beta cell apoptosis and increases insulin gene expression and secretion. J. Biol. Chem. 2010, 285, 33623–33631. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.R.; Keating, D.J.; Chen, C.; Parkington, H.C. Adiponectin increases insulin content and cell proliferation in MIN6 cells via PPARγ-dependent and PPARγ-independent mechanisms. Diabetes Obes. Metab. 2012, 14, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.E.; Conner, A.C.; Digby, J.E.; Ward, K.L.; Ramanjaneya, M.; Randeva, H.S.; Dunmore, S.J. Regulation of beta-cell viability and gene expression by distinct agonist fragments of adiponectin. Peptides 2010, 31, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Dong, W.; Qian, L.; Wu, J.; Peng, Y. Visfatin inhibits apoptosis of pancreatic β-cell line, MIN6, via the mitogen-activated protein kinase/phosphoinositide 3-kinase pathway. J. Mol. Endocrinol. 2011, 47, 13–21. [Google Scholar] [CrossRef]
- Yoshimura, T.; Oppenheim, J.J. Chemokine-like receptor 1 (CMKLR1) and chemokine (C-C motif) receptor-like 2 (CCRL2); two multifunctional receptors with unusual properties. Exp. Cell Res. 2011, 317, 674–684. [Google Scholar] [CrossRef]
- Quoix, N.; Cheng-Xue, R.; Guiot, Y.; Herrera, P.L.; Henquin, J.C.; Gilon, P. The GluCre-ROSA26EYFP mouse: A new model for easy identification of living pancreatic alpha-cells. FEBS Lett. 2007, 581, 4235–4240. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Sequence | Reverse Sequence |
|---|---|---|
| 18S | CGCCGCTAGAGGTGAAATTCT | CATTCTTGGCAAATGCTTTCG |
| Ppia | TGTGCCAGGGTGGTGACTTTAC | TGGGAACCGTTTGTGTTTGG |
| Ins1 | ACCCACCCAGGCTTTTG | CCCAGCTCCAGTTGTTCC |
| Ins2 | ACCCACCCAGGCTTTTG | CCCAGCTCCAGTTGTTCC |
| MafA | CAGCGGCACATTCTGGA | CCCGCCAACTTCTCGTATT |
| Pdx1 | CGTCCAGCTCCCTTTCC | ACGGGTCCTCTTGTTTTCC |
| Ccnd1 | CAGAAGTGCGAAGAGGAGGTC | TCATCTTAGAGGCCACGAACAT |
| Ccnd2 | TGTGGATTGTCTCAAAGCCTG | CAACATCCCGCACGTCTGTA |
| Cdk1 | CTGCAGCTCGGAGCACAGTT | CCAGAACACGGAGGCACTTG |
| Cdk4 | AGACCAGGACCTGAGGACAT | TCAGGTCCCGGTGAACAATG |
| Chemerin | GCCTGGCCTGCATTAAAATGG | CTTGCTTCAGAATTGGGCAGT |
| Cmklr1 | GCCAACATACACGATGTCGC | GGATGTTGGGGTGTAGTGGG |
| Gpr1 | TGAGCTCCTGCTACTTGTGC | AGGCAATGACCACAGACAGG |
| Ccrl2 | GCCCCGGACGATGAATATGAT | CACCAAGATAAACACCGCCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Zhang, R.; Ge, Q.; Yue, L.; Ma, D.; Khattab, F.; Xie, W.; Cui, Y.; Gilon, P.; Zhao, X.; et al. Chemerin as an Inducer of β Cell Proliferation Mediates Mitochondrial Homeostasis and Promotes β Cell Mass Expansion. Int. J. Mol. Sci. 2023, 24, 9136. https://doi.org/10.3390/ijms24119136
Li M, Zhang R, Ge Q, Yue L, Ma D, Khattab F, Xie W, Cui Y, Gilon P, Zhao X, et al. Chemerin as an Inducer of β Cell Proliferation Mediates Mitochondrial Homeostasis and Promotes β Cell Mass Expansion. International Journal of Molecular Sciences. 2023; 24(11):9136. https://doi.org/10.3390/ijms24119136
Chicago/Turabian StyleLi, Min, Ruifan Zhang, Qian Ge, Lingzhi Yue, Dan Ma, Firas Khattab, Wenhua Xie, Yewei Cui, Patrick Gilon, Xueya Zhao, and et al. 2023. "Chemerin as an Inducer of β Cell Proliferation Mediates Mitochondrial Homeostasis and Promotes β Cell Mass Expansion" International Journal of Molecular Sciences 24, no. 11: 9136. https://doi.org/10.3390/ijms24119136
APA StyleLi, M., Zhang, R., Ge, Q., Yue, L., Ma, D., Khattab, F., Xie, W., Cui, Y., Gilon, P., Zhao, X., Li, X., & Cheng, R. (2023). Chemerin as an Inducer of β Cell Proliferation Mediates Mitochondrial Homeostasis and Promotes β Cell Mass Expansion. International Journal of Molecular Sciences, 24(11), 9136. https://doi.org/10.3390/ijms24119136