


Signaling Pathways in the Pathogenesis of Barrett’s Esophagus and Esophageal Adenocarcinoma

 , , , , and
, , , , and 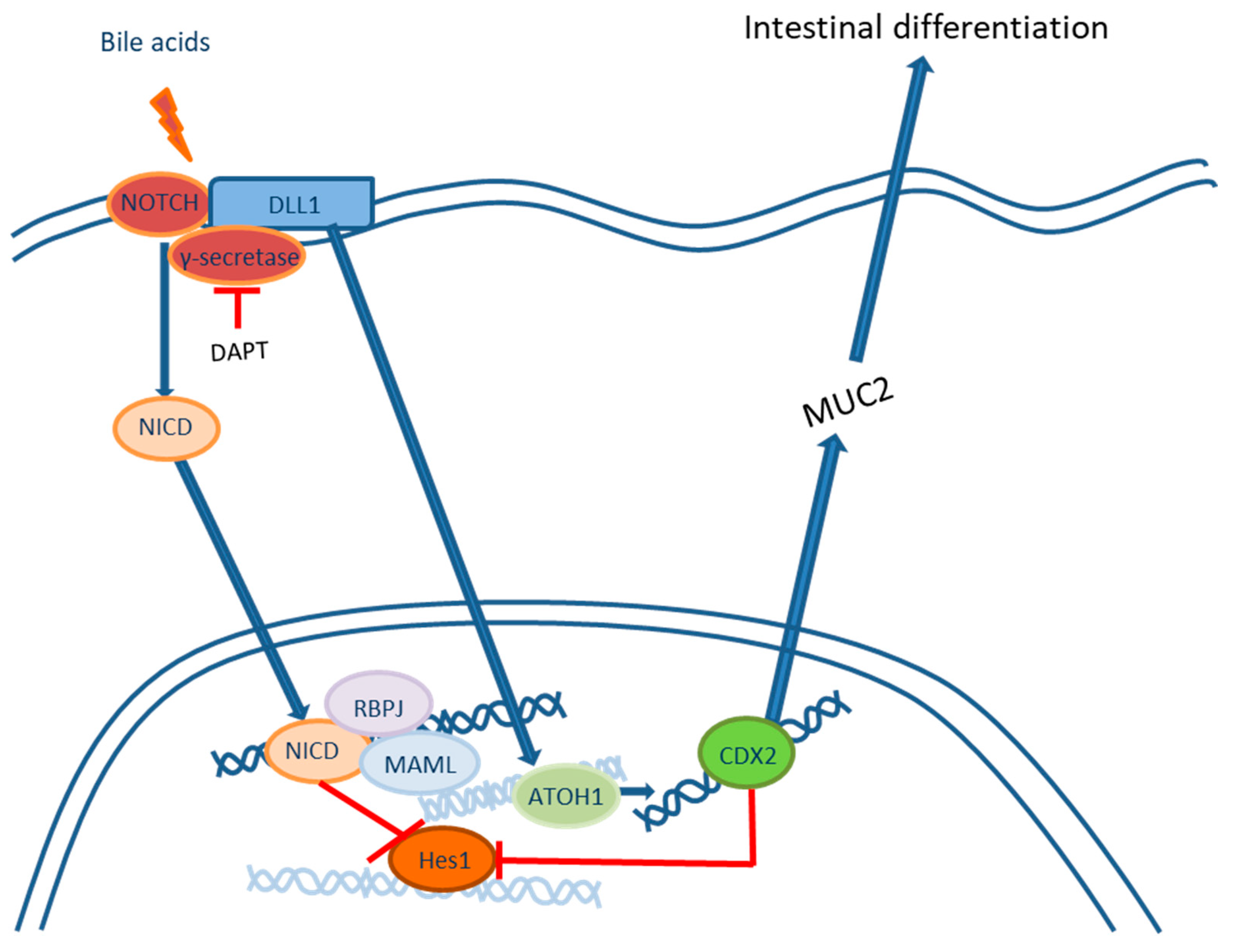{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Gastro-Biliary Reflux as An Inducer of Intestinal Metaplasia
2.1. Roles of the NOTCH Signaling Pathway in the Development of Intestinal Metaplasia
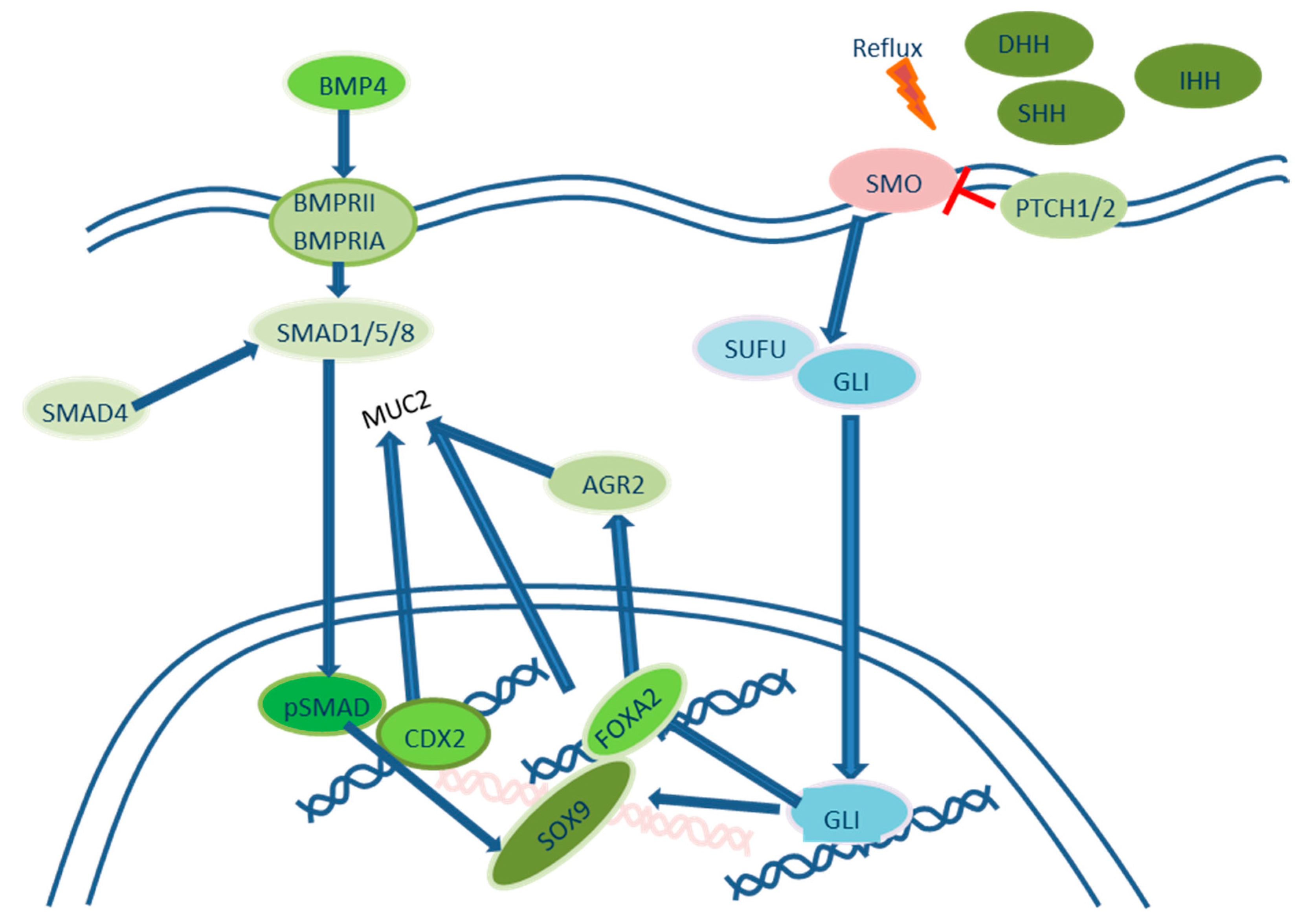
2.2. Roles of Hedgehog Signaling Pathways in the Development of Intestinal Metaplasia
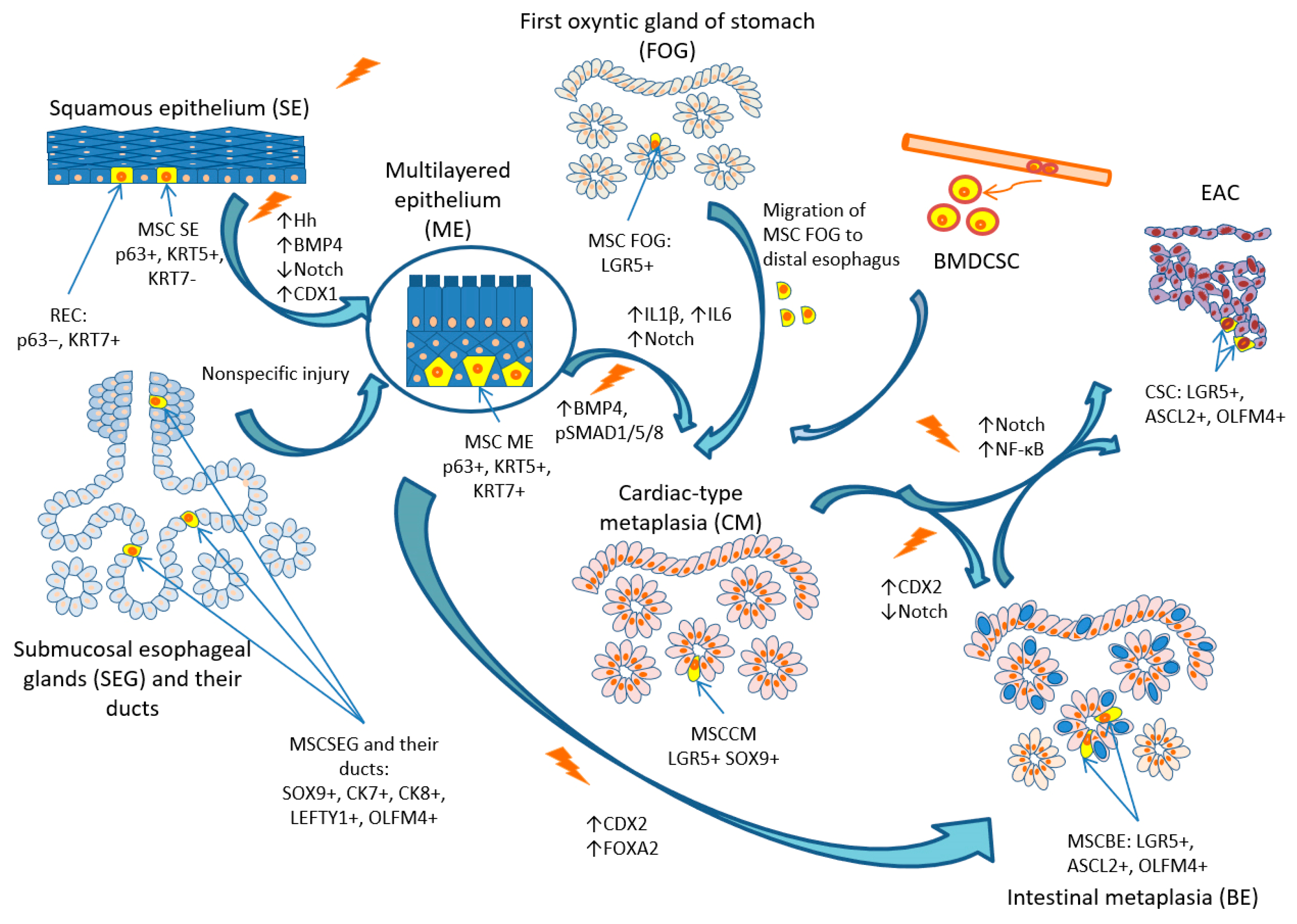
3. Possible Cellular Origin of BE
- SCs and progenitor cells of the squamous epithelium;
- SCs and progenitor cells of the gastro-esophageal junction;
- SCs and progenitor cells of the submucosal glands and their ducts;
- SCs and progenitor cells of the first oxyntic gland of the stomach;
- Residual embryonic cells;
- Circulating bone-marrow-derived multipotent SCs.
3.1. SCs and Progenitor Cells of the Squamous Epithelium
3.2. SCs and Progenitor Cells of the Gastro-Esophageal Junction
3.3. SCs and Progenitor Cells of the Submucosal Glands and Their Ducts
3.4. SCs and Progenitor Cells of the First Oxyntic Gland of the Stomach
4. Repair of Caustic Injury in the Distal Esophagus
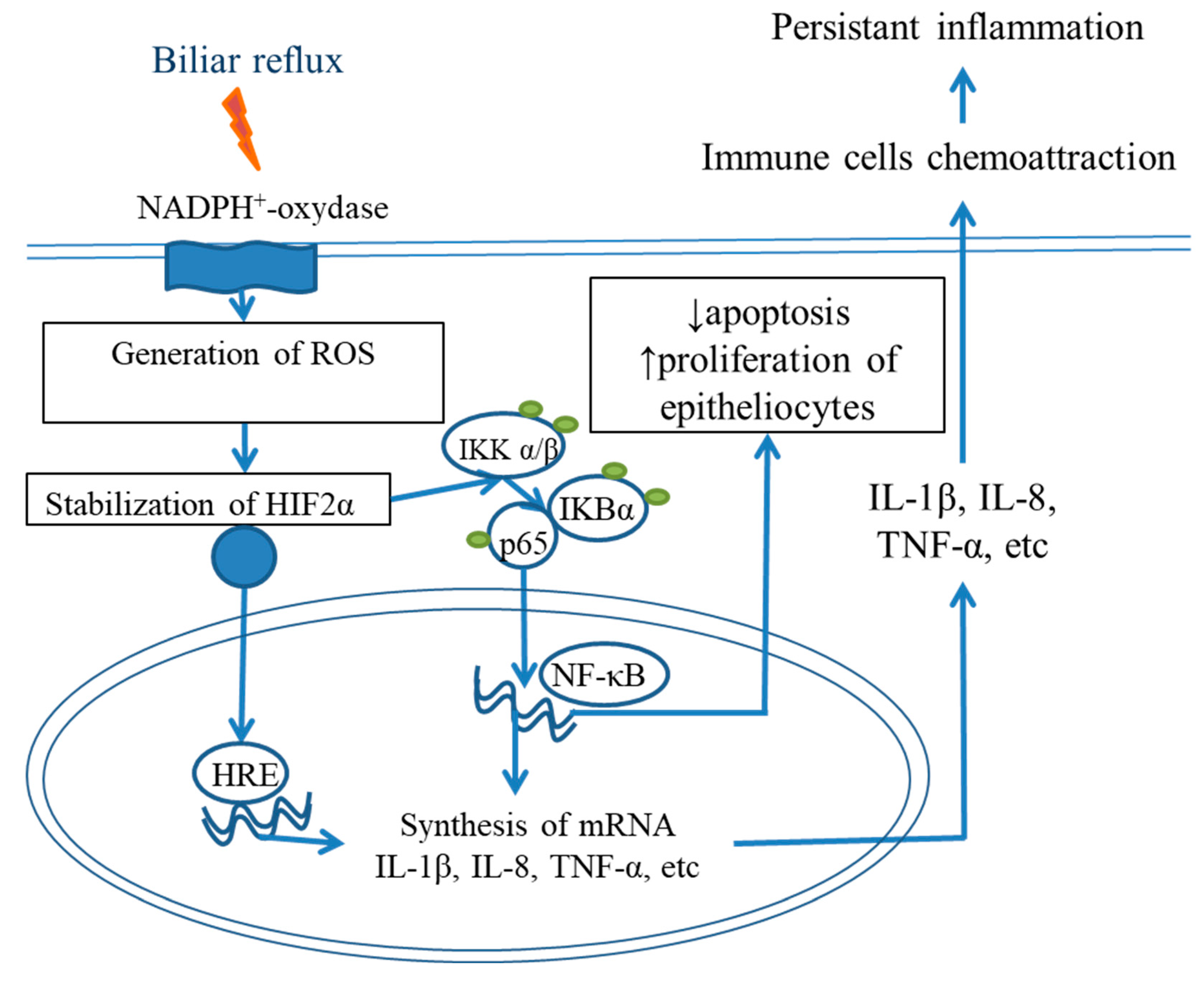
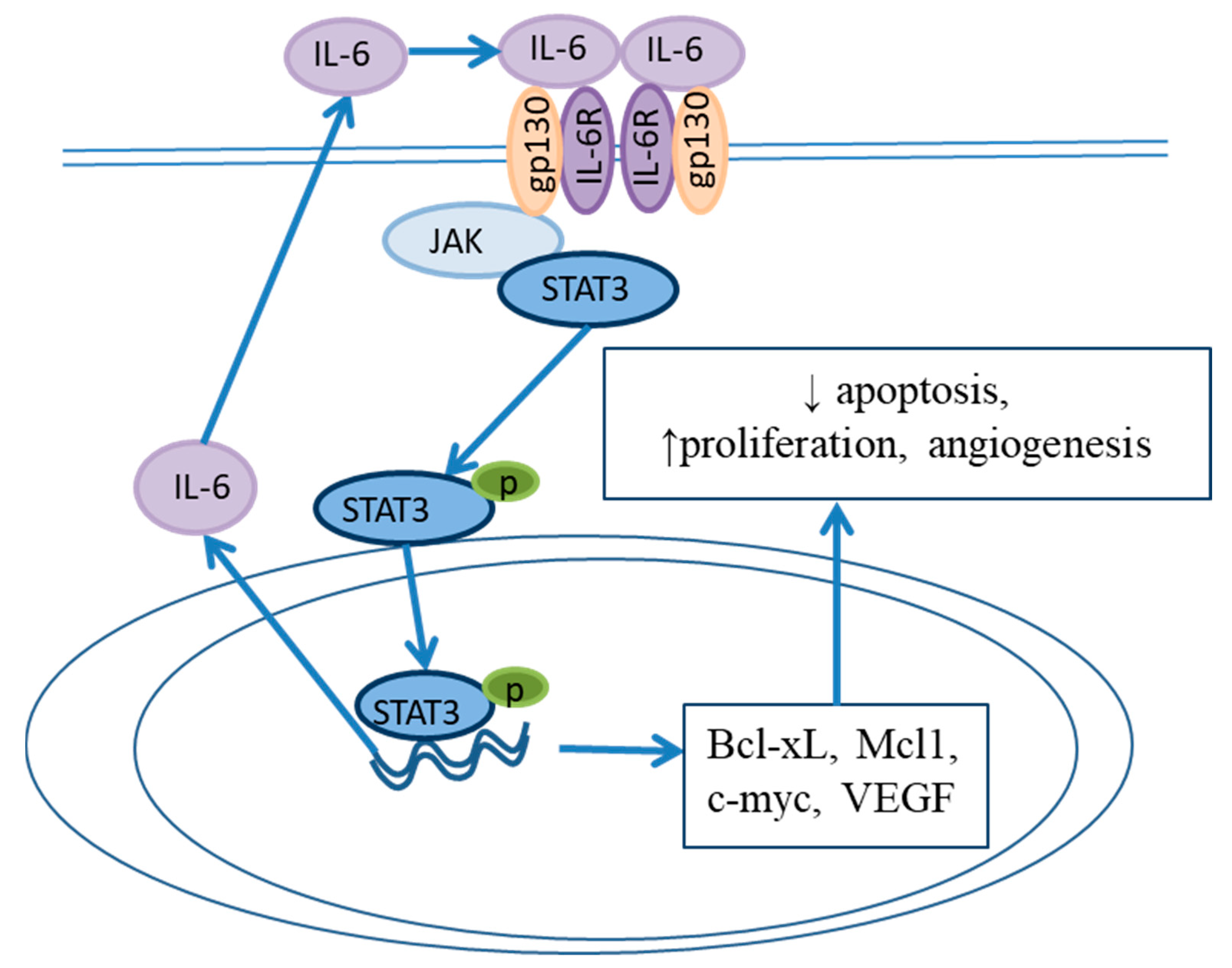
5. Roles of Cytokine Storm and a Proinflammatory Microenvironment in BE Development
NF-κB and IL6/STAT3 Signaling
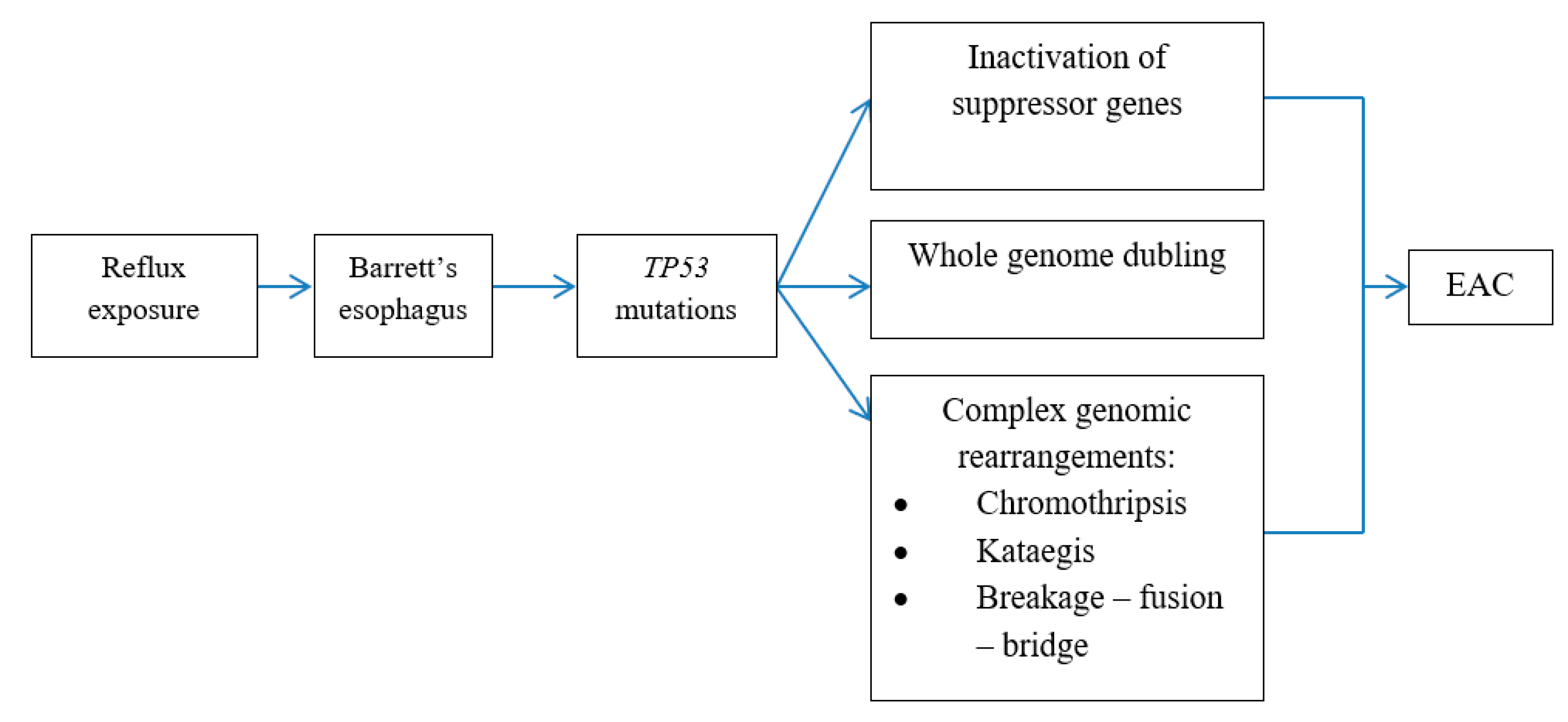
6. Genomic Alterations in BE Carcinogenesis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nicholson, A.M.; Graham, T.A.; Simpson, A.; Humphries, A.; Burch, N.; Rodriguez-Justo, M.; Novelli, M.; Harrison, R.; Wright, N.A.; McDonald, S.A.; et al. Barrett’s metaplasia glands are clonal, contain multiple stem cells and share a common squamous progenitor. Gut 2012, 61, 1380–1389. [Google Scholar] [CrossRef]
- Biswas, S.; Quante, M.; Leedham, S.; Jansen, M. The metaplastic mosaic of Barrett’s oesophagus. Virchows Arch. 2018, 472, 43–54. [Google Scholar] [CrossRef]
- Evans, J.A.; McDonald, S.A. The Complex, Clonal, and Controversial Nature of Barrett’s Esophagus. Adv. Exp. Med. Biol. 2016, 908, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.A.; Carlotti, E.; Lin, M.L.; Hackett, R.J.; Haughey, M.J.; Passman, A.M.; Dunn, L.; Elia, G.; Porter, R.J.; McLean, M.H.; et al. Clonal Transitions and Phenotypic Evolution in Barrett’s Esophagus. Gastroenterology 2022, 162, 1197–1209.e13. [Google Scholar] [CrossRef]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Stachler, M.D.; Camarda, N.D.; Deitrick, C.; Kim, A.; Agoston, A.T.; Odze, R.D.; Hornick, J.L.; Nag, A.; Thorner, A.R.; Ducar, M.; et al. Detection of Mutations in Barrett’s Esophagus Before Progression to High-Grade Dysplasia or Adenocarcinoma. Gastroenterology 2018, 155, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Agoston, A.T.; Pham, T.H.; Odze, R.D.; Wang, D.H.; Das, K.M.; Spechler, S.J.; Souza, R.F. Columnar-Lined Esophagus Develops via Wound Repair in a Surgical Model of Reflux Esophagitis. Cell Mol. Gastroenterol. Hepatol. 2018, 6, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Goldenring, J.R. Pyloric metaplasia, pseudopyloric metaplasia, ulcer-associated cell lineage and spasmolytic polypeptide-expressing metaplasia: Reparative lineages in the gastrointestinal mucosa. J. Pathol. 2018, 245, 132–137. [Google Scholar] [CrossRef]
- Wright, N.A. Aspects of the biology of regeneration and repair in the human gastrointestinal tract. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1998, 353, 925–933. [Google Scholar] [CrossRef]
- Souza, R.F. The role of acid and bile reflux in oesophagitis and Barrett’s metaplasia. Biochem. Soc. Trans. 2010, 38, 348–352. [Google Scholar] [CrossRef]
- Souza, R.F. From Reflux Esophagitis to Esophageal Adenocarcinoma. Dig. Dis. 2016, 34, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.F. Reflux esophagitis and its role in the pathogenesis of Barrett’s metaplasia. J. Gastroenterol. 2017, 52, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.F.; Bayeh, L.; Spechler, S.J.; Tambar, U.K.; Bruick, R.K. A new paradigm for GERD pathogenesis. Not acid injury, but cytokine-mediated inflammation driven by HIF-2alpha: A potential role for targeting HIF-2alpha to prevent and treat reflux esophagitis. Curr. Opin. Pharmacol. 2017, 37, 93–99. [Google Scholar] [CrossRef]
- Souza, R.F.; Spechler, S.J. Oesophagus: A new candidate for the progenitor cell of Barrett metaplasia. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P. Barrett Esophagus: A Review. JAMA 2022, 328, 663–671. [Google Scholar] [CrossRef]
- Eusebi, L.H.; Cirota, G.G.; Zagari, R.M.; Ford, A.C. Global prevalence of Barrett’s oesophagus and oesophageal cancer in individuals with gastro-oesophageal reflux: A systematic review and meta-analysis. Gut 2021, 70, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Eusebi, L.H.; Telese, A.; Cirota, G.G.; Haidry, R.; Zagari, R.M.; Bazzoli, F.; Ford, A.C. Effect of gastro-esophageal reflux symptoms on the risk of Barrett’s esophagus: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2022, 37, 1507–1516. [Google Scholar] [CrossRef]
- Gutschow, C.A.; Bludau, M.; Vallbohmer, D.; Schroder, W.; Bollschweiler, E.; Holscher, A.H. NERD, GERD, and Barrett’s esophagus: Role of acid and non-acid reflux revisited with combined pH-impedance monitoring. Dig. Dis. Sci. 2008, 53, 3076–3081. [Google Scholar] [CrossRef]
- Hak, N.G.; Mostafa, M.; Salah, T.; El-Hemaly, M.; Haleem, M.; Abd El-Raouf, A.; Hamdy, E. Acid and bile reflux in erosive reflux disease, non-erosive reflux disease and Barrett’s esophagus. Hepatogastroenterology 2008, 55, 442–447. [Google Scholar]
- Koek, G.H.; Sifrim, D.; Lerut, T.; Janssens, J.; Tack, J. Multivariate analysis of the association of acid and duodeno-gastro-oesophageal reflux exposure with the presence of oesophagitis, the severity of oesophagitis and Barrett’s oesophagus. Gut 2008, 57, 1056–1064. [Google Scholar] [CrossRef]
- Terabe, F.; Aikou, S.; Aida, J.; Yamamichi, N.; Kaminishi, M.; Takubo, K.; Seto, Y.; Nomura, S. Columnar Metaplasia in Three Types of Surgical Mouse Models of Esophageal Reflux. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Matsui, D.; Omstead, A.N.; Kosovec, J.E.; Komatsu, Y.; Lloyd, E.J.; Raphael, H.; Kelly, R.J.; Zaidi, A.H.; Jobe, B.A. High yield reproducible rat model recapitulating human Barrett’s carcinogenesis. World J. Gastroenterol. 2017, 23, 6077–6087. [Google Scholar] [CrossRef] [PubMed]
- Kazumori, H.; Ishihara, S.; Rumi, M.A.; Kadowaki, Y.; Kinoshita, Y. Bile acids directly augment caudal related homeobox gene Cdx2 expression in oesophageal keratinocytes in Barrett’s epithelium. Gut 2006, 55, 16–25. [Google Scholar] [CrossRef]
- Dvorak, K.; Payne, C.M.; Chavarria, M.; Ramsey, L.; Dvorakova, B.; Bernstein, H.; Holubec, H.; Sampliner, R.E.; Guy, N.; Condon, A.; et al. Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: Relevance to the pathogenesis of Barrett’s oesophagus. Gut 2007, 56, 763–771. [Google Scholar] [CrossRef]
- Song, S.; Guha, S.; Liu, K.; Buttar, N.S.; Bresalier, R.S. COX-2 induction by unconjugated bile acids involves reactive oxygen species-mediated signalling pathways in Barrett’s oesophagus and oesophageal adenocarcinoma. Gut 2007, 56, 1512–1521. [Google Scholar] [CrossRef]
- Morrow, D.J.; Avissar, N.E.; Toia, L.; Redmond, E.M.; Watson, T.J.; Jones, C.; Raymond, D.P.; Litle, V.; Peters, J.H. Pathogenesis of Barrett’s esophagus: Bile acids inhibit the Notch signaling pathway with induction of CDX2 gene expression in human esophageal cells. Surgery 2009, 146, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Tamagawa, Y.; Ishimura, N.; Uno, G.; Yuki, T.; Kazumori, H.; Ishihara, S.; Amano, Y.; Kinoshita, Y. Notch signaling pathway and Cdx2 expression in the development of Barrett’s esophagus. Lab. Investig. 2012, 92, 896–909. [Google Scholar] [CrossRef]
- Tamagawa, Y.; Ishimura, N.; Uno, G.; Aimi, M.; Oshima, N.; Yuki, T.; Sato, S.; Ishihara, S.; Kinoshita, Y. Bile acids induce Delta-like 1 expression via Cdx2-dependent pathway in the development of Barrett’s esophagus. Lab. Investig. 2016, 96, 325–337. [Google Scholar] [CrossRef]
- Reveiller, M.; Ghatak, S.; Toia, L.; Kalatskaya, I.; Stein, L.; D’Souza, M.; Zhou, Z.; Bandla, S.; Gooding, W.E.; Godfrey, T.E.; et al. Bile exposure inhibits expression of squamous differentiation genes in human esophageal epithelial cells. Ann. Surg. 2012, 255, 1113–1120. [Google Scholar] [CrossRef]
- Shen, C.; Zhang, H.; Wang, P.; Feng, J.; Li, J.; Xu, Y.; Zhang, A.; Shao, S.; Yu, X.; Yan, W.; et al. Deoxycholic acid (DCA) confers an intestinal phenotype on esophageal squamous epithelium via induction of the stemness-associated reprogramming factors OCT4 and SOX2. Cell Cycle 2016, 15, 1439–1449. [Google Scholar] [CrossRef]
- Vega, M.E.; Giroux, V.; Natsuizaka, M.; Liu, M.; Klein-Szanto, A.J.; Stairs, D.B.; Nakagawa, H.; Wang, K.K.; Wang, T.C.; Lynch, J.P.; et al. Inhibition of Notch signaling enhances transdifferentiation of the esophageal squamous epithelium towards a Barrett’s-like metaplasia via KLF4. Cell Cycle 2014, 13, 3857–3866. [Google Scholar] [CrossRef] [PubMed]
- Minacapelli, C.D.; Bajpai, M.; Geng, X.; Cheng, C.L.; Chouthai, A.A.; Souza, R.; Spechler, S.J.; Das, K.M. Barrett’s metaplasia develops from cellular reprograming of esophageal squamous epithelium due to gastroesophageal reflux. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G615–G622. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Zhang, H.Y.; Zhang, X.I.; Lynch, J.P.; Strauch, E.D.; Wang, J.Y.; Melton, S.D.; Genta, R.M.; Wang, D.H.; Spechler, S.J.; et al. Acid and bile salt-induced CDX2 expression differs in esophageal squamous cells from patients with and without Barrett’s esophagus. Gastroenterology 2010, 139, 194–203.e1. [Google Scholar] [CrossRef]
- Huo, X.; Dunbar, K.B.; Zhang, X.; Zhang, Q.; Spechler, S.J.; Souza, R.F. In Barrett’s epithelial cells, weakly acidic bile salt solutions cause oxidative DNA damage with response and repair mediated by p38. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G464–G478. [Google Scholar] [CrossRef]
- Theodorou, D.; Ayazi, S.; DeMeester, S.R.; Zehetner, J.; Peyre, C.G.; Grant, K.S.; Augustin, F.; Oh, D.S.; Lipham, J.C.; Chandrasoma, P.T.; et al. Intraluminal pH and goblet cell density in Barrett’s esophagus. J. Gastrointest. Surg. 2012, 16, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Tambunting, L.; Kelleher, D.; Duggan, S.P. The Immune Underpinnings of Barrett’s-Associated Adenocarcinogenesis: A Retrial of Nefarious Immunologic Co-Conspirators. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 1297–1315. [Google Scholar] [CrossRef]
- O’Riordan, J.M.; Abdel-latif, M.M.; Ravi, N.; McNamara, D.; Byrne, P.J.; McDonald, G.S.; Keeling, P.W.; Kelleher, D.; Reynolds, J.V. Proinflammatory cytokine and nuclear factor kappa-B expression along the inflammation-metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am. J. Gastroenterol. 2005, 100, 1257–1264. [Google Scholar] [CrossRef]
- Fitzgerald, R.C.; Abdalla, S.; Onwuegbusi, B.A.; Sirieix, P.; Saeed, I.T.; Burnham, W.R.; Farthing, M.J. Inflammatory gradient in Barrett’s oesophagus: Implications for disease complications. Gut 2002, 51, 316–322. [Google Scholar] [CrossRef]
- Contino, G.; Vaughan, T.L.; Whiteman, D.; Fitzgerald, R.C. The Evolving Genomic Landscape of Barrett’s Esophagus and Esophageal Adenocarcinoma. Gastroenterology 2017, 153, 657–673.e1. [Google Scholar] [CrossRef]
- Peters, Y.; Al-Kaabi, A.; Shaheen, N.J.; Chak, A.; Blum, A.; Souza, R.F.; Di Pietro, M.; Iyer, P.G.; Pech, O.; Fitzgerald, R.C.; et al. Barrett oesophagus. Nat. Rev. Dis. Primers 2019, 5, 35. [Google Scholar] [CrossRef]
- Caspa Gokulan, R.; Garcia-Buitrago, M.T.; Zaika, A.I. From genetics to signaling pathways: Molecular pathogenesis of esophageal adenocarcinoma. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.H.; Clemons, N.J.; Miyashita, T.; Dupuy, A.J.; Zhang, W.; Szczepny, A.; Corcoran-Schwartz, I.M.; Wilburn, D.L.; Montgomery, E.A.; Wang, J.S.; et al. Aberrant epithelial-mesenchymal Hedgehog signaling characterizes Barrett’s metaplasia. Gastroenterology 2010, 138, 1810–1822. [Google Scholar] [CrossRef] [PubMed]
- Milano, F.; van Baal, J.W.; Buttar, N.S.; Rygiel, A.M.; de Kort, F.; DeMars, C.J.; Rosmolen, W.D.; Bergman, J.J.; van Marle, J.; Wang, K.K.; et al. Bone morphogenetic protein 4 expressed in esophagitis induces a columnar phenotype in esophageal squamous cells. Gastroenterology 2007, 132, 2412–2421. [Google Scholar] [CrossRef]
- Mari, L.; Milano, F.; Parikh, K.; Straub, D.; Everts, V.; Hoeben, K.K.; Fockens, P.; Buttar, N.S.; Krishnadath, K.K. A pSMAD/CDX2 complex is essential for the intestinalization of epithelial metaplasia. Cell Rep. 2014, 7, 1197–1210. [Google Scholar] [CrossRef]
- Wang, D.H.; Tiwari, A.; Kim, M.E.; Clemons, N.J.; Regmi, N.L.; Hodges, W.A.; Berman, D.M.; Montgomery, E.A.; Watkins, D.N.; Zhang, X.; et al. Hedgehog signaling regulates FOXA2 in esophageal embryogenesis and Barrett’s metaplasia. J. Clin. Investig. 2014, 124, 3767–3780. [Google Scholar] [CrossRef]
- Que, J.; Garman, K.S.; Souza, R.F.; Spechler, S.J. Pathogenesis and Cells of Origin of Barrett’s Esophagus. Gastroenterology 2019, 157, 349–364.e1. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.H.; Souza, R.F. Transcommitment: Paving the Way to Barrett’s Metaplasia. Adv. Exp. Med. Biol. 2016, 908, 183–212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, D.H. Origins of Metaplasia in Barrett’s Esophagus: Is this an Esophageal Stem or Progenitor Cell Disease? Dig. Dis. Sci. 2018, 63, 2005–2012. [Google Scholar] [CrossRef]
- Wang, D.H. The Esophageal Squamous Epithelial Cell-Still a Reasonable Candidate for the Barrett’s Esophagus Cell of Origin? Cell Mol. Gastroenterol. Hepatol. 2017, 4, 157–160. [Google Scholar] [CrossRef]
- McDonald, S.A.; Lavery, D.; Wright, N.A.; Jansen, M. Barrett oesophagus: Lessons on its origins from the lesion itself. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 50–60. [Google Scholar] [CrossRef]
- Sawhney, R.A.; Shields, H.M.; Allan, C.H.; Boch, J.A.; Trier, J.S.; Antonioli, D.A. Morphological characterization of the squamocolumnar junction of the esophagus in patients with and without Barrett’s epithelium. Dig. Dis. Sci. 1996, 41, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Shields, H.M.; Zwas, F.; Antonioli, D.A.; Doos, W.G.; Kim, S.; Spechler, S.J. Detection by scanning electron microscopy of a distinctive esophageal surface cell at the junction of squamous and Barrett’s epithelium. Dig. Dis. Sci. 1993, 38, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.A.; Shields, H.M.; Antonioli, D.A.; Zwas, F.; Sawhney, R.A.; Trier, J.S. Distribution of cytokeratin markers in Barrett’s specialized columnar epithelium. Gastroenterology 1997, 112, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Chandrasoma, P.T.; Lokuhetty, D.M.; Demeester, T.R.; Bremmer, C.G.; Peters, J.H.; Oberg, S.; Groshen, S. Definition of histopathologic changes in gastroesophageal reflux disease. Am. J. Surg. Pathol. 2000, 24, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Chandrasoma, P. Controversies of the cardiac mucosa and Barrett’s oesophagus. Histopathology 2005, 46, 361–373. [Google Scholar] [CrossRef]
- Chandrasoma, P.; Wijetunge, S.; Ma, Y.; Demeester, S.; Hagen, J.; Demeester, T. The dilated distal esophagus: A new entity that is the pathologic basis of early gastroesophageal reflux disease. Am. J. Surg. Pathol. 2011, 35, 1873–1881. [Google Scholar] [CrossRef]
- Jiang, M.; Li, H.; Zhang, Y.; Yang, Y.; Lu, R.; Liu, K.; Lin, S.; Lan, X.; Wang, H.; Wu, H.; et al. Transitional basal cells at the squamous-columnar junction generate Barrett’s oesophagus. Nature 2017, 550, 529–533. [Google Scholar] [CrossRef]
- Glickman, J.N.; Chen, Y.Y.; Wang, H.H.; Antonioli, D.A.; Odze, R.D. Phenotypic characteristics of a distinctive multilayered epithelium suggests that it is a precursor in the development of Barrett’s esophagus. Am. J. Surg. Pathol. 2001, 25, 569–578. [Google Scholar] [CrossRef]
- Coad, R.A.; Woodman, A.C.; Warner, P.J.; Barr, H.; Wright, N.A.; Shepherd, N.A. On the histogenesis of Barrett’s oesophagus and its associated squamous islands: A three-dimensional study of their morphological relationship with native oesophageal gland ducts. J. Pathol. 2005, 206, 388–394. [Google Scholar] [CrossRef]
- Kruger, L.; Gonzalez, L.M.; Pridgen, T.A.; McCall, S.J.; von Furstenberg, R.J.; Harnden, I.; Carnighan, G.E.; Cox, A.M.; Blikslager, A.T.; Garman, K.S. Ductular and proliferative response of esophageal submucosal glands in a porcine model of esophageal injury and repair. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G180–G191. [Google Scholar] [CrossRef]
- Owen, R.P.; White, M.J.; Severson, D.T.; Braden, B.; Bailey, A.; Goldin, R.; Wang, L.M.; Ruiz-Puig, C.; Maynard, N.D.; Green, A.; et al. Single cell RNA-seq reveals profound transcriptional similarity between Barrett’s oesophagus and oesophageal submucosal glands. Nat. Commun. 2018, 9, 4261. [Google Scholar] [CrossRef] [PubMed]
- Leedham, S.J.; Preston, S.L.; McDonald, S.A.; Elia, G.; Bhandari, P.; Poller, D.; Harrison, R.; Novelli, M.R.; Jankowski, J.A.; Wright, N.A. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut 2008, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Bhagat, G.; Abrams, J.A.; Marache, F.; Good, P.; Lee, M.D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Kunze, B.; Wein, F.; Fang, H.Y.; Anand, A.; Baumeister, T.; Strangmann, J.; Gerland, S.; Ingermann, J.; Munch, N.S.; Wiethaler, M.; et al. Notch Signaling Mediates Differentiation in Barrett’s Esophagus and Promotes Progression to Adenocarcinoma. Gastroenterology 2020, 159, 575–590. [Google Scholar] [CrossRef]
- Schellnegger, R.; Quante, A.; Rospleszcz, S.; Schernhammer, M.; Hohl, B.; Tobiasch, M.; Pastula, A.; Brandtner, A.; Abrams, J.A.; Strauch, K.; et al. Goblet Cell Ratio in Combination with Differentiation and Stem Cell Markers in Barrett Esophagus Allow Distinction of Patients with and without Esophageal Adenocarcinoma. Cancer Prev. Res. 2017, 10, 55–66. [Google Scholar] [CrossRef]
- Becker, L.; Huang, Q.; Mashimo, H. Lgr5, an intestinal stem cell marker, is abnormally expressed in Barrett’s esophagus and esophageal adenocarcinoma. Dis. Esophagus 2010, 23, 168–174. [Google Scholar] [CrossRef]
- Von Rahden, B.H.; Kircher, S.; Lazariotou, M.; Reiber, C.; Stuermer, L.; Otto, C.; Germer, C.T.; Grimm, M. LgR5 expression and cancer stem cell hypothesis: Clue to define the true origin of esophageal adenocarcinomas with and without Barrett’s esophagus? J. Exp. Clin. Cancer Res. 2011, 30, 23. [Google Scholar] [CrossRef]
- Lavery, D.L.; Nicholson, A.M.; Poulsom, R.; Jeffery, R.; Hussain, A.; Gay, L.J.; Jankowski, J.A.; Zeki, S.S.; Barr, H.; Harrison, R.; et al. The stem cell organisation, and the proliferative and gene expression profile of Barrett’s epithelium, replicates pyloric-type gastric glands. Gut 2014, 63, 1854–1863. [Google Scholar] [CrossRef]
- Jang, B.G.; Lee, B.L.; Kim, W.H. Intestinal Stem Cell Markers in the Intestinal Metaplasia of Stomach and Barrett’s Esophagus. PLoS ONE 2015, 10, e0127300. [Google Scholar] [CrossRef]
- Wang, X.; Ouyang, H.; Yamamoto, Y.; Kumar, P.A.; Wei, T.S.; Dagher, R.; Vincent, M.; Lu, X.; Bellizzi, A.M.; Ho, K.Y.; et al. Residual embryonic cells as precursors of a Barrett’s-like metaplasia. Cell 2011, 145, 1023–1035. [Google Scholar] [CrossRef]
- Xian, W.; Duleba, M.; Zhang, Y.; Yamamoto, Y.; Ho, K.Y.; Crum, C.; McKeon, F. The Cellular Origin of Barrett’s Esophagus and Its Stem Cells. Adv. Exp. Med. Biol. 2019, 1123, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Sarosi, G.; Brown, G.; Jaiswal, K.; Feagins, L.A.; Lee, E.; Crook, T.W.; Souza, R.F.; Zou, Y.S.; Shay, J.W.; Spechler, S.J. Bone marrow progenitor cells contribute to esophageal regeneration and metaplasia in a rat model of Barrett’s esophagus. Dis. Esophagus 2008, 21, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, L.; Stenstrom, B.; Chen, D.; Piperdi, B.; Levey, S.; Lyle, S.; Wang, T.C.; Houghton, J. Human Barrett’s adenocarcinoma of the esophagus, associated myofibroblasts, and endothelium can arise from bone marrow-derived cells after allogeneic stem cell transplant. Stem Cells Dev. 2011, 20, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Goldenring, J.R. Mind the Gap: Crossing Boundaries to Establish Reparative Metaplasia. Cell Mol. Gastroenterol. Hepatol. 2018, 6, 468–469. [Google Scholar] [CrossRef]
- Zhang, Q.; Agoston, A.T.; Pham, T.H.; Zhang, W.; Zhang, X.; Huo, X.; Peng, S.; Bajpai, M.; Das, K.; Odze, R.D.; et al. Acidic Bile Salts Induce Epithelial to Mesenchymal Transition via VEGF Signaling in Non-Neoplastic Barrett’s Cells. Gastroenterology 2019, 156, 130–144.e10. [Google Scholar] [CrossRef]
- Phipps, S.M.; Garry, C.E.; Kamal, S.; Johnson, J.D.; Gilmer, J.; Long, A.; Kelleher, D.; Duggan, S.P. High Content Imaging of Barrett’s-Associated High-Grade Dysplasia Cells After siRNA Library Screening Reveals Acid-Responsive Regulators of Cellular Transitions. Cell Mol. Gastroenterol. Hepatol. 2020, 10, 601–622. [Google Scholar] [CrossRef]
- Souza, R.F.; Huo, X.; Mittal, V.; Schuler, C.M.; Carmack, S.W.; Zhang, H.Y.; Zhang, X.; Yu, C.; Hormi-Carver, K.; Genta, R.M.; et al. Gastroesophageal reflux might cause esophagitis through a cytokine-mediated mechanism rather than caustic acid injury. Gastroenterology 2009, 137, 1776–1784. [Google Scholar] [CrossRef]
- Dunbar, K.B.; Agoston, A.T.; Odze, R.D.; Huo, X.; Pham, T.H.; Cipher, D.J.; Castell, D.O.; Genta, R.M.; Souza, R.F.; Spechler, S.J. Association of Acute Gastroesophageal Reflux Disease with Esophageal Histologic Changes. JAMA 2016, 315, 2104–2112. [Google Scholar] [CrossRef]
- Huo, X.; Agoston, A.T.; Dunbar, K.B.; Cipher, D.J.; Zhang, X.; Yu, C.; Cheng, E.; Zhang, Q.; Pham, T.H.; Tambar, U.K.; et al. Hypoxia-inducible factor-2alpha plays a role in mediating oesophagitis in GORD. Gut 2017, 66, 1542–1554. [Google Scholar] [CrossRef]
- Spechler, S.J.; Merchant, J.L.; Wang, T.C.; Chandrasoma, P.; Fox, J.G.; Genta, R.M.; Goldenring, J.R.; Hayakawa, Y.; Kuipers, E.J.; Lund, P.K.; et al. A Summary of the 2016 James W. Freston Conference of the American Gastroenterological Association: Intestinal Metaplasia in the Esophagus and Stomach: Origins, Differences, Similarities and Significance. Gastroenterology 2017, 153, e6–e13. [Google Scholar] [CrossRef]
- Feagins, L.A.; Zhang, H.Y.; Zhang, X.; Hormi-Carver, K.; Thomas, T.; Terada, L.S.; Spechler, S.J.; Souza, R.F. Mechanisms of oxidant production in esophageal squamous cell and Barrett’s cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G411–G417. [Google Scholar] [CrossRef] [PubMed]
- Haddad, J.J.; Harb, H.L. Cytokines and the regulation of hypoxia-inducible factor (HIF)-1alpha. Int. Immunopharmacol. 2005, 5, 461–483. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Gong, J.; Geng, J.; Song, Y. Deoxycholic acid induces the overexpression of intestinal mucin, MUC2, via NF-kB signaling pathway in human esophageal adenocarcinoma cells. BMC Cancer 2008, 8, 333. [Google Scholar] [CrossRef]
- Dvorakova, K.; Payne, C.M.; Ramsey, L.; Holubec, H.; Sampliner, R.; Dominguez, J.; Dvorak, B.; Bernstein, H.; Bernstein, C.; Prasad, A.; et al. Increased expression and secretion of interleukin-6 in patients with Barrett’s esophagus. Clin. Cancer Res. 2004, 10, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Zhang, Q.; Zhang, X.; Yu, C.; Huo, X.; Cheng, E.; Wang, D.H.; Spechler, S.J.; Souza, R.F. Cancer-related inflammation and Barrett’s carcinogenesis: Interleukin-6 and STAT3 mediate apoptotic resistance in transformed Barrett’s cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G454–G460. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, K.; Dvorak, B. Role of interleukin-6 in Barrett’s esophagus pathogenesis. World J. Gastroenterol. 2013, 19, 2307–2312. [Google Scholar] [CrossRef]
- Clemons, N.J.; McColl, K.E.; Fitzgerald, R.C. Nitric oxide and acid induce double-strand DNA breaks in Barrett’s esophagus carcinogenesis via distinct mechanisms. Gastroenterology 2007, 133, 1198–1209. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Hormi-Carver, K.; Zhang, X.; Spechler, S.J.; Souza, R.F. In benign Barrett’s epithelial cells, acid exposure generates reactive oxygen species that cause DNA double-strand breaks. Cancer Res. 2009, 69, 9083–9089. [Google Scholar] [CrossRef]
- Suzuki, H.; Iijima, K.; Scobie, G.; Fyfe, V.; McColl, K.E. Nitrate and nitrosative chemistry within Barrett’s oesophagus during acid reflux. Gut 2005, 54, 1527–1535. [Google Scholar] [CrossRef]
- Ishiyama, F.; Iijima, K.; Asanuma, K.; Ara, N.; Yoshitake, J.; Abe, Y.; Koike, T.; Imatani, A.; Ohara, S.; Shimosegawa, T. Exogenous luminal nitric oxide exacerbates esophagus tissue damage in a reflux esophagitis model of rats. Scand. J. Gastroenterol. 2009, 44, 527–537. [Google Scholar] [CrossRef]
- Jolly, A.J.; Wild, C.P.; Hardie, L.J. Sodium deoxycholate causes nitric oxide mediated DNA damage in oesophageal cells. Free Radic. Res. 2009, 43, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Eluri, S.; Brugge, W.R.; Daglilar, E.S.; Jackson, S.A.; Styn, M.A.; Callenberg, K.M.; Welch, D.C.; Barr, T.M.; Duits, L.C.; Bergman, J.J.; et al. The Presence of Genetic Mutations at Key Loci Predicts Progression to Esophageal Adenocarcinoma in Barrett’s Esophagus. Am. J. Gastroenterol. 2015, 110, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Eluri, S.; Klaver, E.; Duits, L.C.; Jackson, S.A.; Bergman, J.J.; Shaheen, N.J. Validation of a biomarker panel in Barrett’s esophagus to predict progression to esophageal adenocarcinoma. Dis. Esophagus 2018, 31, doy026. [Google Scholar] [CrossRef]
- Newell, F.; Patel, K.; Gartside, M.; Krause, L.; Brosda, S.; Aoude, L.G.; Loffler, K.A.; Bonazzi, V.F.; Patch, A.M.; Kazakoff, S.H.; et al. Complex structural rearrangements are present in high-grade dysplastic Barrett’s oesophagus samples. BMC Med. Genom. 2019, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Ellsworth, E.; Jackson, S.A.; Thakkar, S.J.; Smith, D.M., Jr.; Finkelstein, S. Correlation of the presence and extent of loss of heterozygosity mutations with histological classifications of Barrett’s esophagus. BMC Gastroenterol. 2012, 12, 181. [Google Scholar] [CrossRef]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef]
- Nones, K.; Waddell, N.; Wayte, N.; Patch, A.M.; Bailey, P.; Newell, F.; Holmes, O.; Fink, J.L.; Quinn, M.C.J.; Tang, Y.H.; et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat. Commun. 2014, 5, 5224. [Google Scholar] [CrossRef]
- Trindade, A.J.; McKinley, M.J.; Alshelleh, M.; Levi, G.; Stewart, M.; Quinn, K.J.; Thomas, R.M. Mutational load may predict risk of progression in patients with Barrett’s oesophagus and indefinite for dysplasia: A pilot study. BMJ Open Gastroenterol. 2019, 6, e000268. [Google Scholar] [CrossRef]
- Weaver, J.M.J.; Ross-Innes, C.S.; Shannon, N.; Lynch, A.G.; Forshew, T.; Barbera, M.; Murtaza, M.; Ong, C.J.; Lao-Sirieix, P.; Dunning, M.J.; et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat. Genet. 2014, 46, 837–843. [Google Scholar] [CrossRef]
- Tanaka, T.; Watanabe, M.; Yamashita, K. Potential therapeutic targets of TP53 gene in the context of its classically canonical functions and its latest non-canonical functions in human cancer. Oncotarget 2018, 9, 16234–16247. [Google Scholar] [CrossRef]
- Secrier, M.; Li, X.; de Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Jones, D.T.; Zapatka, M.; Stutz, A.M.; Zichner, T.; Weischenfeldt, J.; Jager, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef]
- Harris, R.S.; Petersen-Mahrt, S.K.; Neuberger, M.S. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 2002, 10, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef]
- Murnane, J.P. Telomeres and chromosome instability. DNA Repair 2006, 5, 1082–1092. [Google Scholar] [CrossRef]
- Panda, A.; Shin, M.R.; Cheng, C.; Bajpai, M. Barrett’s Epithelium to Esophageal Adenocarcinoma: Is There a “Point of No Return”? Front. Genet. 2021, 12, 706706. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maslenkina, K.; Mikhaleva, L.; Naumenko, M.; Vandysheva, R.; Gushchin, M.; Atiakshin, D.; Buchwalow, I.; Tiemann, M. Signaling Pathways in the Pathogenesis of Barrett’s Esophagus and Esophageal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 9304. https://doi.org/10.3390/ijms24119304
Maslenkina K, Mikhaleva L, Naumenko M, Vandysheva R, Gushchin M, Atiakshin D, Buchwalow I, Tiemann M. Signaling Pathways in the Pathogenesis of Barrett’s Esophagus and Esophageal Adenocarcinoma. International Journal of Molecular Sciences. 2023; 24(11):9304. https://doi.org/10.3390/ijms24119304
Chicago/Turabian StyleMaslenkina, Ksenia, Liudmila Mikhaleva, Maxim Naumenko, Rositsa Vandysheva, Michail Gushchin, Dmitri Atiakshin, Igor Buchwalow, and Markus Tiemann. 2023. "Signaling Pathways in the Pathogenesis of Barrett’s Esophagus and Esophageal Adenocarcinoma" International Journal of Molecular Sciences 24, no. 11: 9304. https://doi.org/10.3390/ijms24119304
APA StyleMaslenkina, K., Mikhaleva, L., Naumenko, M., Vandysheva, R., Gushchin, M., Atiakshin, D., Buchwalow, I., & Tiemann, M. (2023). Signaling Pathways in the Pathogenesis of Barrett’s Esophagus and Esophageal Adenocarcinoma. International Journal of Molecular Sciences, 24(11), 9304. https://doi.org/10.3390/ijms24119304