
GM1 Ganglioside as a Disease-Modifying Therapeutic for Parkinson’s Disease: A Multi-Functional Glycosphingolipid That Targets Multiple Parkinson’s Disease-Relevant Pathogenic Mechanisms

{kind=link}
{kind=link}
Abstract
:1. Introduction
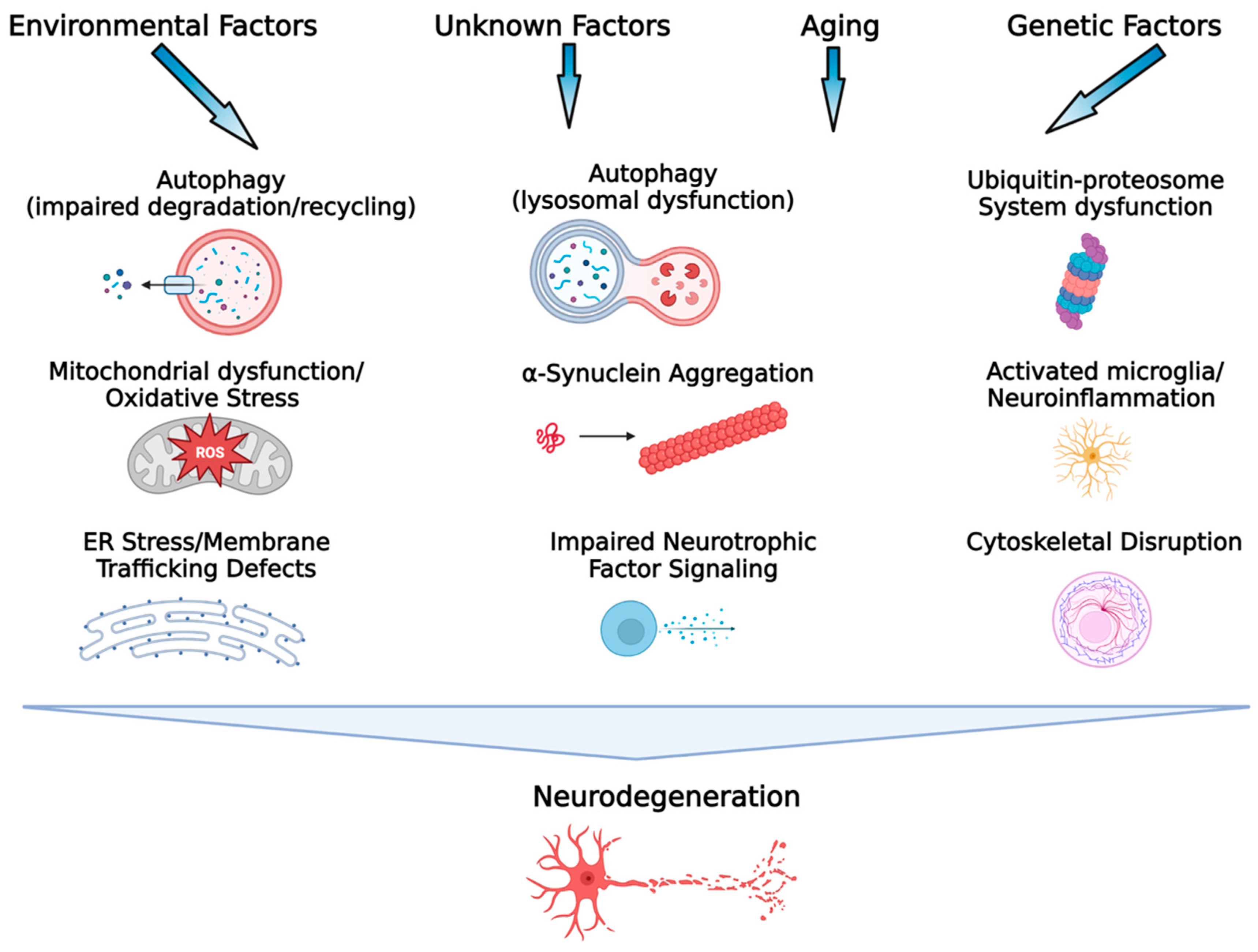
2. Multiple and Complex Pathogenic Mechanisms in Parkinson’s Disease
3. GM1 Ganglioside: A Multi-Functional Glycosphingolipid Important for the Development and Function of the Nervous System
4. The Relationship of GM1 Ganglioside to Parkinson’s Disease
5. Why Is GM1 Ganglioside a Potentially Effective Disease-Modifying Therapeutic for Parkinson’s Disease?
6. The Future of GM1 Ganglioside as a Disease-Modifying Therapeutic for PD
Funding
Conflicts of Interest
References
- Marras, C.; Tanner, C.M. The Epidemiology of Parkinson’s Disease. In Movememnt Disorders. Neurological Principles and Practice; Watts, R.L., Koller, W.C., Eds.; Mcgraw-Hill: New York, NY, USA, 2002; pp. 177–196. [Google Scholar]
- Dorsey, E.R.; Constantinescu, R.; Thompson, J.P.; Biglan, K.M.; Holloway, R.G.; Kieburtz, K.; Marshall, F.J.; Ravina, B.M.; Schifitto, G.; Siderowf, A.; et al. Projected Number of People with Parkinson Disease in The Most Populous Nations, 2005 Through 2030. Neurology 2007, 68, 384–386. [Google Scholar] [CrossRef] [PubMed]
- Geneva World Health Organization. Global Health Estimates 2020: Disease Burden By Cause, Age, Sex By Country and By Region, 2000–2019; Geneva World Health Organization: Geneva, Switzerland, 2020; Available online: Https://Apps.Who.Int/Iris/Bitstream/Handle/10665/332070/9789240005105-Eng.Pdf (accessed on 14 May 2023).
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of The Parkinson Pandemic. J. Parkinsons. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Lenka, A.; Jankovic, J. How Should Future Clinical Trials Be Designed in The Search for Disease-Modifying Therapies for Parkinson’s Disease? Expert. Rev. Neurother. 2023, 23, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Jenner, P.; Przedborski, S. Pathogenesis of Parkinson’s Disease. Mov. Disord. 2013, 28, 24–30. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondria in The Aetiology and Pathogenesis of Parkinson’s Disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Guzman, J.N.; Sanchez-Padilla, J.; Schumacker, P.T. The Role of Calcium and Mitochondrial Oxidant Stress in The Loss of Substantia Nigra Pars Compacta Dopaminergic Neurons in Parkinson’s Disease. Neuroscience 2011, 198, 221–231. [Google Scholar] [CrossRef]
- Mcnaught, K.S.; Jenner, P. Proteasomal Function Is Impaired in Substantia Nigra in Parkinson’s Disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Lansbury, P.T. Is There a Cause-And-Effect Relationship between α-Synuclein Fibrillization and Parkinson’s Disease? Nat. Cell Biol. 2000, 2, 115–119. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. Alpha-Synuclein Is Phosphorylated in Synucleinopathy Lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; De Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 Is the Dominant Pathological Modification of Alpha-Synuclein in Familial and Sporadic Lewy Body Disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-Mediated Autophagy Rescues Midbrain Dopamine Neurons from Alpha-Synuclein Toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cuervo, A.M. Autophagy Gone Awry in Neurodegenerative Diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Senkevich, K.; Gan-Or, Z. Autophagy Lysosomal Pathway Dysfunction in Parkinson’s Disease; Evidence from Human Genetics. Parkinsonism. Relat. Disord. 2020, 73, 60–71. [Google Scholar] [CrossRef]
- Costa, C.A.D.; Manaa, W.E.; Duplan, E.; Checler, F. The Endoplasmic Reticulum Stress/Unfolded Protein Response and Their Contributions to Parkinson’s Disease Physiopathology. Cells 2020, 9, 2495. [Google Scholar] [CrossRef] [PubMed]
- Mou, Z.; Yuan, Y.H.; Zhang, Z.; Song, L.K.; Chen, N.H. Endoplasmic Reticulum Stress, an Important Factor in The Development of Parkinson’s Disease. Toxicol. Lett. 2020, 324, 20–29. [Google Scholar] [CrossRef]
- Pellegrini, L.; Wetzel, A.; Granno, S.; Heaton, G.; Harvey, K. Back to The Tubule: Microtubule Dynamics in Parkinson’s Disease. Cell Mol. Life Sci. 2017, 74, 409–434. [Google Scholar] [CrossRef]
- Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as A Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. [Google Scholar] [CrossRef]
- Kramer, E.R.; Liss, B. GDNF-Ret Signaling in Midbrain Dopaminergic Neurons and Its Implication for Parkinson Disease. Febs. Lett. 2015, 589, 3760–3772. [Google Scholar] [CrossRef]
- Mori, F.; Nishie, M.; Kakita, A.; Yoshimoto, M.; Takahashi, H.; Wakabayashi, K. Relationship Among Alpha-Synuclein Accumulation, Dopamine Synthesis, and Neurodegeneration in Parkinson Disease Substantia Nigra. J. Neuropathol. Exp. Neurol. 2006, 65, 808–815. [Google Scholar] [CrossRef]
- Le, W.; Pan, T.; Huang, M.; Xu, P.; Xie, W.; Zhu, W.; Zhang, X.; Deng, H.; Jankovic, J. Decreased Nurr1 Gene Expression in Patients with Parkinson’s Disease. J. Neurol. Sci. 2008, 273, 29–33. [Google Scholar] [CrossRef]
- Jankovic, J.; Chen, S.; Le, W.D. The Role of Nurr1 in The Development of Dopaminergic Neurons and Parkinson’s Disease. Prog. Neurobiol. 2005, 77, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Long-Smith, C.M.; Sullivan, A.M.; Nolan, Y.M. The Influence of Microglia on The Pathogenesis of Parkinson’s Disease. Prog. Neurobiol. 2009, 89, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, Y.; Yagi, S.; Yokokura, M.; Sakamoto, M. Neuroinflammation in The Living Brain of Parkinson’s Disease. Parkinsonism. Relat. Disord. 2009, 15 (Suppl. S3), S200–S204. [Google Scholar] [CrossRef]
- Schneider, J.S. A Critical Role for GM1 Ganglioside in The Pathophysiology and Potential Treatment of Parkinson’s Disease. Glycoconj. J. 2021, 39, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Schengrund, C.L. Lipid Rafts: Keys to Neurodegeneration. Brain. Res. Bull. 2010, 82, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Schengrund, C.L. Gangliosides: Glycosphingolipids Essential for Normal Neural Development and Function. Trends Biochem. Sci. 2015, 40, 397–406. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G. The Multi-Tasked Life of GM1 Ganglioside, A True Factotum of Nature. Trends Biochem. Sci. 2015, 40, 407–418. [Google Scholar] [CrossRef]
- Hadaczek, P.; Wu, G.; Sharma, N.; Ciesielska, A.; Bankiewicz, K.; Davidow, A.L.; Lu, Z.H.; Forsayeth, J.; Ledeen, R.W. GDNF Signaling Implemented by GM1 Ganglioside; Failure in Parkinson’s Disease and GM1-Deficient Murine Model. Exp. Neurol. 2015, 263, 177–189. [Google Scholar] [CrossRef]
- Bachis, A.; Rabin, S.J.; Del Fiacco, M.; Mocchetti, I. Gangliosides Prevent Excitotoxicity Through Activation of TrkB Receptor. Neurotox. Res. 2002, 4, 225–234. [Google Scholar] [CrossRef]
- Mo, L.; Ren, Q.; Duchemin, A.M.; Neff, N.H.; Hadjiconstantinou, M. GM1 and ERK Signaling in The Aged Brain. Brain. Res. 2005, 1054, 125–134. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, W.; Shen, M.; Ma, X.; Li, R.; Jin, X.; Bai, H.; Gao, L. Protective Effect of GM1 Attenuates Hippocampus and Cortex Apoptosis After Ketamine Exposure in Neonatal Rat Via Pi3k/Akt/Gsk3beta Pathway. Mol. Neurobiol. 2021, 58, 3471–3483. [Google Scholar] [CrossRef] [PubMed]
- Martinez, Z.; Zhu, M.; Han, S.; Fink, A.L. GM1 Specifically Interacts with Alpha-Synuclein and Inhibits Fibrillation. Biochemistry 2007, 46, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Kim, N.C.; Luth, E.S.; Selkoe, D.J. N-Alpha-Acetylation of Alpha-Synuclein Increases Its Helical Folding Propensity, GM1 Binding Specificity and Resistance to Aggregation. PLoS ONE 2014, 9, E103727. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Aras, R.; Williams, C.K.; Koprich, J.B.; Brotchie, J.M.; Singh, V. GM1 Ganglioside Modifies Alpha-Synuclein Toxicity and Is Neuroprotective in A Rat Alpha-Synuclein Model of Parkinson’s Disease. Sci. Rep. 2019, 9, 8362. [Google Scholar] [CrossRef]
- Wei, J.; Fujita, M.; Sekigawa, A.; Sekiyama, K.; Waragai, M.; Hashimoto, M. Gangliosides’ Protection Against Lysosomal Pathology of Synucleinopathies. Autophagy 2009, 5, 860–861. [Google Scholar] [CrossRef]
- Dai, R.; Zhang, S.; Duan, W.; Wei, R.; Chen, H.; Cai, W.; Yang, L.; Wang, Q. Enhanced Autophagy Contributes to Protective Effects of GM1 Ganglioside Against Abeta1-42-Induced Neurotoxicity and Cognitive Deficits. Neurochem. Res. 2017, 42, 2417–2426. [Google Scholar] [CrossRef]
- Guo, Y.L.; Duan, W.J.; Lu, D.H.; Ma, X.H.; Li, X.X.; Li, Z.; Bi, W.; Kurihara, H.; Liu, H.Z.; Li, Y.F.; et al. Autophagy-Dependent Removal of Alpha-Synuclein: A Novel Mechanism of GM1 Ganglioside Neuroprotection Against Parkinson’s Disease. Acta. Pharmacol. Sin. 2021, 42, 518–528. [Google Scholar] [CrossRef]
- Hatano, T.; Kubo, S.; Imai, S.; Maeda, M.; Ishikawa, K.; Mizuno, Y.; Hattori, N. Leucine-Rich Repeat Kinase 2 Associates with Lipid Rafts. Hum. Mol. Genet. 2007, 16, 678–690. [Google Scholar] [CrossRef]
- Finsterwald, C.; Dias, S.; Magistretti, P.J.; Lengacher, S. Ganglioside GM1 Targets Astrocytes to Stimulate Cerebral Energy Metabolism. Front. Pharmacol. 2021, 12, 653842. [Google Scholar] [CrossRef]
- Yang, R.; Wang, Q.; Min, L.; Sui, R.; Li, J.; Liu, X. Monosialoganglioside Improves Memory Deficits and Relieves Oxidative Stress in The Hippocampus of Rat Model of Alzheimer’s Disease. Neurol. Sci. 2013, 34, 1447–1451. [Google Scholar] [CrossRef]
- Gong, G.; Yin, L.; Yuan, L.; Sui, D.; Sun, Y.; Fu, H.; Chen, L.; Wang, X. Ganglioside GM1 Protects Against High Altitude Cerebral Edema in Rats by Suppressing the Oxidative Stress and Inflammatory Response Via the PI3k/AKT-Nrf2 Pathway. Mol. Immunol. 2018, 95, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Favaron, M.; Manev, H.; Alho, H.; Bertolino, M.; Ferret, B.; Guidotti, A.; Costa, E. Gangliosides Prevent Glutamate and Kainate Neurotoxicity in Primary Neuronal Cultures of Neonatal Rat Cerebellum and Cortex. Proc. Natl. Acad. Sci. USA 1988, 85, 7351–7355. [Google Scholar] [CrossRef] [PubMed]
- Lipartiti, M.; Lazzaro, A.; Zanoni, R.; Bonvento, G.; Mazzari, S. Effects of Monosialoganglioside GM1 in Experimental Models of Ischemic Brain Damage. Ital. J. Neurol. Sci. 1991, 12, 11–13. [Google Scholar]
- Lipartiti, M.; Lazzaro, A.; Zanoni, R.; Mazzari, S.; Toffano, G.; Leon, A. Monosialoganglioside GM1 Reduces NMDA Neurotoxicity in Neonatal Rat Brain. Exp. Neurol. 1991, 113, 301–305. [Google Scholar] [CrossRef]
- Park, J.Y.; Kim, K.S.; Lee, S.B.; Ryu, J.S.; Chung, K.C.; Choo, Y.K.; Jou, I.; Kim, J.; Park, S.M. On The Mechanism of Internalization of Alpha-Synuclein into Microglia: Roles of Ganglioside GM1 and Lipid Raft. J. Neurochem. 2009, 110, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Singh, G.; Williams, C.K.; Singh, V. GM1 Ganglioside Modifies Microglial and Neuroinflammatory Responses to Alpha-Synuclein in The Rat AAV-A53t Alpha-Synuclein Model of Parkinson’s Disease. Mol. Cell Neurosci. 2022, 120, 103729. [Google Scholar] [CrossRef] [PubMed]
- Galleguillos, D.; Wang, Q.; Steinberg, N.; Zaidi, A.; Shrivastava, G.; Dhami, K.; Daskhan, G.C.; Schmidt, E.N.; Dworsky-Fried, Z.; Giuliani, F.; et al. Anti-Inflammatory Role of GM1 and Other Gangliosides on Microglia. J. Neuroinflamm. 2022, 19, 9. [Google Scholar] [CrossRef]
- Fazzari, M.; Di Biase, E.; Lunghi, G.; Mauri, L.; Chiricozzi, E.; Sonnino, S. Novel Insights on GM1 and Parkinson’s Disease: A Critical Review. Glycoconj. J. 2022, 39, 27–38. [Google Scholar] [CrossRef]
- Chowdhury, S.; Ledeen, R. The Key Role of GM1 Ganglioside in Parkinson’s Disease. Biomolecules 2022, 12, 173. [Google Scholar] [CrossRef]
- Sonnino, S. The Relationship between Depletion of Brain GM1 Ganglioside and Parkinson’s Disease. Febs. Open. Bio. 2023. [Google Scholar] [CrossRef]
- Svennerholm, L.; Bostrom, K.; Fredman, P.; Mansson, J.E.; Rosengren, B.; Rynmark, B.M. Human Brain Gangliosides: Developmental Changes from Early Fetal Stage to Advanced Age. Biochim. Biophys. Acta 1989, 1005, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Svennerholm, L.; Bostrom, K.; Jungbjer, B.; Olsson, L. Membrane Lipids of Adult Human Brain: Lipid Composition of Frontal and Temporal Lobe in Subjects of Age 20 to 100 Years. J. Neurochem. 1994, 63, 1802–1811. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, T.; Yamaguchi, K. Mammalian Sialidases: Physiological and Pathological Roles in Cellular Functions. Glycobiology 2012, 22, 880–896. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Wu, G.; Lu, Z.H.; Kumar, R.; Ledeen, R. Age-Related Decline in Gangliosides GM1 and GD1a in Non-CNS Tissues of Normal Mice: Implications for Peripheral Symptoms of Parkinson’s Disease. Biomedicines 2023, 11, 209. [Google Scholar] [CrossRef]
- Verma, M.; Schneider, J.S. SiRNA-Mediated Knockdown of B3GALT4 Decreases GM1 Ganglioside Expression and Enhances Vulnerability for Neurodegeneration. Mol. Cell Neurosci. 2019, 95, 25–30. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Choi, H.; Chevalier, A.; Hogan, D.; Akgoc, Z.; Schneider, J.S. Sex-Related Abnormalities in Substantia Nigra Lipids in Parkinson’s Disease. Asn. Neuro 2018, 10, 1759091418781889. [Google Scholar] [CrossRef]
- Huebecker, M.; Moloney, E.B.; Van Der Spoel, A.C.; Priestman, D.A.; Isacson, O.; Hallett, P.J.; Platt, F.M. Reduced Sphingolipid Hydrolase Activities, Substrate Accumulation and Ganglioside Decline in Parkinson’s Disease. Mol. Neurodegener. 2019, 14, 40. [Google Scholar] [CrossRef]
- Schneider, J.S. Altered Expression of Genes Involved in Ganglioside Biosynthesis in Substantia Nigra Neurons in Parkinson’s Disease. PLoS ONE 2018, 13, E0199189. [Google Scholar] [CrossRef]
- Niimi, Y.; Ito, S.; Mizutani, Y.; Murate, K.; Shima, S.; Ueda, A.; Satake, W.; Hattori, N.; Toda, T.; Mutoh, T. Altered Regulation of Serum Lysosomal Acid Hydrolase Activities in Parkinson’s Disease: A Potential Peripheral Biomarker? Parkinsonism. Relat. Disord. 2019, 61, 132–137. [Google Scholar] [CrossRef]
- Niimi, Y.; Mizutani, Y.; Akiyama, H.; Watanabe, H.; Shiroki, R.; Hirabayashi, Y.; Hoshinaga, K.; Mutoh, T. Cerebrospinal Fluid Profiles in Parkinson’s Disease: No Accumulation of Glucosylceramide, But Significant Downregulation of Active Complement C5 Fragment. J. Parkinsons. Dis. 2021, 11, 221–232. [Google Scholar] [CrossRef]
- Ledeen, R.; Chowdhury, S.; Lu, Z.H.; Chakraborty, M.; Wu, G. Systemic Deficiency of GM1 Ganglioside in Parkinson’s Disease Tissues and Its Relation to The Disease Etiology. Glycoconj. J. 2022, 39, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Lu, Z.H.; Kulkarni, N.; Ledeen, R.W. Deficiency of Ganglioside GM1 Correlates with Parkinson’s Disease in Mice and Humans. J. Neurosci. Res. 2012, 90, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Gollomp, S.M.; Sendek, S.; Colcher, A.; Cambi, F.; Du, W. A Randomized, Controlled, Delayed Start Trial of GM1 Ganglioside in Treated Parkinson’s Disease Patients. J. Neurol. Sci. 2013, 324, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Cambi, F.; Gollomp, S.M.; Kuwabara, H.; Brasic, J.R.; Leiby, B.; Sendek, S.; Wong, D.F. GM1 Ganglioside in Parkinson’s Disease: Pilot Study of Effects on Dopamine Transporter Binding. J. Neurol. Sci. 2015, 356, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Lungu, C.; Cedarbaum, J.M.; Dawson, T.M.; Dorsey, E.R.; Faraco, C.; Federoff, H.J.; Fiske, B.; Fox, R.; Goldfine, A.M.; Kieburtz, K.; et al. Seeking Progress in Disease Modification in Parkinson Disease. Parkinsonism. Relat. Disord. 2021, 90, 134–141. [Google Scholar] [CrossRef]
- Mcfarthing, K.; Rafaloff, G.; Baptista, M.; Mursaleen, L.; Fuest, R.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in The Clinical Trial Pipeline: 2022 Update. J. Parkinsons. Dis. 2022, 12, 1073–1082. [Google Scholar] [CrossRef]
- Tikhonova, M.A.; Chang, H.M.; Singh, S.K. Editorial: Experimental and Innovative Approaches to Multi-Target Treatment of Parkinson’s and Alzheimer’s Diseases—Volume II. Front. Neurosci. 2023, 17, 1171866. [Google Scholar] [CrossRef]
- Schneider, J.S.; Sendek, S.; Daskalakis, C.; Cambi, F. GM1 Ganglioside in Parkinson’s Disease: Results of a Five Year Open Study. J. Neurol. Sci. 2010, 292, 45–51. [Google Scholar] [CrossRef]
- Di Martino, A. Elimination of Scrapie-Agent Infectivity in Naturally Derived Biologics. In Bovine Spongiform Encephalopathy; Gibbs, C.J., Ed.; Springer: New York, NY, USA, 1996; pp. 325–337. [Google Scholar]
- Wang, B.; Brand-Miller, J. The Role and Potential of Sialic Acid in Human Nutrition. Eur. J. Clin. Nutr. 2003, 57, 1351–1369. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Seo, J.H.; Alselehdar, S.K.; Defrees, S.; Ledeen, R.W. Mice Deficient in GM1 Manifest Both Motor and Non-Motor Symptoms of Parkinson’s Disease; Successful Treatment with Synthetic GM1 Ganglioside. Exp. Neurol. 2020, 329, 113284. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, L.; Yang, X.; Bai, Y.; Chen, X. Process Engineering and Glycosyltransferase Improvement for Short Route Chemoenzymatic Total Synthesis of GM1 Gangliosides. Chem. Eur. J. 2023, 29, e202300005. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, J.; Jiao, S.; Ji, T. Ganglioside-Monosialic Acid (GM1) Prevents Oxaliplatin-Induced Peripheral Neurotoxicity in Patients with Gastrointestinal Tumors. World J. Surg. Oncol. 2013, 11, 19. [Google Scholar] [CrossRef]
- Ghidoni, R.; Fiorilli, A.; Trinchera, M.; Venerando, B.; Chigorno, V.; Tettamanti, G. Uptake, Cell Penetration and Metabolic Processing of Exogenously Administered GM1 Ganglioside in Rat Brain. Neurochem. Int. 1989, 15, 455–465. [Google Scholar] [CrossRef]
- Revunov, E.; Johnstrom, P.; Arakawa, R.; Malmquist, J.; Jucaite, A.; Defay, T.; Takano, A.; Schou, M. First Radiolabeling of a Ganglioside with a Positron Emitting Radionuclide: In Vivo PET Demonstrates Low Exposure of Radiofluorinated GM1 in Non-Human Primate Brain. Acs. Chem. Neurosci. 2020, 11, 1245–1249. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.S.; Monahan, A.J.; Carvey, P.M.; Hendey, B. Blood-Brain Barrier Pathology in Alzheimer’s and Parkinson’s Disease: Implications for Drug Therapy. Cell Transplant. 2007, 16, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal Blood-Brain Barrier Permeability in Parkinson’s Disease. J. Cereb. Blood. Flow. Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef]
- Kumbale, R.; Frey, W.H.; Wilson, S.; Rahman, Y.E. GM1 Delivery to The CSF Via the Olfactory Pathway. Drug Deliv. 1999, 6, 23–30. [Google Scholar] [CrossRef]
- Itokazu, Y.; Fuchigami, T.; Morgan, J.C.; Yu, R.K. Intranasal Infusion of GD3 and GM1 Gangliosides Downregulates Alpha-Synuclein and Controls Tyrosine Hydroxylase Gene in a PD Model Mouse. Mol. Ther. 2021, 29, 3059–3071. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, J.S. GM1 Ganglioside as a Disease-Modifying Therapeutic for Parkinson’s Disease: A Multi-Functional Glycosphingolipid That Targets Multiple Parkinson’s Disease-Relevant Pathogenic Mechanisms. Int. J. Mol. Sci. 2023, 24, 9183. https://doi.org/10.3390/ijms24119183
Schneider JS. GM1 Ganglioside as a Disease-Modifying Therapeutic for Parkinson’s Disease: A Multi-Functional Glycosphingolipid That Targets Multiple Parkinson’s Disease-Relevant Pathogenic Mechanisms. International Journal of Molecular Sciences. 2023; 24(11):9183. https://doi.org/10.3390/ijms24119183
Chicago/Turabian StyleSchneider, Jay S. 2023. "GM1 Ganglioside as a Disease-Modifying Therapeutic for Parkinson’s Disease: A Multi-Functional Glycosphingolipid That Targets Multiple Parkinson’s Disease-Relevant Pathogenic Mechanisms" International Journal of Molecular Sciences 24, no. 11: 9183. https://doi.org/10.3390/ijms24119183
APA StyleSchneider, J. S. (2023). GM1 Ganglioside as a Disease-Modifying Therapeutic for Parkinson’s Disease: A Multi-Functional Glycosphingolipid That Targets Multiple Parkinson’s Disease-Relevant Pathogenic Mechanisms. International Journal of Molecular Sciences, 24(11), 9183. https://doi.org/10.3390/ijms24119183