
Narrative Review: Update on the Molecular Diagnosis of Fragile X Syndrome
 , , , and
, , , and
Abstract
:1. Introduction
2. Genetics, Epidemiology, and Aetiology
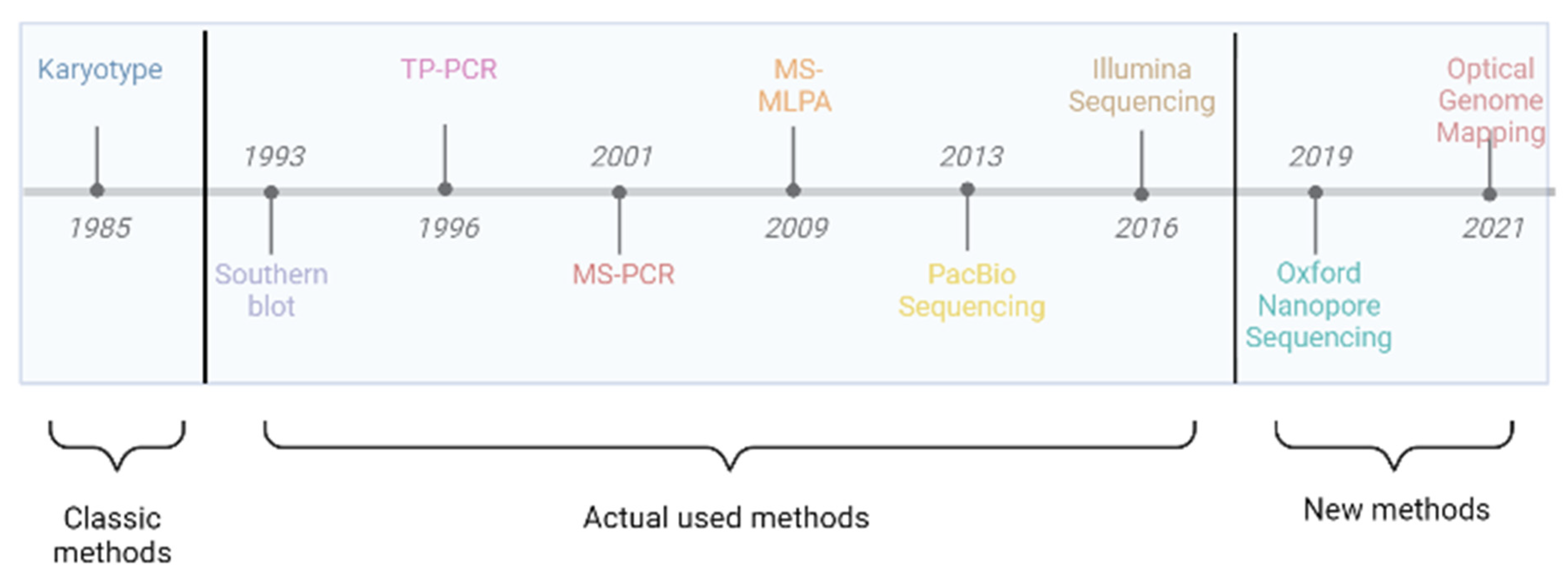
3. Cytogenetic and Molecular Diagnosis for Fragile X Syndrome
3.1. Karyotyping
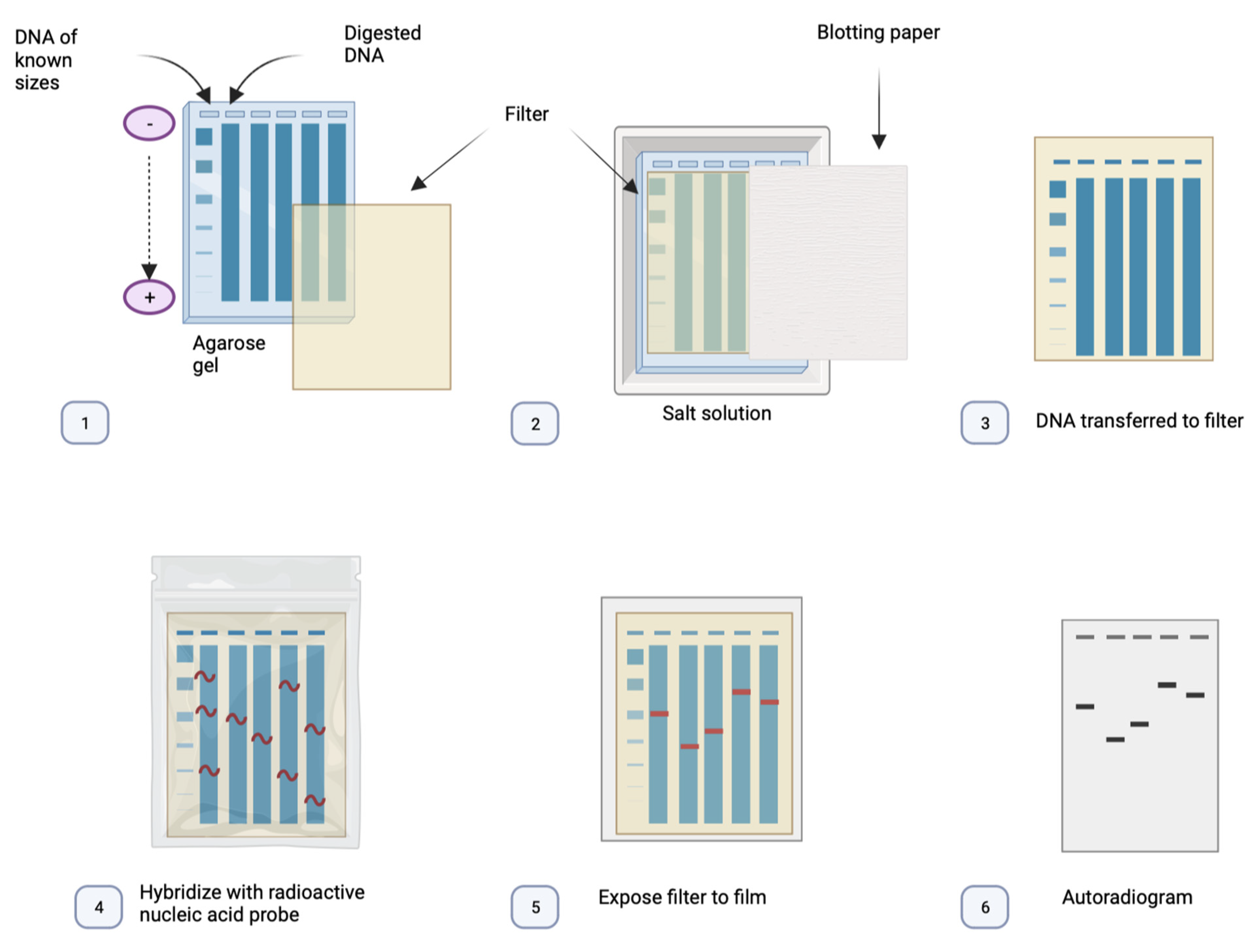
3.2. Southern Blot
3.3. Triplet Repeat Primed PCR (TP-PCR)
3.4. Digital PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TRN | MS | PM | AGG Int | MOS | Del Ins | P. mut | Advantages | Disadvantages | |
|---|---|---|---|---|---|---|---|---|---|
| Karyotype | – | – | – | – | + | ± | – | Cheap. | Outdated. Inaccurate. Faulty. |
| Southern blot | ± | + | ± | – | + | + | – | Golden standard. (repeat expansion and methylation status) [55]. | Very labour-intensive. Time-consuming. Not in routine diagnostic settings [55,56]. |
| TP-PCR | + | – | + | ± | ± | – | – | Rapid. Facile. Routine diagnostic. High sample throughput [43]. | Does not provide the size of expanded CGG repeats. |
| D-PCR | + | – | + | – | + | + | – | Low costs. High number of fragments. | Costs with equipment and consumables. |
| MS-PCR | – | + | – | – | ± | – | – | High sensitivity and specificity. | False-positive results. |
| MS-MLPA | – | ± | – | – | ± | ± | ± | Cheap. Golden standard (copy number). | False-positive results. Not for females. |
| Illumina seq | ± | – | + | + | ± | + | + | Standardized technique. Ideal for point mutations. | Not for large expansions. |
| PacBio seq | + | + | + | + | + | + | + | High coverage and accuracy [57]. A single assay. | High costs for equipment. |
| Nanopore seq | + | + | + | + | + | + | + | Scalable (Flongle for few patients and PromethION for large numbers). Time and space efficient [58]. All in a single assay. | Only for research use. Complex bioinformatics interpretation. |
| OGM | ± | – | – | – | + | + | – | High-resolution genome-wide analysis of all structural variants [59]. | Low throughput. Not for PM. Only for research use. |
3.5. Methylation-Specific PCR
3.6. Methylation-Specific Multiplex Ligation-Dependent Probe Amplification
3.7. Optical Genome Mapping
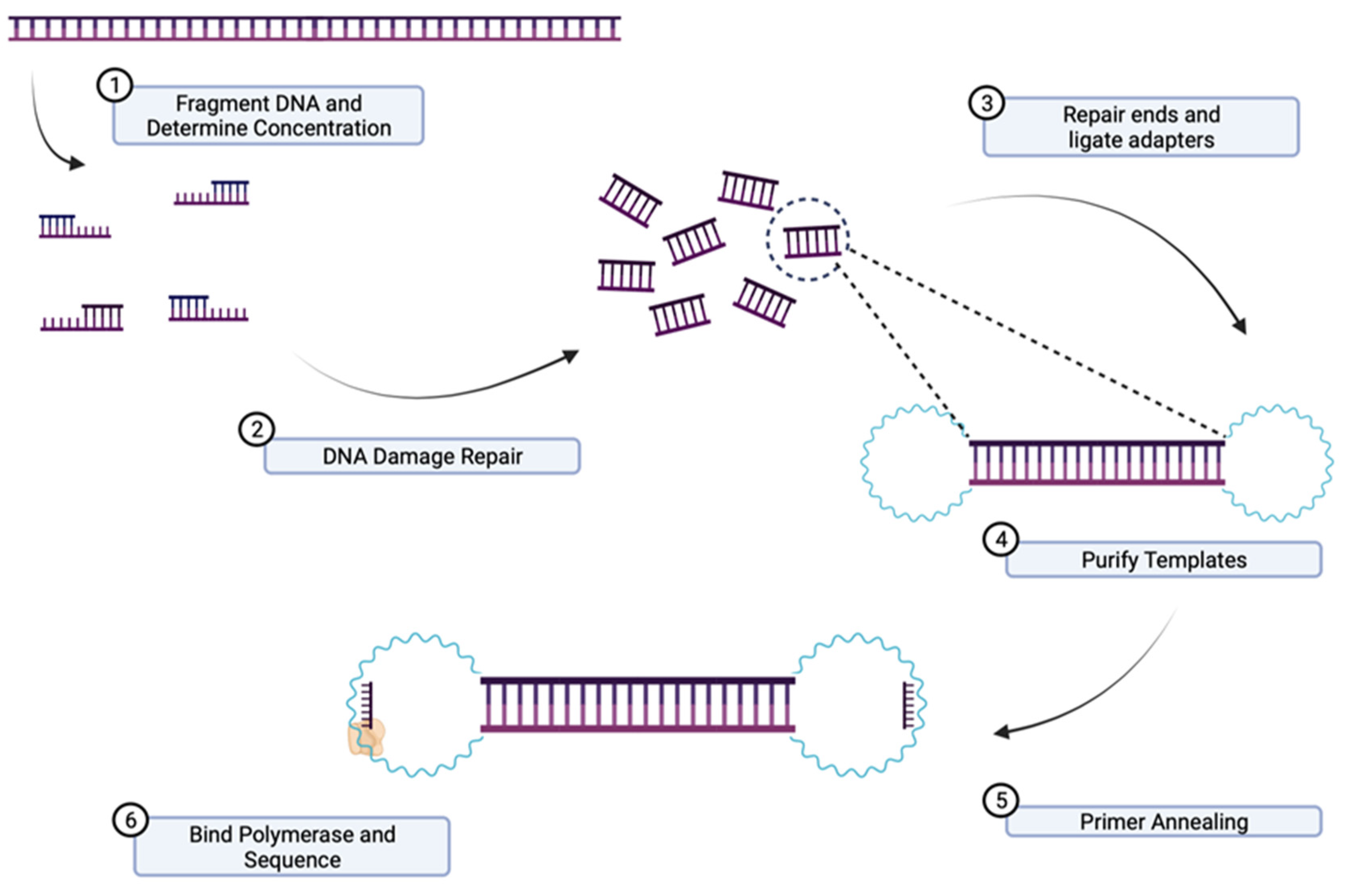
3.8. Short-Read Sequencing
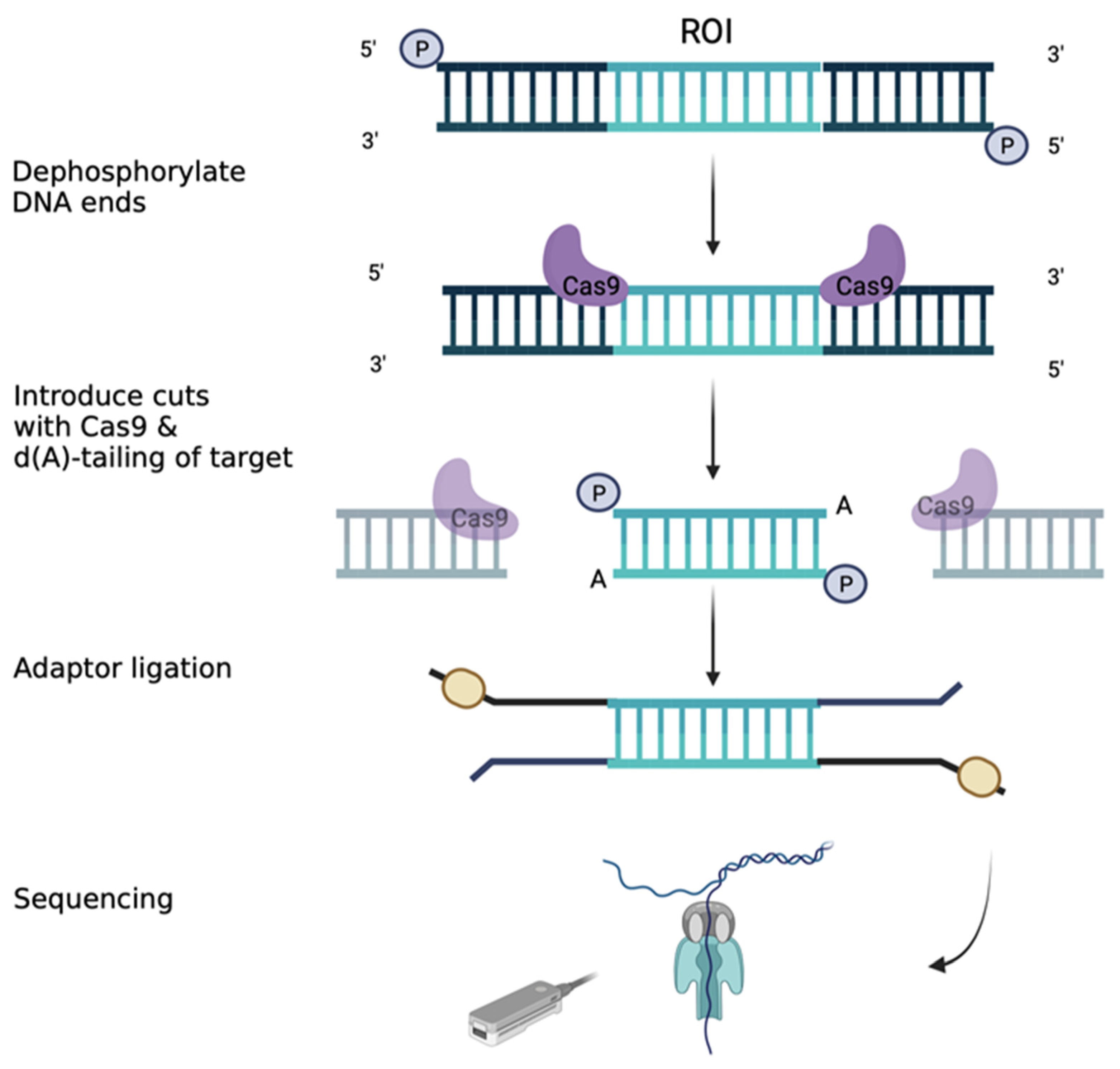
3.9. Long-Range Sequencing
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stone, W.L.; Basit, H.; Shah, M.; Los, E. Fragile X Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Nakahori, Y.; Knight, S.J.L.; Holland, J.; Schwartz, C.; Roche, A.; Tarleton, J.; Wong, S.; Flint, T.J.; Froster-Iskenius, U.; Bentley, D.; et al. Molecular Heterogeneity of the Fragile X Syndrome. Nucleic Acids Res. 1991, 19, 4355–4359. [Google Scholar] [CrossRef] [PubMed]
- Devys, D.; Lutz, Y.; Rouyer, N.; Bellocq, J.-P.; Mandel, J.-L. The FMR–1 Protein Is Cytoplasmic, Most Abundant in Neurons and Appears Normal in Carriers of a Fragile X Premutation. Nat. Genet. 1993, 4, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Ciaccio, C.; Fontana, L.; Milani, D.; Tabano, S.; Miozzo, M.; Esposito, S. Fragile X Syndrome: A Review of Clinical and Molecular Diagnoses. Ital. J. Pediatr. 2017, 43, 39. [Google Scholar] [CrossRef] [PubMed]
- Reches, A. Fragile X Syndrome: Introduction. In Fragile-X Syndrome; Ben-Yosef, D., Mayshar, Y., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1942, pp. 3–10. ISBN 978-1-4939-9079-5. [Google Scholar]
- Hagerman, R.J.; Hagerman, P.J. Fragile X Syndrome. In Outcomes in Neurodevelopmental and Genetic Disorders; Howlin, P., Udwin, O., Eds.; Cambridge University Press: Cambridge, UK, 2002; pp. 198–219. ISBN 978-0-521-79721-4. [Google Scholar]
- Oakes, A.; Thurman, A.J.; McDuffie, A.; Bullard, L.M.; Hagerman, R.J.; Abbeduto, L. Characterising Repetitive Behaviours in Young Boys with Fragile X Syndrome: Repetitive Behaviours in FXS. J. Intellect. Disabil. Res. 2016, 60, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Crawford, H.; Scerif, G.; Wilde, L.; Beggs, A.; Stockton, J.; Sandhu, P.; Shelley, L.; Oliver, C.; McCleery, J. Genetic Modifiers in Rare Disorders: The Case of Fragile X Syndrome. Eur. J. Hum. Genet. 2021, 29, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Cabal-Herrera, A.M.; Tassanakijpanich, N.; Salcedo-Arellano, M.J.; Hagerman, R.J. Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS): Pathophysiology and Clinical Implications. Int. J. Mol. Sci. 2020, 21, 4391. [Google Scholar] [CrossRef]
- Deng, P.-Y.; Klyachko, V.A. Channelopathies in Fragile X Syndrome. Nat. Rev. Neurosci. 2021, 22, 275–289. [Google Scholar] [CrossRef]
- Kumari, D.; Usdin, K. Molecular Analysis of FMR1 Alleles for Fragile X Syndrome Diagnosis and Patient Stratification. Expert Rev. Mol. Diagn. 2020, 20, 363–365. [Google Scholar] [CrossRef]
- Spector, E.; Behlmann, A.; Kronquist, K.; Rose, N.C.; Lyon, E.; Reddi, H.V. Laboratory Testing for Fragile X, 2021 Revision: A Technical Standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 799–812. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X Syndrome. Nat. Rev. Dis. Primer 2017, 3, 17065. [Google Scholar] [CrossRef]
- Owens, K.M.; Dohany, L.; Holland, C.; DaRe, J.; Mann, T.; Settler, C.; Longman, R.E. FMR1 Premutation Frequency in a Large, Ethnically Diverse Population Referred for Carrier Testing. Am. J. Med. Genet. A. 2018, 176, 1304–1308. [Google Scholar] [CrossRef] [PubMed]
- Nobile, V.; Pucci, C.; Chiurazzi, P.; Neri, G.; Tabolacci, E. DNA Methylation, Mechanisms of FMR1 Inactivation and Therapeutic Perspectives for Fragile X Syndrome. Biomolecules 2021, 11, 296. [Google Scholar] [CrossRef]
- Verkerk, A.J.M.H.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.A.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a Gene (FMR-1) Containing a CGG Repeat Coincident with a Breakpoint Cluster Region Exhibiting Length Variation in Fragile X Syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Naumann, A.; Hochstein, N.; Weber, S.; Fanning, E.; Doerfler, W. A Distinct DNA-Methylation Boundary in the 5′- Upstream Sequence of the FMR1 Promoter Binds Nuclear Proteins and Is Lost in Fragile X Syndrome. Am. J. Hum. Genet. 2009, 85, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Tortora, N.; Allen, E.; Macpherson, J.; Mila, M.; Vianna-Morgante, A.M.; Sherman, S.L.; Dobkin, C.; Latham, G.J.; et al. Expansions and Contractions of the FMR1 CGG Repeat in 5,508 Transmissions of Normal, Intermediate, and Premutation Alleles. Am. J. Med. Genet. A. 2019, 179, 1148–1156. [Google Scholar] [CrossRef]
- Tabolacci, E.; Nobile, V.; Pucci, C.; Chiurazzi, P. Mechanisms of the FMR1 Repeat Instability: How Does the CGG Sequence Expand? Int. J. Mol. Sci. 2022, 23, 5425. [Google Scholar] [CrossRef]
- Johnson, K.; Herring, J.; Richstein, J. Fragile X Premutation Associated Conditions (FXPAC). Front. Pediatr. 2020, 8, 266. [Google Scholar] [CrossRef] [PubMed]
- Valor, L.M.; Morales, J.C.; Hervás-Corpión, I.; Marín, R. Molecular Pathogenesis and Peripheral Monitoring of Adult Fragile X-Associated Syndromes. Int. J. Mol. Sci. 2021, 22, 8368. [Google Scholar] [CrossRef]
- Tassanakijpanich, N.; Hagerman, R.J.; Worachotekamjorn, J. Fragile X Premutation and Associated Health Conditions: A Review. Clin. Genet. 2021, 99, 751–760. [Google Scholar] [CrossRef]
- Fink, D.A.; Nelson, L.M.; Pyeritz, R.; Johnson, J.; Sherman, S.L.; Cohen, Y.; Elizur, S.E. Fragile X Associated Primary Ovarian Insufficiency (FXPOI): Case Report and Literature Review. Front. Genet. 2018, 9, 529. [Google Scholar] [CrossRef]
- Eichler, E.E.; Holden, J.J.A.; Popovich, B.W.; Reiss, A.L.; Snow, K.; Thibodeau, S.N.; Richards, C.S.; Ward, P.A.; Nelson, D.L. Length of Uninterrupted CGG Repeats Determines Instability in the FMR1 Gene. Nat. Genet. 1994, 8, 88–94. [Google Scholar] [CrossRef]
- Fu, Y.-H.; Kuhl, D.P.A.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkert, A.J.M.H.; Holden, J.J.A.; Fenwick, R.G.; Warren, S.T.; et al. Variation of the CGG Repeat at the Fragile X Site Results in Genetic Instability: Resolution of the Sherman Paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Yrigollen, C.M.; Durbin-Johnson, B.; Gane, L.; Nelson, D.L.; Hagerman, R.; Hagerman, P.J.; Tassone, F. AGG Interruptions within the Maternal FMR1 Gene Reduce the Risk of Offspring with Fragile X Syndrome. Genet. Med. 2012, 14, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.K.; Tassone, F.; Dyer, P.N.; Hersch, S.M.; Harris, J.B.; Greenough, W.T.; Hagerman, R.J. Tissue Heterogeneity of TheFMR1 Mutation in a High-Functioning Male with Fragile X Syndrome. Am. J. Med. Genet. 1999, 84, 233–239. [Google Scholar] [CrossRef]
- Bjerregaard, V.A.; Garribba, L.; McMurray, C.T.; Hickson, I.D.; Liu, Y. Folate Deficiency Drives Mitotic Missegregation of the Human FRAXA Locus. Proc. Natl. Acad. Sci. 2018, 115, 13003–13008. [Google Scholar] [CrossRef] [PubMed]
- Hoogsteen, K. The Crystal and Molecular Structure of a Hydrogen-Bonded Complex between 1-Methylthymine and 9-Methyladenine. Acta Crystallogr. 1963, 16, 907–916. [Google Scholar] [CrossRef]
- Ajjugal, Y.; Kolimi, N.; Rathinavelan, T. Secondary Structural Choice of DNA and RNA Associated with CGG/CCG Trinucleotide Repeat Expansion Rationalizes the RNA Misprocessing in FXTAS. Sci. Rep. 2021, 11, 8163. [Google Scholar] [CrossRef]
- Weisman-Shomer, P. Interruption of the Fragile X Syndrome Expanded Sequence d(CGG)n by Interspersed d(AGG) Trinucleotides Diminishes the Formation and Stability of d(CGG)n Tetrahelical Structures. Nucleic Acids Res. 2000, 28, 1535–1541. [Google Scholar] [CrossRef]
- Richter, J.D.; Zhao, X. The Molecular Biology of FMRP: New Insights into Fragile X Syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [CrossRef]
- D’Antoni, S.; Spatuzza, M.; Bonaccorso, C.M.; Musumeci, S.A.; Ciranna, L.; Nicoletti, F.; Huber, K.M.; Catania, M.V. Dysregulation of Group-I Metabotropic Glutamate (MGlu) Receptor Mediated Signalling in Disorders Associated with Intellectual Disability and Autism. Neurosci. Biobehav. Rev. 2014, 46, 228–241. [Google Scholar] [CrossRef]
- Hsu, P.J.; Shi, H.; Zhu, A.C.; Lu, Z.; Miller, N.; Edens, B.M.; Ma, Y.C.; He, C. The RNA-Binding Protein FMRP Facilitates the Nuclear Export of N6-Methyladenosine–Containing MRNAs. J. Biol. Chem. 2019, 294, 19889–19895. [Google Scholar] [CrossRef]
- Alpatov, R.; Lesch, B.J.; Nakamoto-Kinoshita, M.; Blanco, A.; Chen, S.; Stützer, A.; Armache, K.J.; Simon, M.D.; Xu, C.; Ali, M.; et al. A Chromatin-Dependent Role of the Fragile X Mental Retardation Protein FMRP in the DNA Damage Response. Cell 2014, 157, 869–881. [Google Scholar] [CrossRef]
- Shah, S.; Molinaro, G.; Liu, B.; Wang, R.; Huber, K.M.; Richter, J.D. FMRP Control of Ribosome Translocation Promotes Chromatin Modifications and Alternative Splicing of Neuronal Genes Linked to Autism. Cell Rep. 2020, 30, 4459–4472.e6. [Google Scholar] [CrossRef] [PubMed]
- Krawczun, M.S.; Jenkins, E.C.; Brown, W.T. Analysis of the Fragile-X Chromosome: Localization and Detection of the Fragile Site in High Resolution Preparations. Hum. Genet. 1985, 69, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Sambrook, J. Analysis of DNA by Southern Blotting. Cold Spring Harb. Protoc. 2021, 7, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hadd, A.G.; Sah, S.; Houghton, J.F.; Filipovic-Sadic, S.; Zhang, W.; Hagerman, P.J.; Tassone, F.; Latham, G.J. High-Resolution Methylation Polymerase Chain Reaction for Fragile X Analysis: Evidence for Novel FMR1 Methylation Patterns Undetected in Southern Blot Analyses. Genet. Med. 2011, 13, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.K.; Arpone, M.; Bui, M.; Kraan, C.M.; Ling, L.; Francis, D.; Hunter, M.F.; Rogers, C.; Field, M.J.; Santa María, L.; et al. Tissue Mosaicism, FMR1 Expression and Intellectual Functioning in Males with Fragile X Syndrome. Am. J. Med. Genet. A. 2023, 191, 357–369. [Google Scholar] [CrossRef]
- Rajan-Babu, I.-S.; Chong, S.S. Triplet-Repeat Primed PCR and Capillary Electrophoresis for Characterizing the Fragile X Mental Retardation 1 CGG Repeat Hyperexpansions. In Clinical Applications of Capillary Electrophoresis; Phillips, T.M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1972, pp. 199–210. ISBN 978-1-4939-9212-6. [Google Scholar]
- Warner, J.P.; Barron, L.H.; Goudie, D.; Kelly, K.; Dow, D.; Fitzpatrick, D.R.; Brock, D.J. A General Method for the Detection of Large CAG Repeat Expansions by Fluorescent PCR. J. Med. Genet. 1996, 33, 1022–1026. [Google Scholar] [CrossRef]
- Ciotti, P.; Di Maria, E.; Bellone, E.; Ajmar, F.; Mandich, P. Triplet Repeat Primed PCR (TP PCR) in Molecular Diagnostic Testing for Friedreich Ataxia. J. Mol. Diagn. 2004, 6, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Li, N.; Wan, H.; Luo, L.; Wu, Y.; Li, S.; An, Y.; Wu, B.-L. Developing a One-Step Triplet-Repeat Primed PCR Assay for Diagnosing Myotonic Dystrophy. J. Genet. Genomics 2018, 45, 549–552. [Google Scholar] [CrossRef]
- Chheda, P.; Chanekar, M.; Salunkhe, Y.; Dama, T.; Pais, A.; Pande, S.; Bendre, R.; Shah, N. A Study of Triplet-Primed PCR for Identification of CAG Repeat Expansion in the HTT Gene in a Cohort of 503 Indian Cases with Huntington’s Disease Symptoms. Mol. Diagn. Ther. 2018, 22, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Filipovic-Sadic, S.; Sah, S.; Chen, L.; Krosting, J.; Sekinger, E.; Zhang, W.; Hagerman, P.J.; Stenzel, T.T.; Hadd, A.G.; Latham, G.J.; et al. A Novel FMR1 PCR Method for the Routine Detection of Low Abundance Expanded Alleles and Full Mutations in Fragile X Syndrome. Clin. Chem. 2010, 56, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F. Advanced Technologies for the Molecular Diagnosis of Fragile X Syndrome. Expert Rev. Mol. Diagn. 2015, 15, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Curtis-Cioffi, K.M.C.; Rodrigueiro, D.A.; Rodrigues, V.C.; Cicarelli, R.M.B.; Scarel-Caminaga, R.M. Comparison Between the Polymerase Chain Reaction-Based Screening and the Southern Blot Methods for Identification of Fragile X Syndrome. Genet. Test. Mol. Biomark. 2012, 16, 1303–1308. [Google Scholar] [CrossRef]
- Rajan-Babu, I.; Lian, M.; Chong, S.S. Triplet-Primed PCR Assays for Accurate Screening of FMR1 CGG Repeat Expansion and Genotype Verification. Curr. Protoc. 2022, 2, e427. [Google Scholar] [CrossRef]
- Bizouarn, F. Introduction to Digital PCR. In Quantitative Real-Time PCR.; Biassoni, R., Raso, A., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; Volume 1160, pp. 27–41. ISBN 978-1-4939-0732-8. [Google Scholar]
- Mao, X.; Liu, C.; Tong, H.; Chen, Y.; Liu, K. Principles of Digital PCR and Its Applications in Current Obstetrical and Gynecological Diseases. Am. J. Transl. Res. 2019, 11, 7209–7222. [Google Scholar] [PubMed]
- Quan, P.-L.; Sauzade, M.; Brouzes, E. DPCR: A Technology Review. Sensors 2018, 18, 1271. [Google Scholar] [CrossRef]
- Nyaruaba, R.; Mwaliko, C.; Kering, K.K.; Wei, H. Droplet Digital PCR Applications in the Tuberculosis World. Tuberculosis 2019, 117, 85–92. [Google Scholar] [CrossRef]
- Campomenosi, P.; Gini, E.; Noonan, D.M.; Poli, A.; D’Antona, P.; Rotolo, N.; Dominioni, L.; Imperatori, A. A Comparison between Quantitative PCR and Droplet Digital PCR Technologies for Circulating MicroRNA Quantification in Human Lung Cancer. BMC Biotechnol. 2016, 16, 60. [Google Scholar] [CrossRef]
- Gu, H.; Kim, M.J.; Yang, D.; Song, J.Y.; Cho, S.I.; Park, S.S.; Seong, M.-W. Accuracy and Performance Evaluation of Triplet Repeat Primed PCR as an Alternative to Conventional Diagnostic Methods for Fragile X Syndrome. Ann. Lab. Med. 2021, 41, 394–400. [Google Scholar] [CrossRef]
- Silva, C.; Maia, N.; Santos, F.; Rodrigues, B.; Marques, I.; Santos, R.; Jorge, P. Development and Validation in 500 Female Samples of a TP-PCR Assay to Identify AFF2 GCC Expansions. Sci. Rep. 2021, 11, 14676. [Google Scholar] [CrossRef]
- Jeon, M.-S.; Jeong, D.M.; Doh, H.; Kang, H.A.; Jung, H.; Eyun, S. A Practical Comparison of the Next-Generation Sequencing Platform and Assemblers Using Yeast Genome. Life Sci. Alliance 2023, 6, e202201744. [Google Scholar] [CrossRef]
- Pervez, M.T.; ul Hasnain, M.J.; Abbas, S.H.; Moustafa, M.F.; Aslam, N.; Shah, S.S.M. A Comprehensive Review of Performance of Next-Generation Sequencing Platforms. BioMed Res. Int. 2022, 2022, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lestringant, V.; Duployez, N.; Penther, D.; Luquet, I.; Derrieux, C.; Lutun, A.; Preudhomme, C.; West, M.; Ouled-Haddou, H.; Devoldere, C.; et al. Optical Genome Mapping, a Promising Alternative to Gold Standard Cytogenetic Approaches in a Series of Acute Lymphoblastic Leukemias. Genes. Chromosomes Cancer 2021, 60, 657–667. [Google Scholar] [CrossRef]
- Kojabad, A.A.; Farzanehpour, M.; Galeh, H.E.G.; Dorostkar, R.; Jafarpour, A.; Bolandian, M.; Nodooshan, M.M. Droplet Digital PCR of Viral DNA/RNA, Current Progress, Challenges, and Future Perspectives. J. Med. Virol. 2021, 93, 4182–4197. [Google Scholar] [CrossRef]
- Galimberti, S.; Balducci, S.; Guerrini, F.; Del Re, M.; Cacciola, R. Digital Droplet PCR in Hematologic Malignancies: A New Useful Molecular Tool. Diagnostics 2022, 12, 1305. [Google Scholar] [CrossRef]
- Jin, P.; Gao, X.; Wang, M.; Qian, Y.; Yang, J.; Yang, Y.; Xu, Y.; Xu, Y.; Dong, M. Case Report: Identification of Maternal Low-Level Mosaicism in the Dystrophin Gene by Droplet Digital Polymerase Chain Reaction. Front. Genet. 2021, 12, 686993. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Mora, M.I.; Agusti, I.; Wijngaard, R.; Martinez-Barrios, E.; Barcos, T.; Borras, A.; Peralta, S.; Guimera, M.; Goday, A.; Manau, D.; et al. Evaluation of FMR4, FMR5 and FMR6 Expression Levels as Non-Invasive Biomarkers for the Diagnosis of Fragile X-Associated Primary Ovarian Insufficiency (FXPOI). J. Clin. Med. 2022, 11, 2186. [Google Scholar] [CrossRef] [PubMed]
- Godler, D.E.; Christodoulou, J.; Bruno, D.; Li, X.; Inaba, Y.; Bui, Q.M.; Francis, D.; Elliot, J.; Wotton, T.; Cohen, J.; et al. The Use of Droplet Digital PCR and High Resolution Melt for Detection of Low Level Mosaicism. Pathology 2018, 50, S30. [Google Scholar] [CrossRef]
- Ku, J.-L.; Jeon, Y.-K.; Park, J.-G. Methylation-Specific PCR. In Epigenetics Protocols; Tollefsbol, T.O., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; Volume 791, pp. 23–32. ISBN 978-1-61779-315-8. [Google Scholar]
- Khodadadi, E.; Fahmideh, L.; Khodadadi, E.; Dao, S.; Yousefi, M.; Taghizadeh, S.; Asgharzadeh, M.; Yousefi, B.; Kafil, H.S. Current Advances in DNA Methylation Analysis Methods. BioMed Res. Int. 2021, 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Trinh, B.N.; Campan, M.; Sharma, S.; Long, T.I.; Ananthnarayan, S.; Liang, G.; Esteva, F.J.; Hortobagyi, G.N.; McCormick, F.; et al. DNA Methylation Analysis by Digital Bisulfite Genomic Sequencing and Digital MethyLight. Nucleic Acids Res. 2008, 36, 4689–4698. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Zhou, L.; Jackson, J.; Tassone, F. Diagnostic Profile of the AmplideX Fragile X Dx and Carrier Screen Kit for Diagnosis and Screening of Fragile X Syndrome and Other FMR1-Related Disorders. Expert Rev. Mol. Diagn. 2021, 21, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Ersalesi, N.; Dobkin, C.; Ted Brown, W.; Cao, R.; Blatt, E.; Sah, S.; Latham, G.J.; Hadd, A.G. Fragile X Full Mutation Expansions Are Inhibited by One or More AGG Interruptions in Premutation Carriers. Genet. Med. 2015, 17, 358–364. [Google Scholar] [CrossRef]
- Nygren, A.O.H. Methylation-Specific MLPA (MS-MLPA): Simultaneous Detection of CpG Methylation and Copy Number Changes of up to 40 Sequences. Nucleic Acids Res. 2005, 33, e128. [Google Scholar] [CrossRef]
- Gatta, V.; Gennaro, E.; Franchi, S.; Cecconi, M.; Antonucci, I.; Tommasi, M.; Palka, G.; Coviello, D.; Stuppia, L.; Grasso, M. MS-MLPA Analysis for FMR1 Gene: Evaluation in a Routine Diagnostic Setting. BMC Med. Genet. 2013, 14, 79. [Google Scholar] [CrossRef]
- Delpu, Y.; Barseghyan, H.; Bocklandt, S.; Hastie, A.; Chaubey, A. Next-Generation Cytogenomics: High-Resolution Structural Variation Detection by Optical Genome Mapping. In Cytogenomics; Elsevier: Amsterdam, The Netherlands, 2021; pp. 123–146. ISBN 978-0-12-823579-9. [Google Scholar]
- Barseghyan, H.; Chun Pang, A.W.; Chaubey, A.; Hastie, A. EP317: Comparative Benchmarking of Optical Genome Mapping and Chromosomal Microarray Reveals High Technological Concordance in CNV Identification and Structural Variant Refinement. Genet. Med. 2022, 24, S199. [Google Scholar] [CrossRef]
- Muggli, M.; Ramandi, B.; Miller, N.; Zhang, D.; Lam, E.; Wang, J.; Wang, T.; Lee, J.; Pang, A.; Sadowski, H.; et al. EP379: Optical Genome Mapping for High Throughput Analysis of Repeat Expansion Disorders. Genet. Med. 2022, 24, S238. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Broeckel, U.; Levy, B.; Skinner, S.; Sahajpal, N.S.; Rodriguez, V.; Stence, A.; Awayda, K.; Scharer, G.; Skinner, C.; et al. Multisite Assessment of Optical Genome Mapping for Analysis of Structural Variants in Constitutional Postnatal Cases. J. Mol. Diagn. 2023, 25, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Barseghyan, H.; Kolhe, R.; Hastie, A.; Chaubey, A. Optical Genome Mapping as a Next-Generation Cytogenomic Tool for Detection of Structural and Copy Number Variations for Prenatal Genomic Analyses. Genes 2021, 12, 398. [Google Scholar] [CrossRef] [PubMed]
- Heather, J.M.; Chain, B. The Sequence of Sequencers: The History of Sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, 9–10. [Google Scholar] [CrossRef]
- Sitzmann, A.F.; Hagelstrom, R.T.; Tassone, F.; Hagerman, R.J.; Butler, M.G. Rare FMR1 Gene Mutations Causing Fragile X Syndrome: A Review. Am. J. Med. Genet. A. 2018, 176, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.C.; Bray, S.M.; Suhl, J.A.; Cutler, D.J.; Coffee, B.; Zwick, M.E.; Warren, S.T. Identification of Novel FMR1 Variants by Massively Parallel Sequencing in Developmentally Delayed Males. Am. J. Med. Genet. A. 2010, 152A, 2512–2520. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Travers, K.J.; Chin, C.-S.; Rank, D.R.; Eid, J.S.; Turner, S.W. A Flexible and Efficient Template Format for Circular Consensus Sequencing and SNP Detection. Nucleic Acids Res. 2010, 38, e159. [Google Scholar] [CrossRef]
- Chintalaphani, S.R.; Pineda, S.S.; Deveson, I.W.; Kumar, K.R. An Update on the Neurological Short Tandem Repeat Expansion Disorders and the Emergence of Long-Read Sequencing Diagnostics. Acta Neuropathol. Commun. 2021, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate Circular Consensus Long-Read Sequencing Improves Variant Detection and Assembly of a Human Genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single Molecule Real-Time (SMRT) Sequencing Comes of Age: Applications and Utilities for Medical Diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef]
- Chakraborty, S.; Vatta, M.; Bachinski, L.L.; Krahe, R.; Dlouhy, S.; Bai, S. Molecular Diagnosis of Myotonic Dystrophy. Curr. Protoc. Hum. Genet. 2016, 91, 9–29. [Google Scholar] [CrossRef]
- Liang, Q.; Liu, Y.; Liu, Y.; Duan, R.; Meng, W.; Zhan, J.; Xia, J.; Mao, A.; Liang, D.; Wu, L. Comprehensive Analysis of Fragile X Syndrome: Full Characterization of the FMR1 Locus by Long-Read Sequencing. Clin. Chem. 2022, 68, 1529–1540. [Google Scholar] [CrossRef]
- Tsai, Y.-C.; Greenberg, D.; Powell, J.; Höijer, I.; Ameur, A.; Strahl, M.; Ellis, E.; Jonasson, I.; Pinto, R.M.; Vanessa, C. Amplification-Free, CRISPR-Cas9 Targeted Enrichment and SMRT Sequencing of Repeat-Expansion Disease Causative Genomic Regions. bioRxiv 2017. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, Y.; Ying, C.; Wang, D.; Du, C. Nanopore-Based Fourth-Generation DNA Sequencing Technology. Genom. Proteom. Bioinform. 2015, 13, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Kono, N.; Arakawa, K. Nanopore Sequencing: Review of Potential Applications in Functional Genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.; Holmes, N.; Clarke, T.; Munro, R.; Debebe, B.J.; Loose, M. Readfish Enables Targeted Nanopore Sequencing of Gigabase-Sized Genomes. Nat. Biotechnol. 2021, 39, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Stevanovski, I.; Chintalaphani, S.R.; Gamaarachchi, H.; Ferguson, J.M.; Pineda, S.S.; Scriba, C.K.; Tchan, M.; Fung, V.; Ng, K.; Cortese, A.; et al. Comprehensive Genetic Diagnosis of Tandem Repeat Expansion Disorders with Programmable Targeted Nanopore Sequencing. Sci. Adv. 2022, 8, eabm5386. [Google Scholar] [CrossRef] [PubMed]
- Giesselmann, P.; Brändl, B.; Raimondeau, E.; Bowen, R.; Rohrandt, C.; Tandon, R.; Kretzmer, H.; Assum, G.; Galonska, C.; Siebert, R.; et al. Analysis of Short Tandem Repeat Expansions and Their Methylation State with Nanopore Sequencing. Nat. Biotechnol. 2019, 37, 1478–1481. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ge, C.; Malachowski, T.; Kim, J.H.; Chandradoss, K.R.; Su, C.; Wu, H.; Rojas, A.; Wallace, O.; Titus, K.R.; et al. Spatially Coordinated Heterochromatinization of Distal Short Tandem Repeats in Fragile X Syndrome. bioRxiv 2021. [Google Scholar] [CrossRef]
- Grosso, V.; Marcolungo, L.; Maestri, S.; Alfano, M.; Lavezzari, D.; Iadarola, B.; Salviati, A.; Mariotti, B.; Botta, A.; D’Apice, M.R.; et al. Characterization of FMR1 Repeat Expansion and Intragenic Variants by Indirect Sequence Capture. Front. Genet. 2021, 12, 743230. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciobanu, C.-G.; Nucă, I.; Popescu, R.; Antoci, L.-M.; Caba, L.; Ivanov, A.V.; Cojocaru, K.-A.; Rusu, C.; Mihai, C.-T.; Pânzaru, M.-C. Narrative Review: Update on the Molecular Diagnosis of Fragile X Syndrome. Int. J. Mol. Sci. 2023, 24, 9206. https://doi.org/10.3390/ijms24119206
Ciobanu C-G, Nucă I, Popescu R, Antoci L-M, Caba L, Ivanov AV, Cojocaru K-A, Rusu C, Mihai C-T, Pânzaru M-C. Narrative Review: Update on the Molecular Diagnosis of Fragile X Syndrome. International Journal of Molecular Sciences. 2023; 24(11):9206. https://doi.org/10.3390/ijms24119206
Chicago/Turabian StyleCiobanu, Cristian-Gabriel, Irina Nucă, Roxana Popescu, Lucian-Mihai Antoci, Lavinia Caba, Anca Viorica Ivanov, Karina-Alexandra Cojocaru, Cristina Rusu, Cosmin-Teodor Mihai, and Monica-Cristina Pânzaru. 2023. "Narrative Review: Update on the Molecular Diagnosis of Fragile X Syndrome" International Journal of Molecular Sciences 24, no. 11: 9206. https://doi.org/10.3390/ijms24119206
APA StyleCiobanu, C. -G., Nucă, I., Popescu, R., Antoci, L. -M., Caba, L., Ivanov, A. V., Cojocaru, K. -A., Rusu, C., Mihai, C. -T., & Pânzaru, M. -C. (2023). Narrative Review: Update on the Molecular Diagnosis of Fragile X Syndrome. International Journal of Molecular Sciences, 24(11), 9206. https://doi.org/10.3390/ijms24119206