

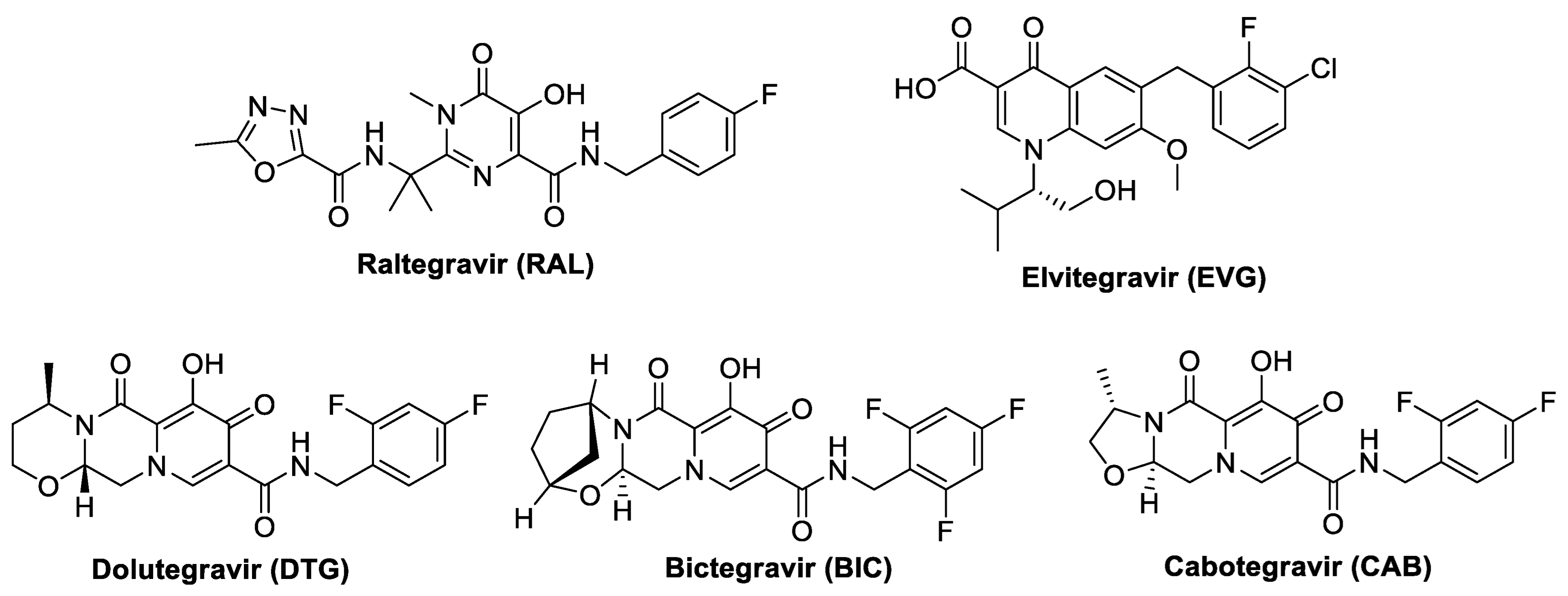
Recent Developments in the Synthesis of HIV-1 Integrase Strand Transfer Inhibitors Incorporating Pyridine Moiety



{kind=link}
{kind=link}
{kind=link}
{kind=link}
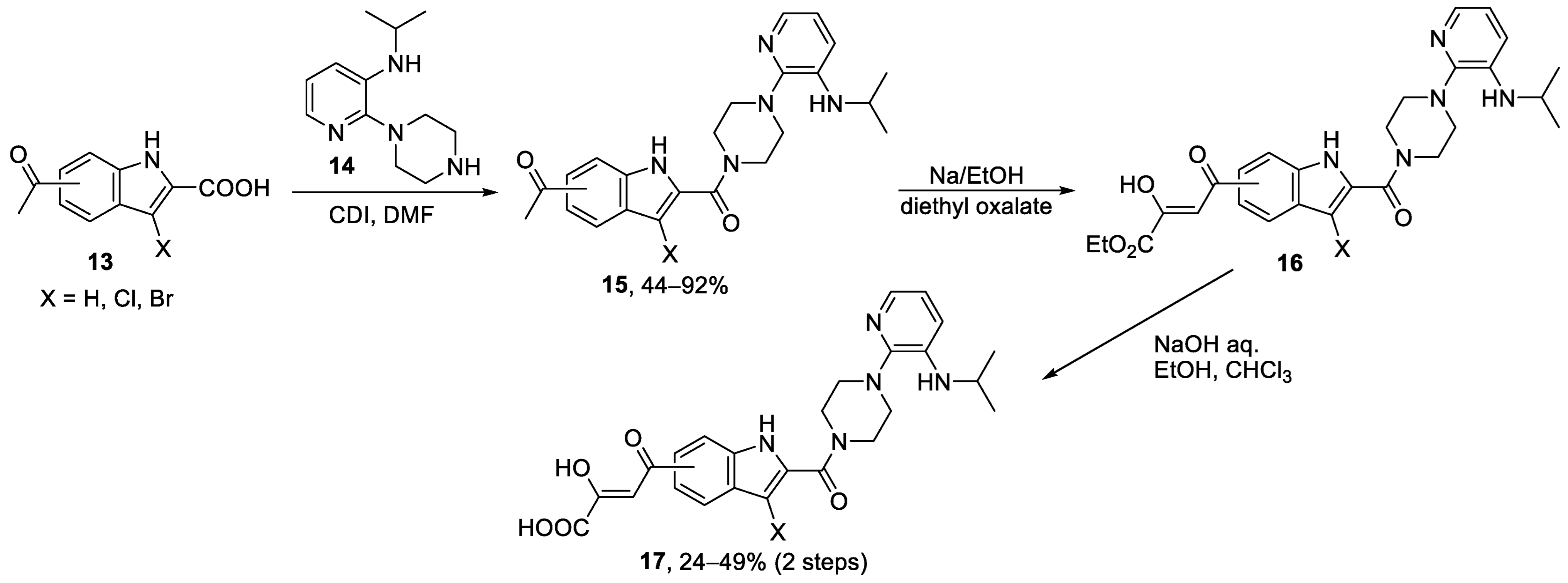{kind=link}
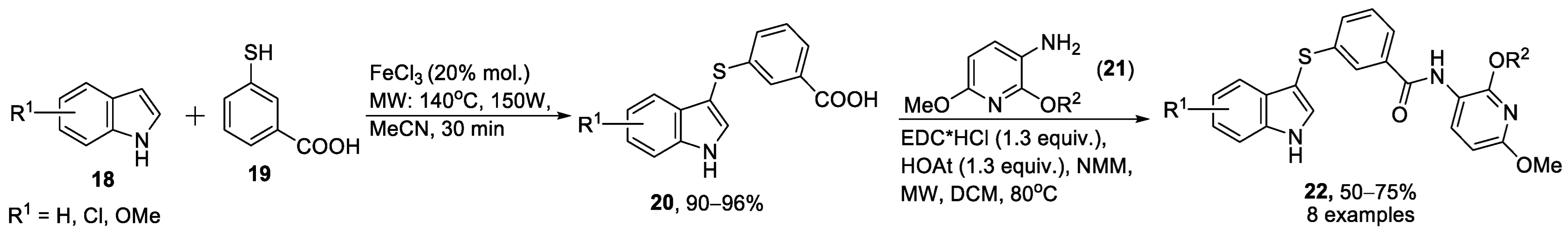{kind=link}
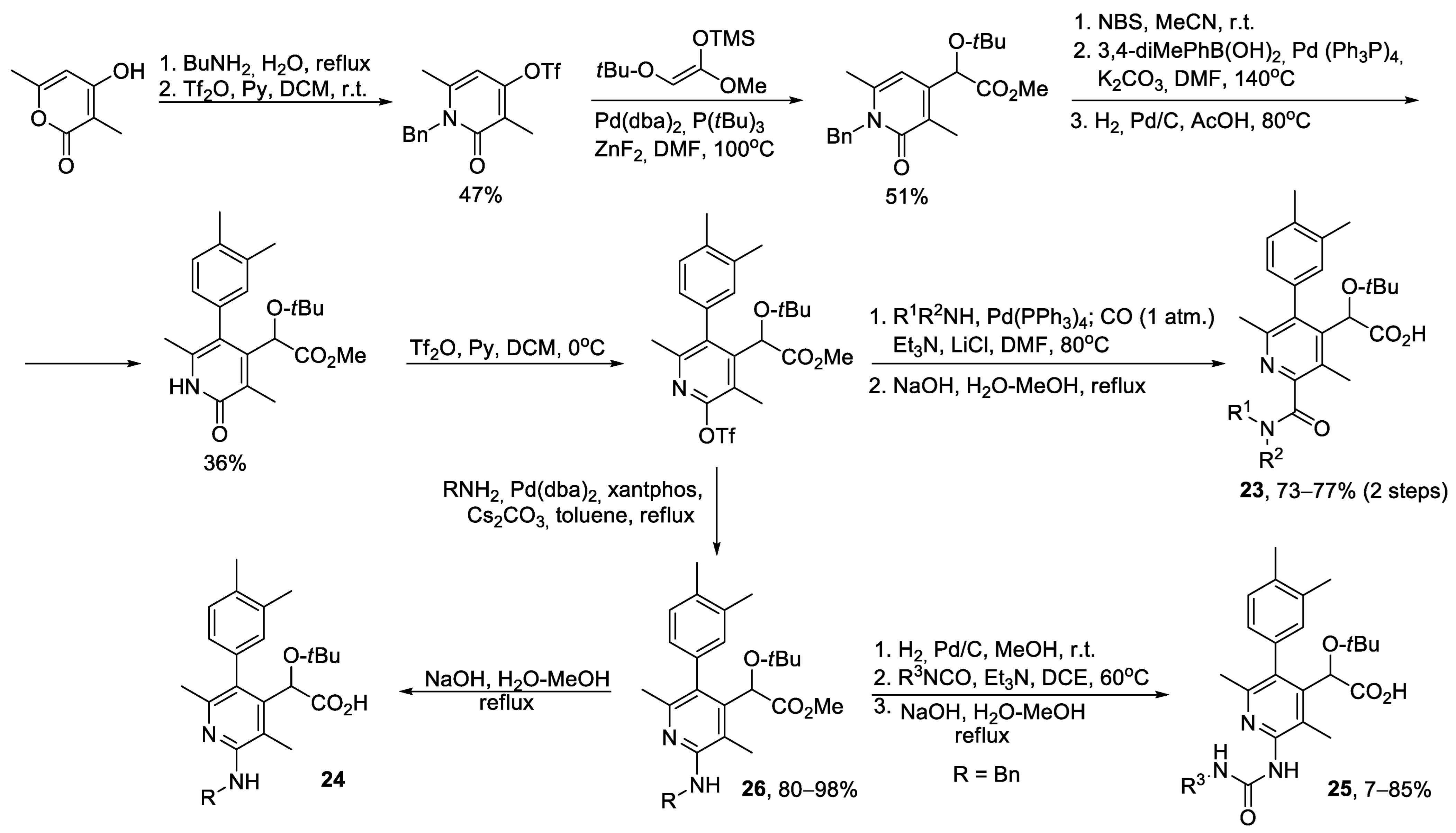{kind=link}
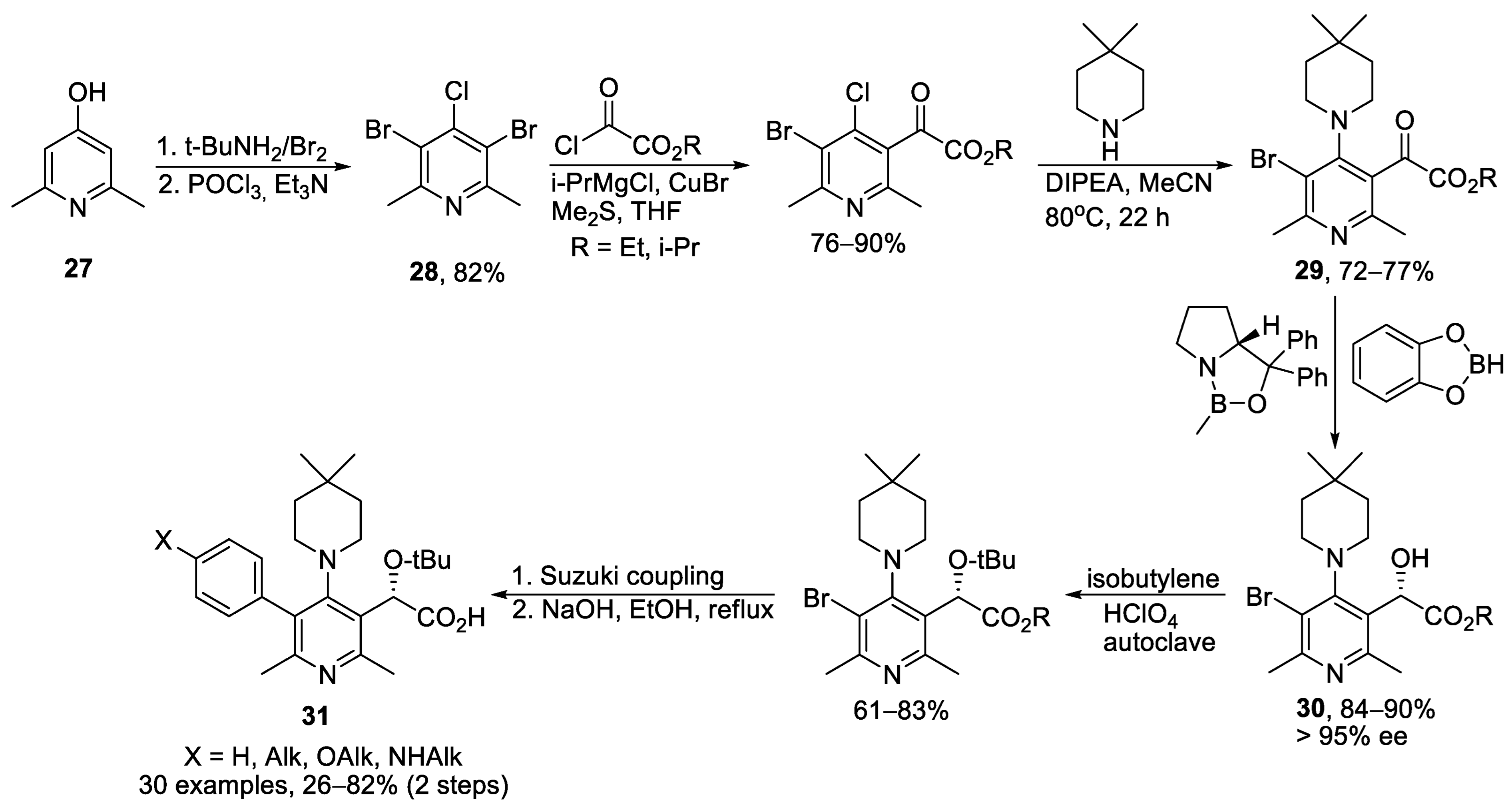{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
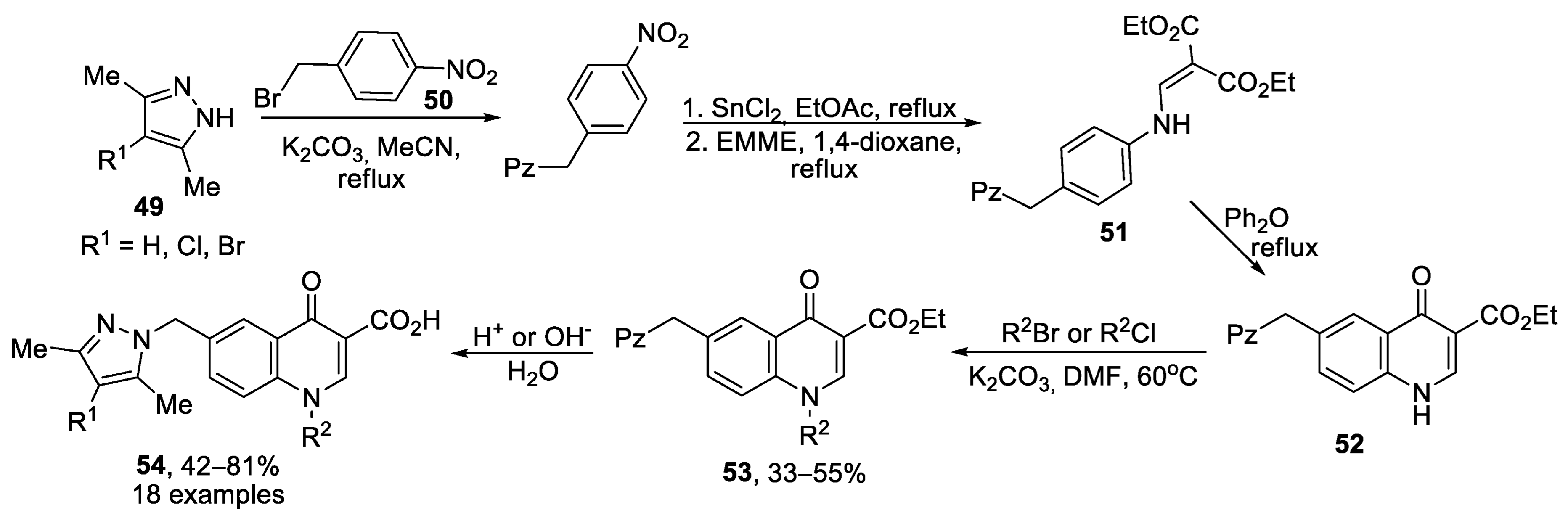{kind=link}
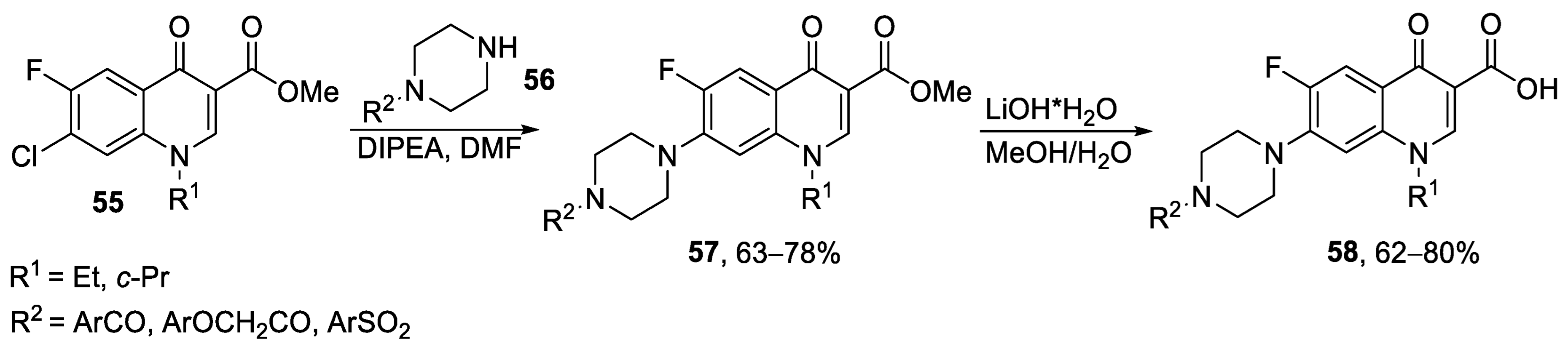{kind=link}
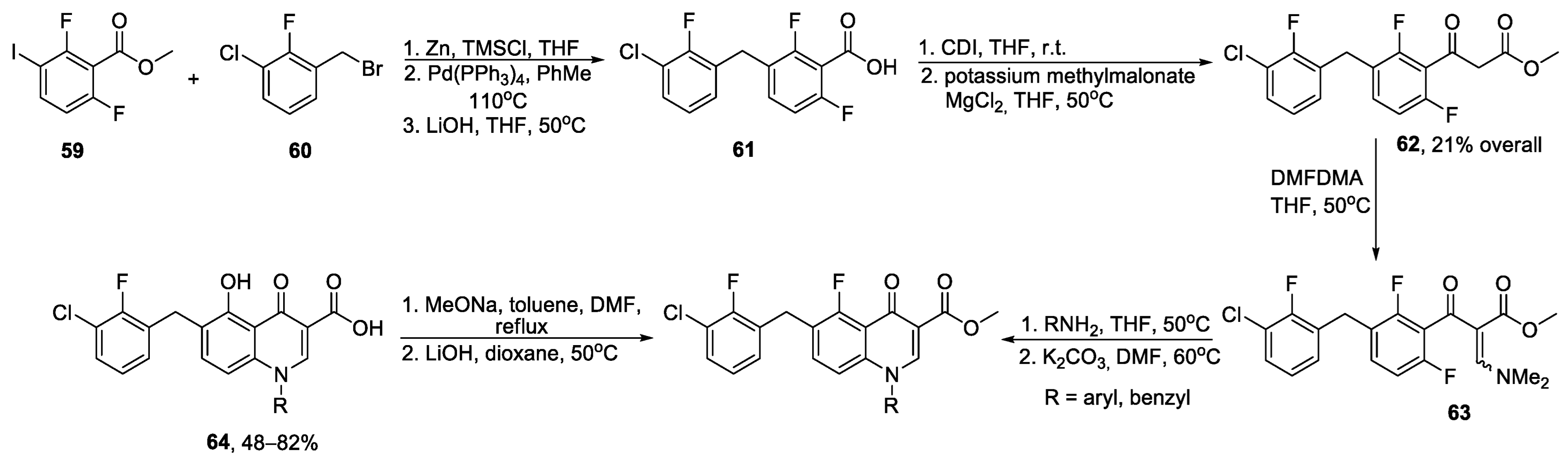{kind=link}
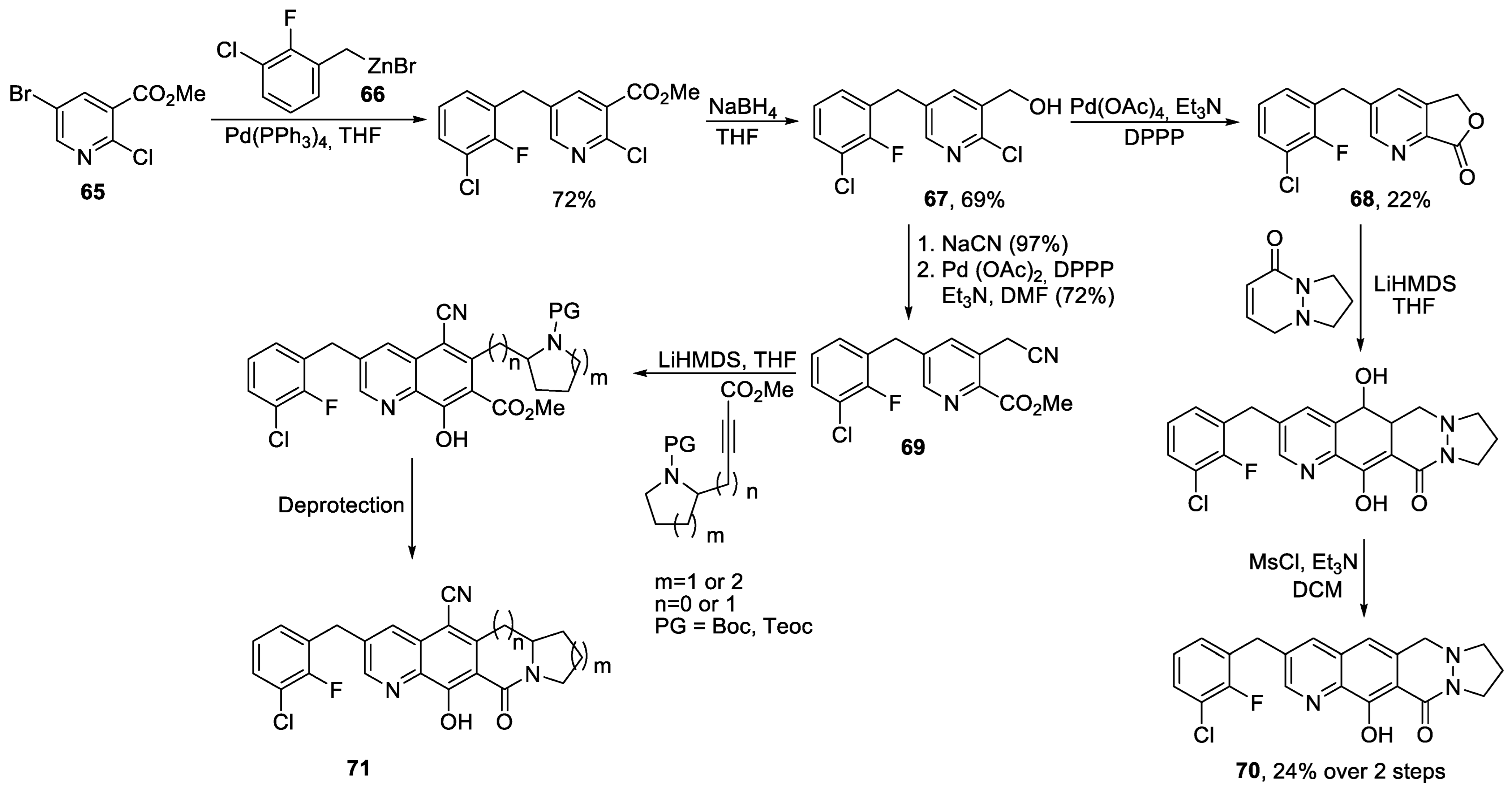{kind=link}
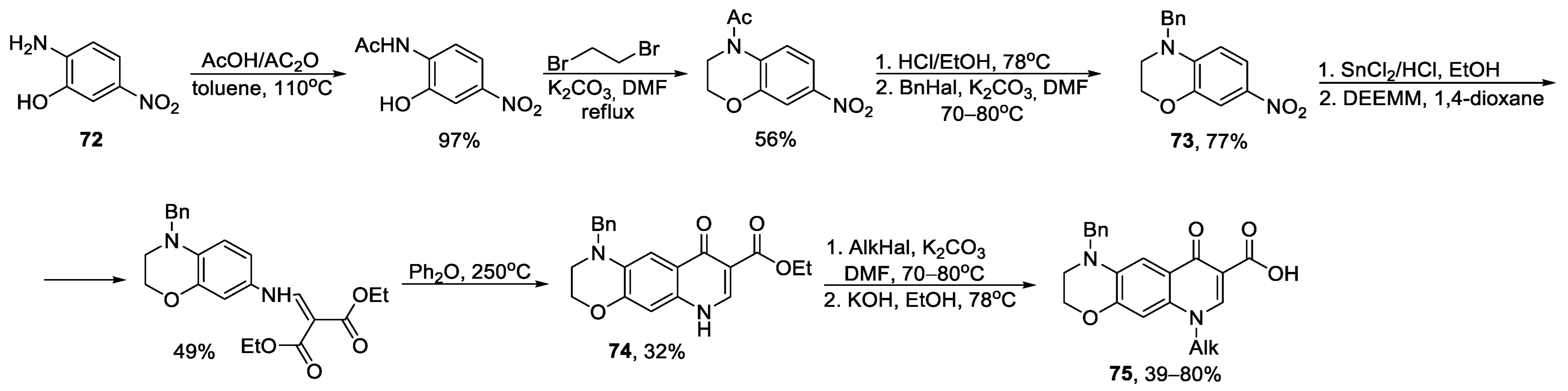{kind=link}
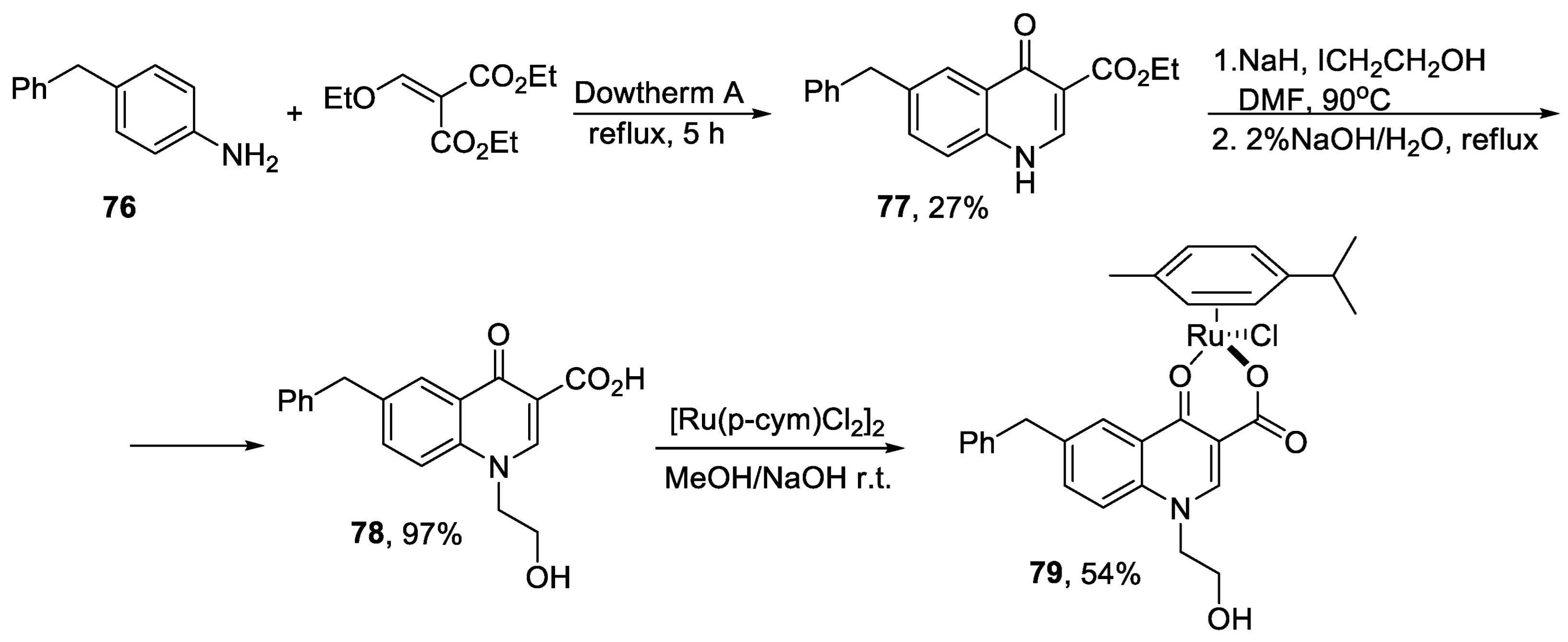{kind=link}
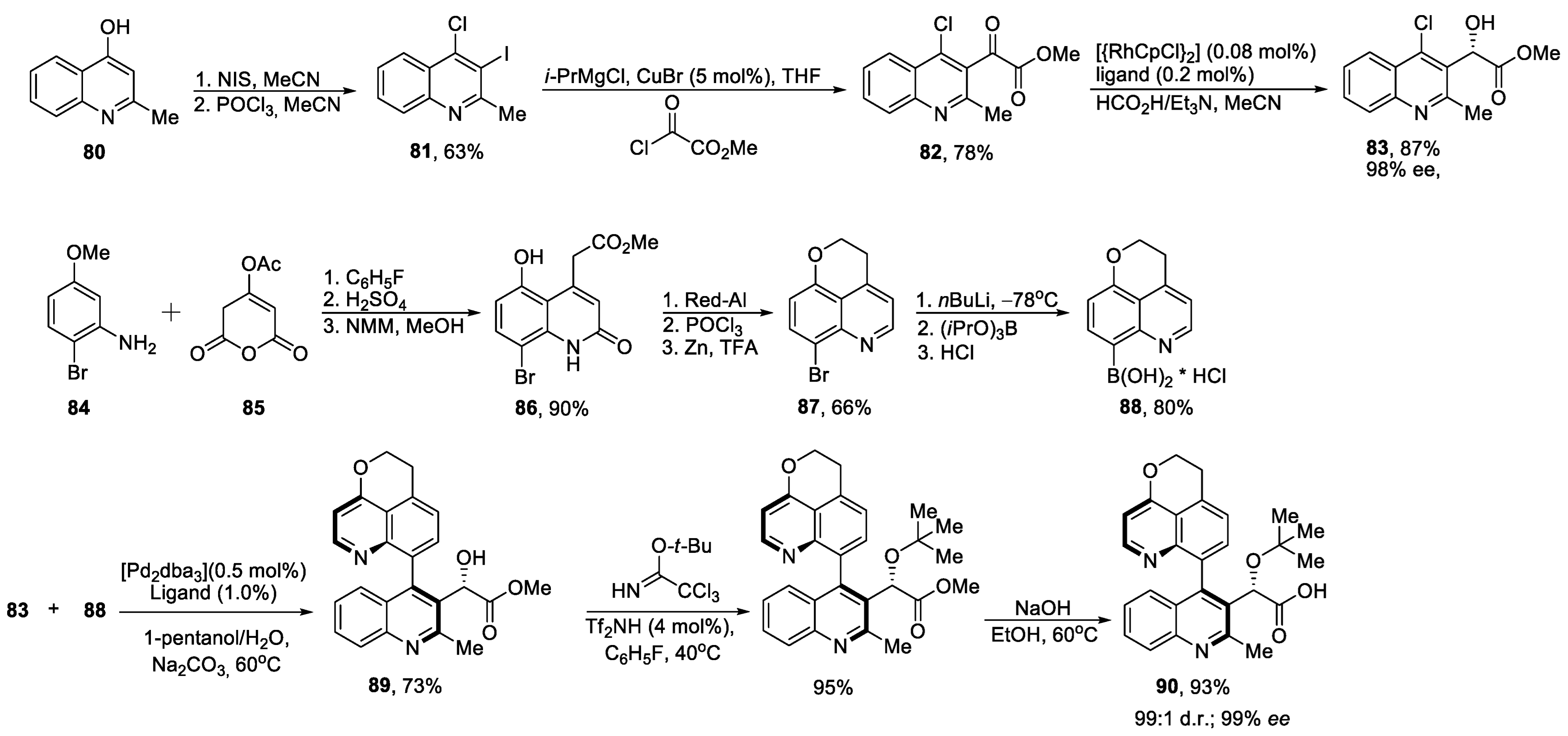{kind=link}
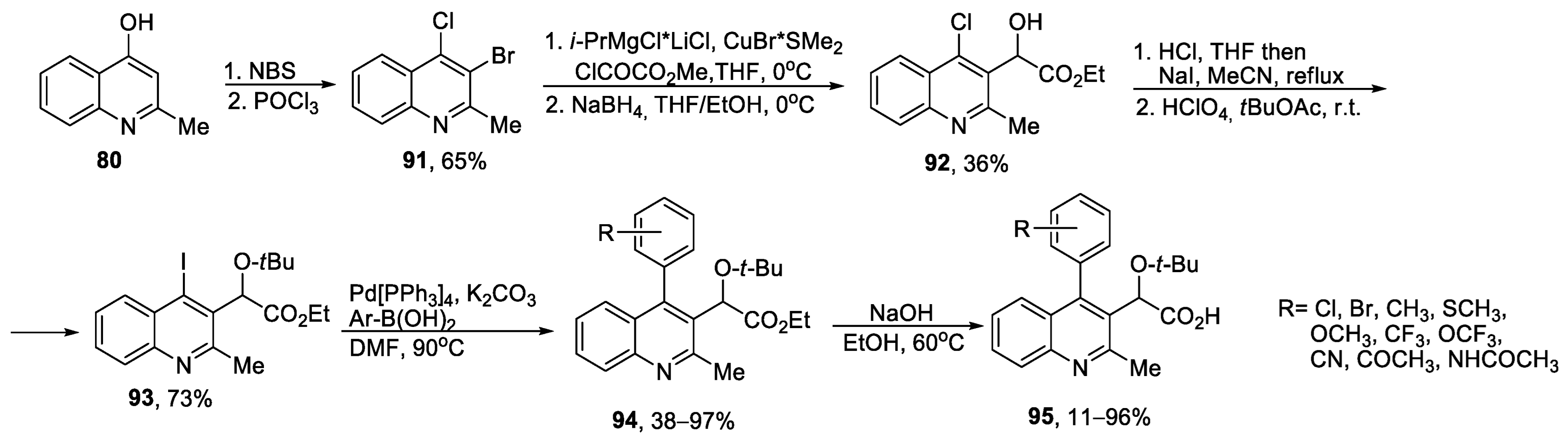{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
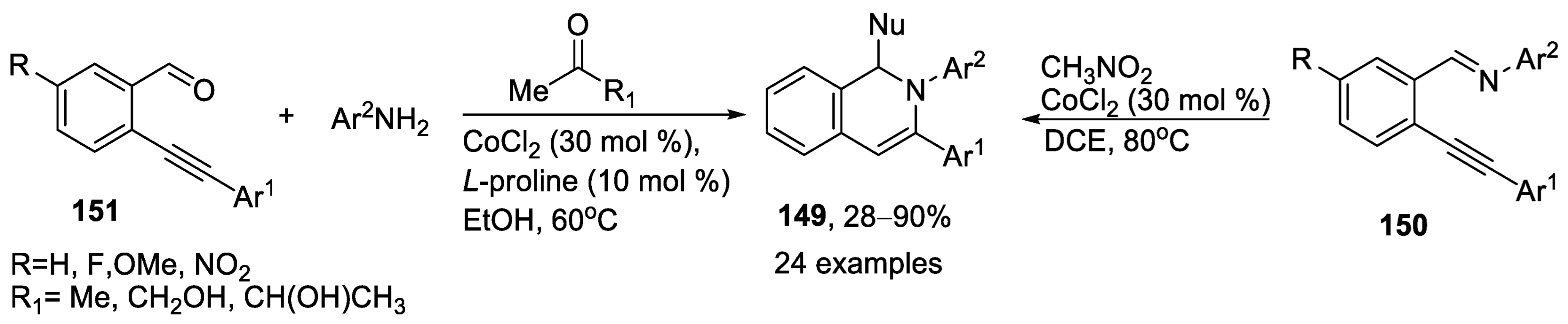{kind=link}
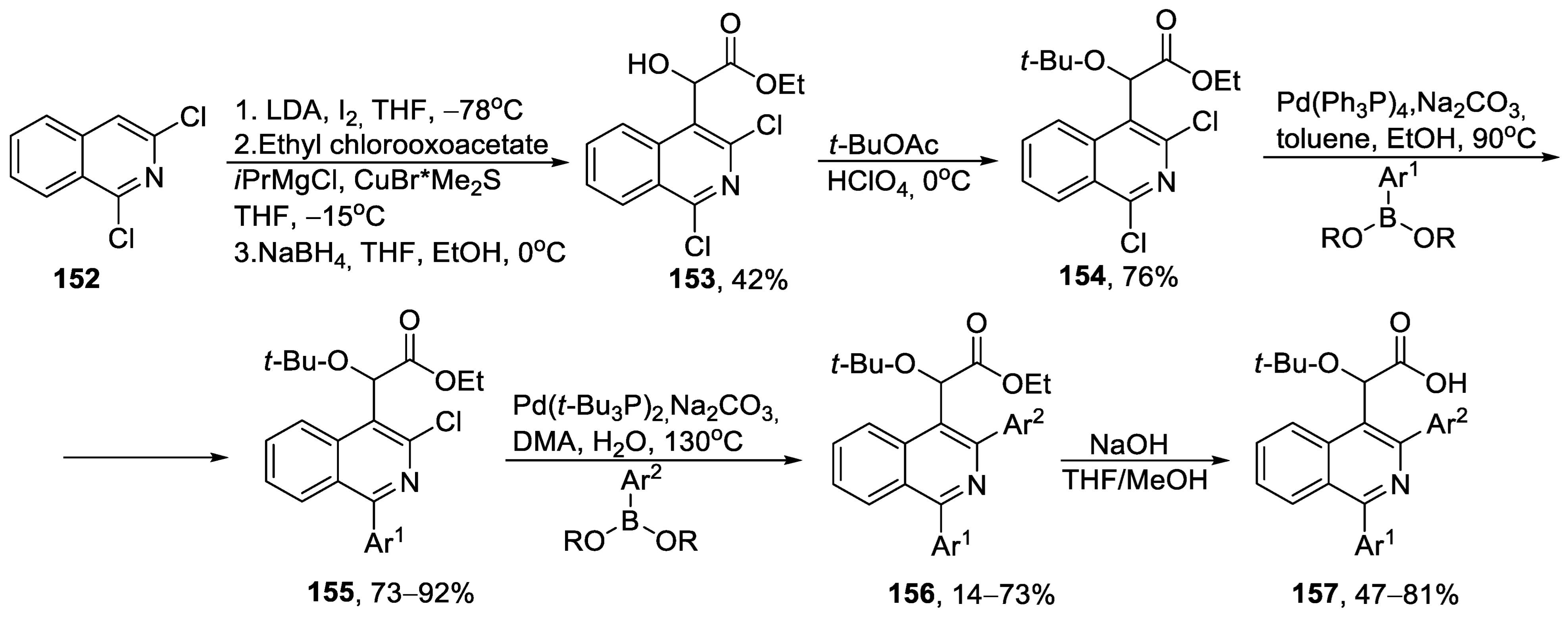{kind=link}
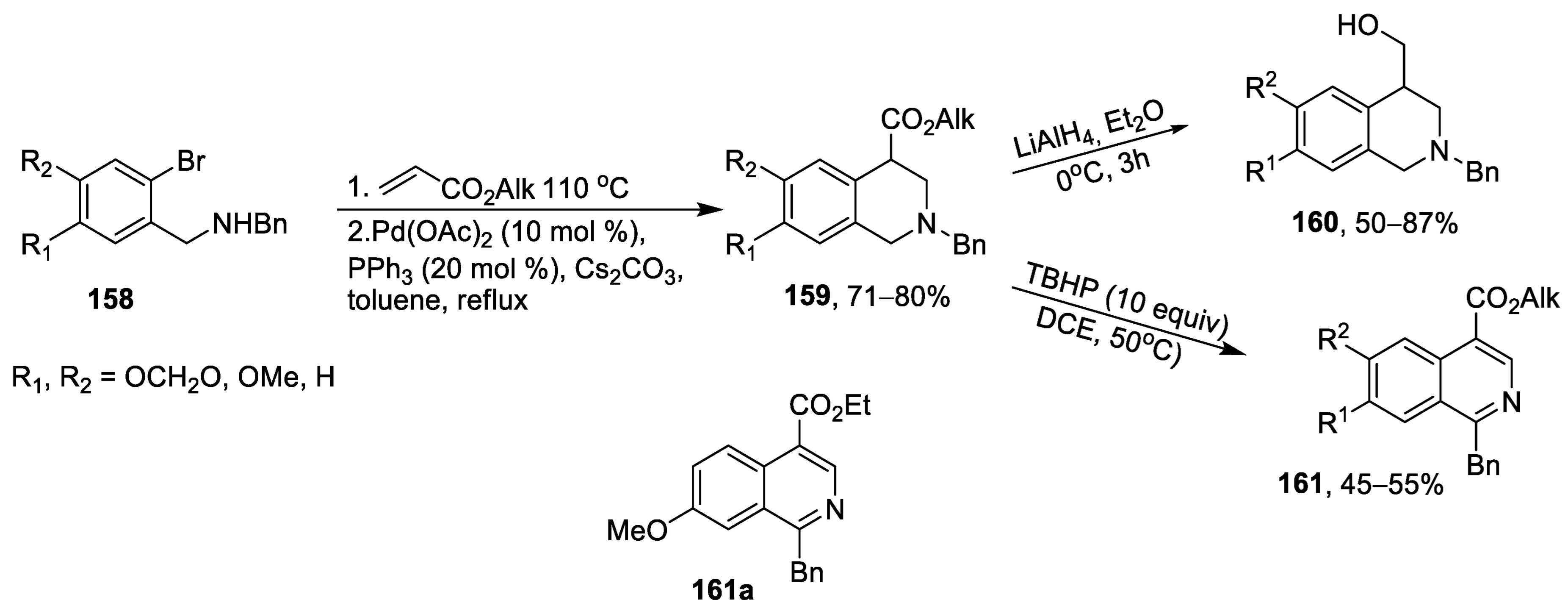{kind=link}
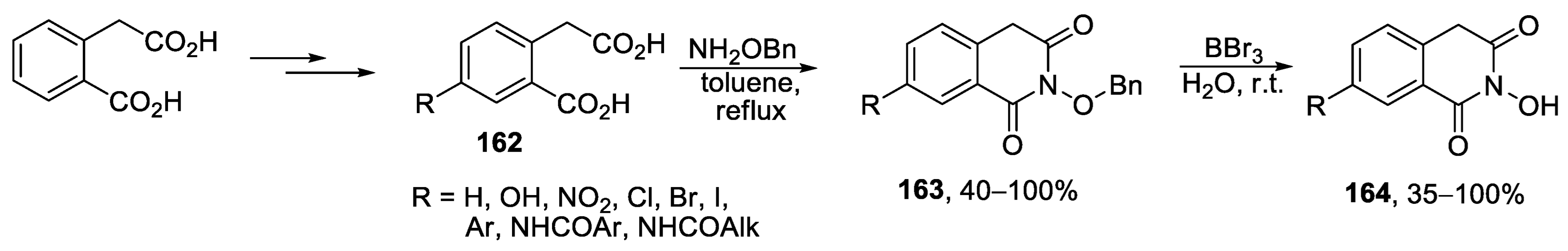{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
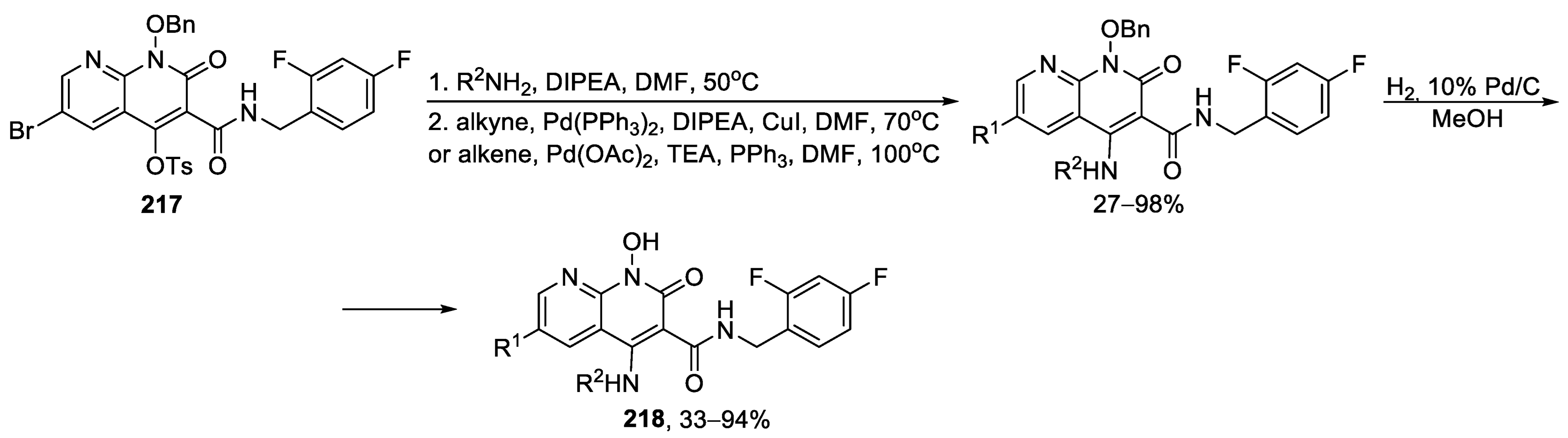{kind=link}
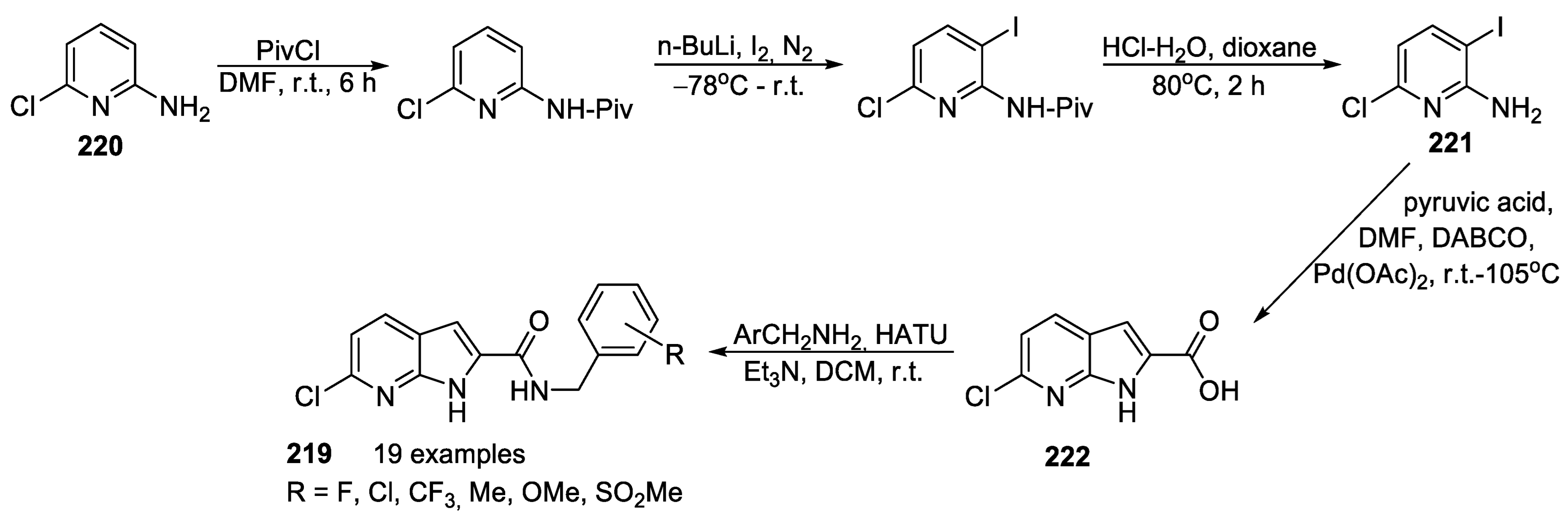{kind=link}
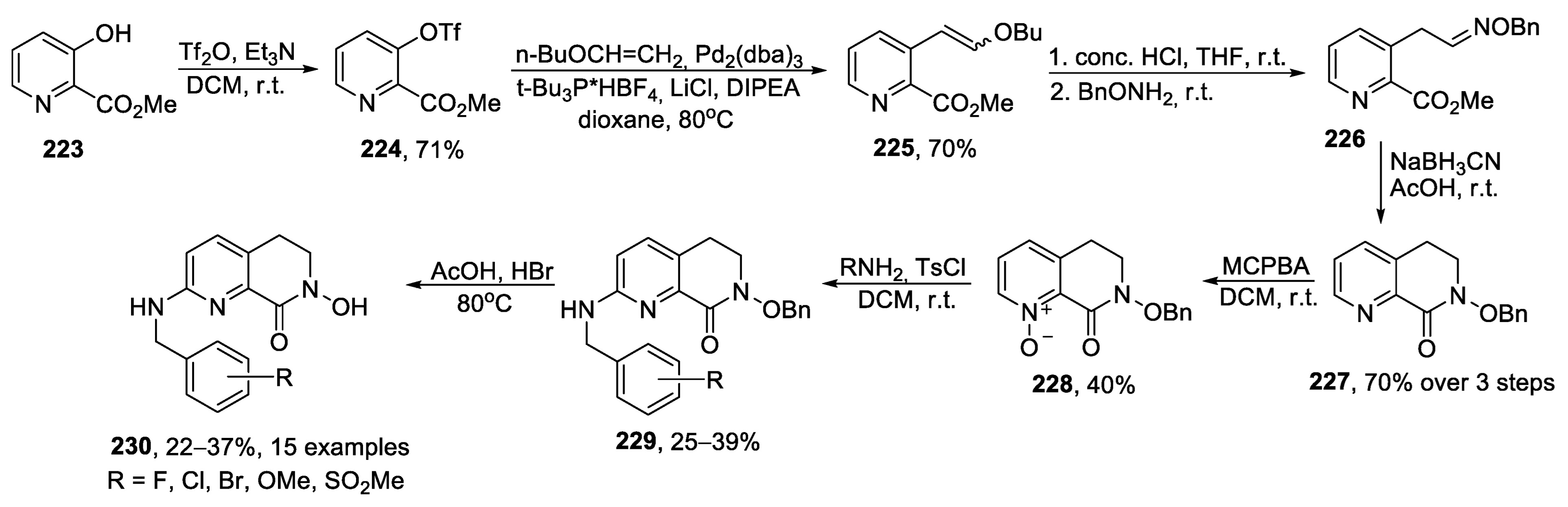{kind=link}
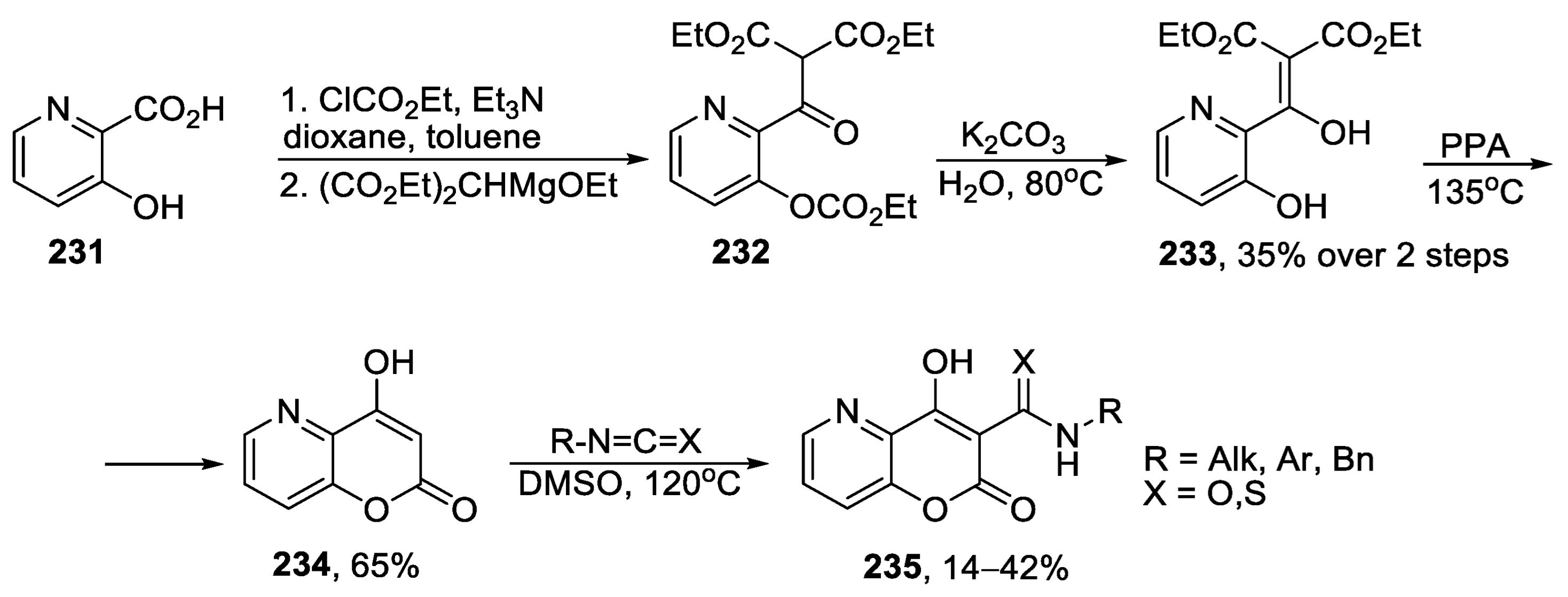{kind=link}
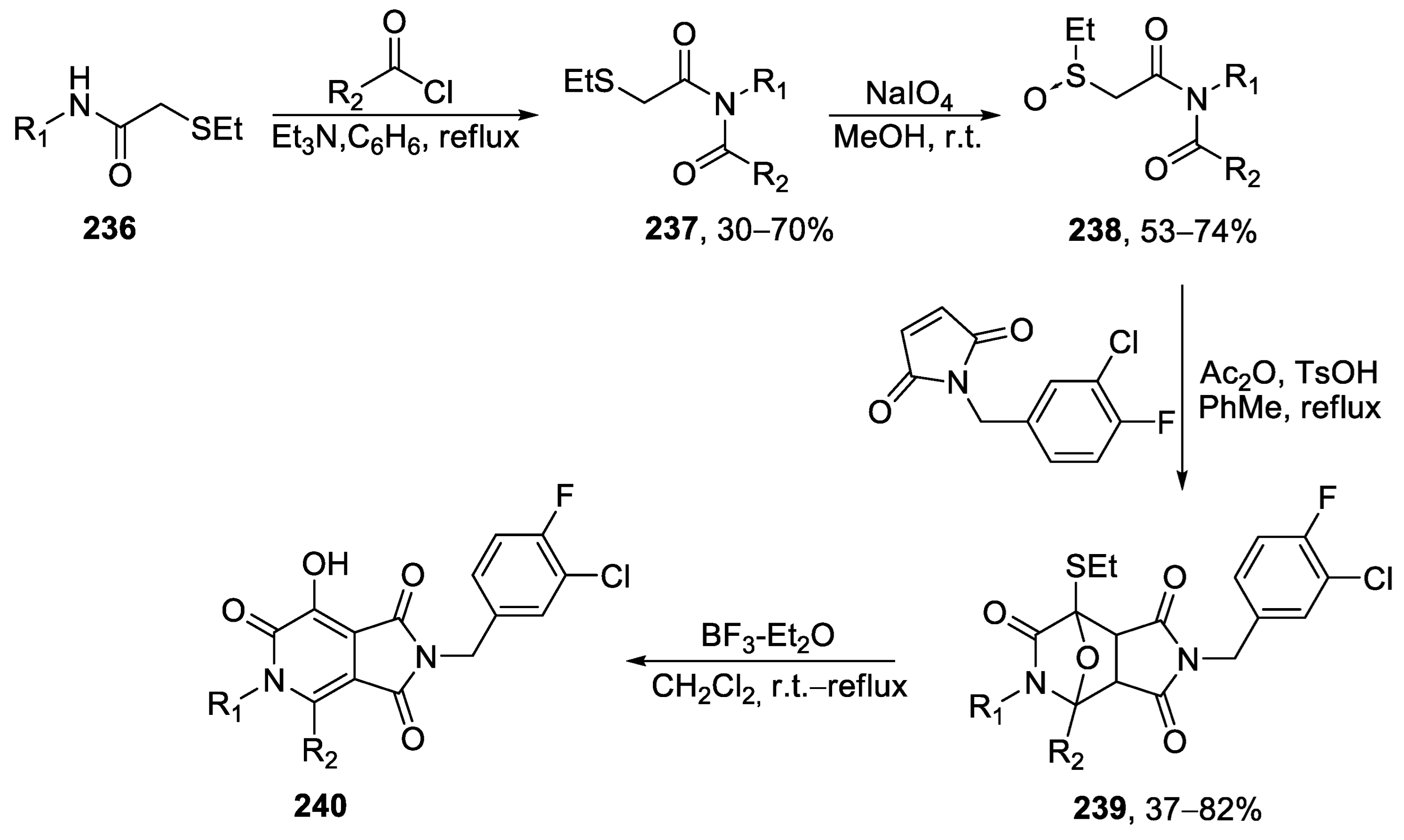{kind=link}
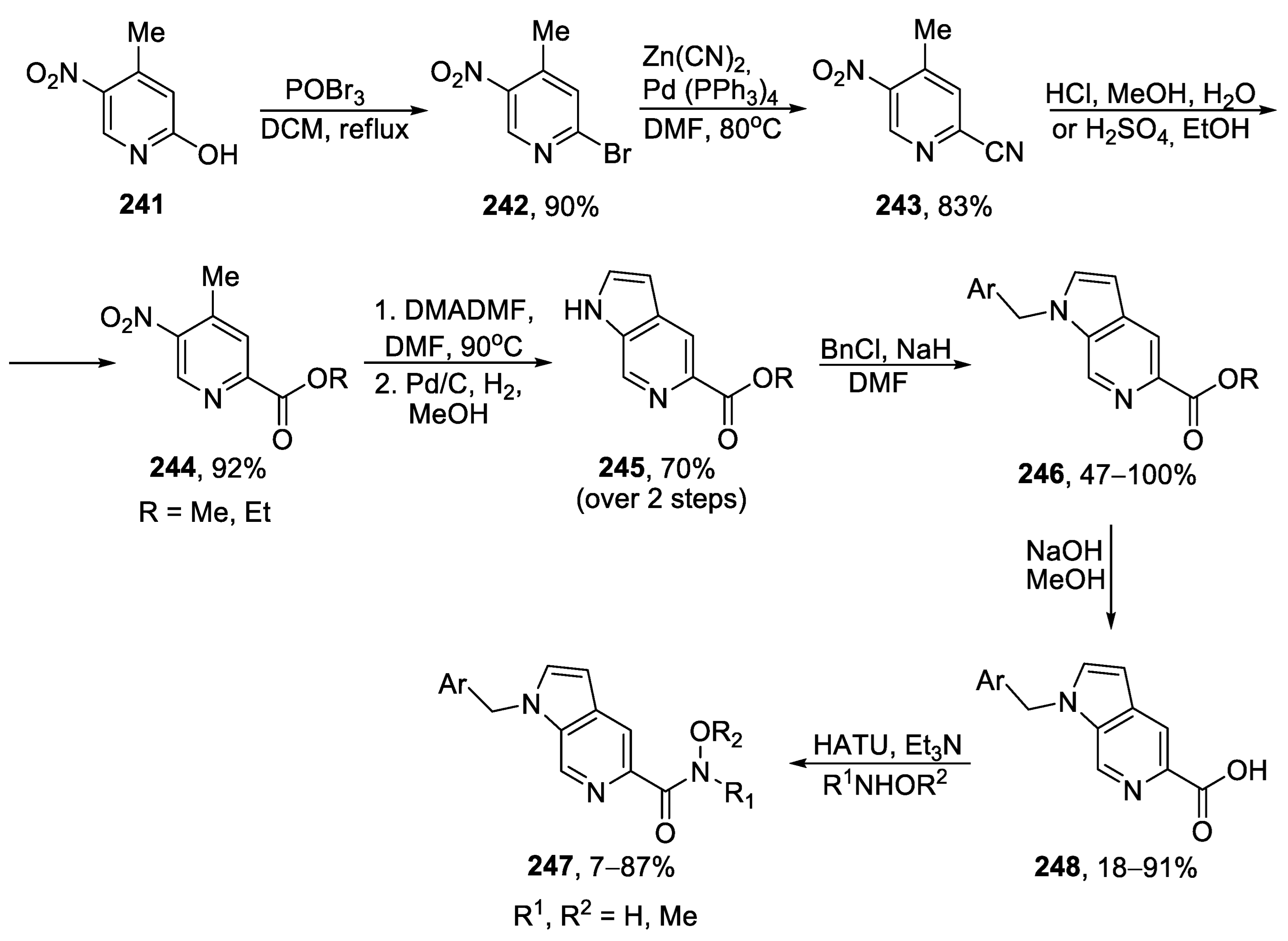{kind=link}
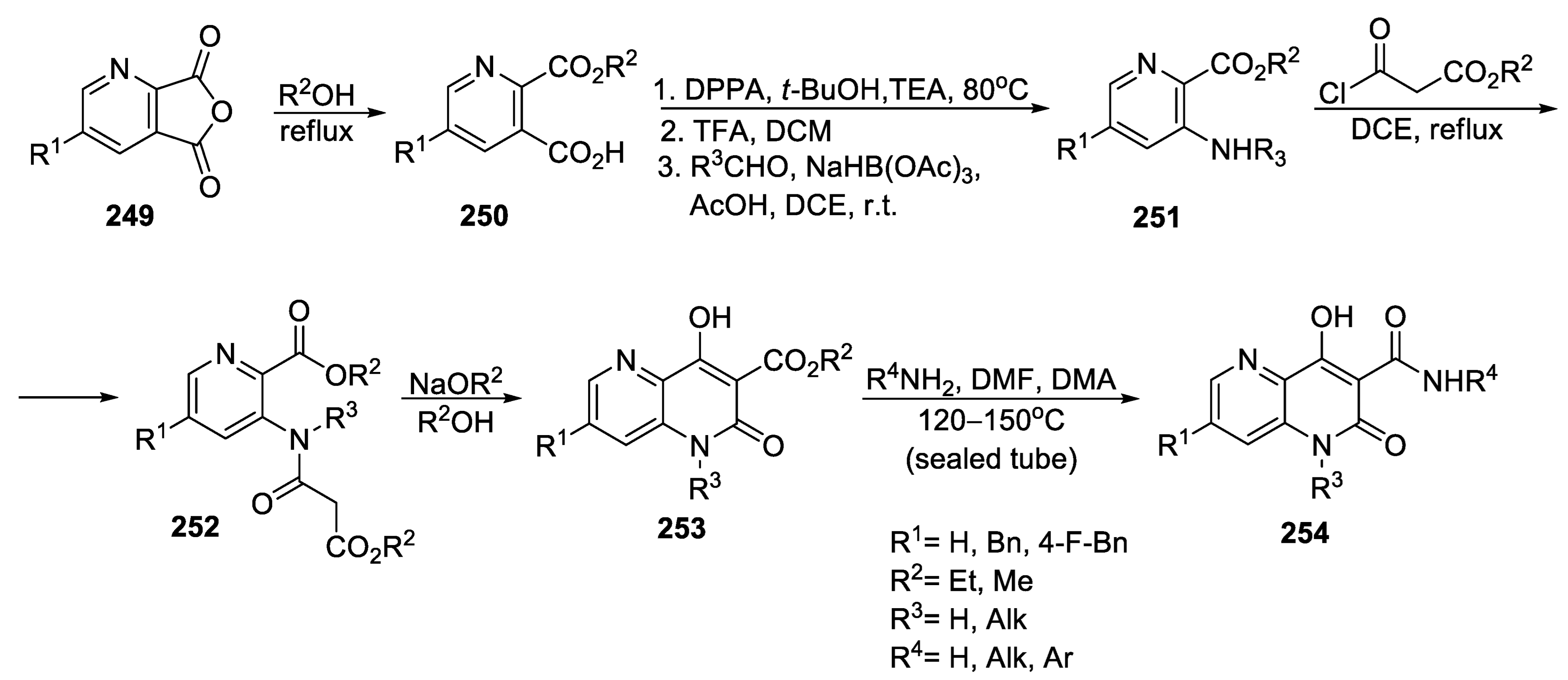{kind=link}
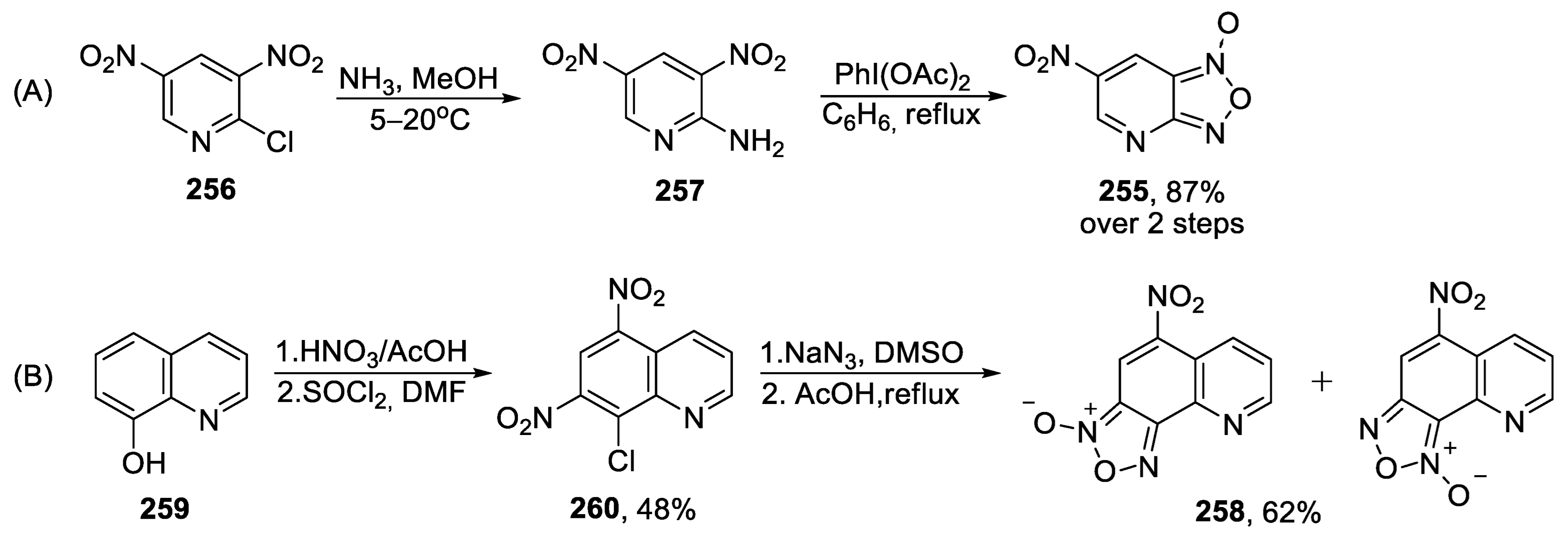{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Synthesis of Pyridine-Based HIV-1 Integrase Inhibitors
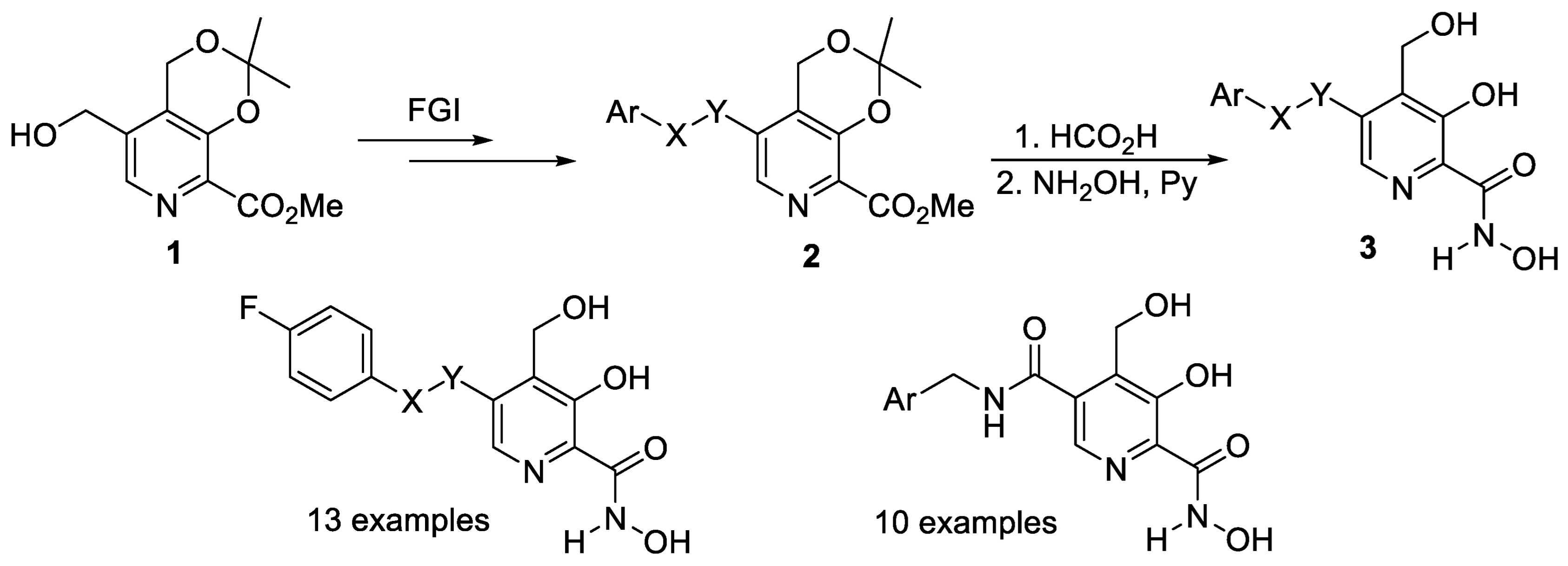
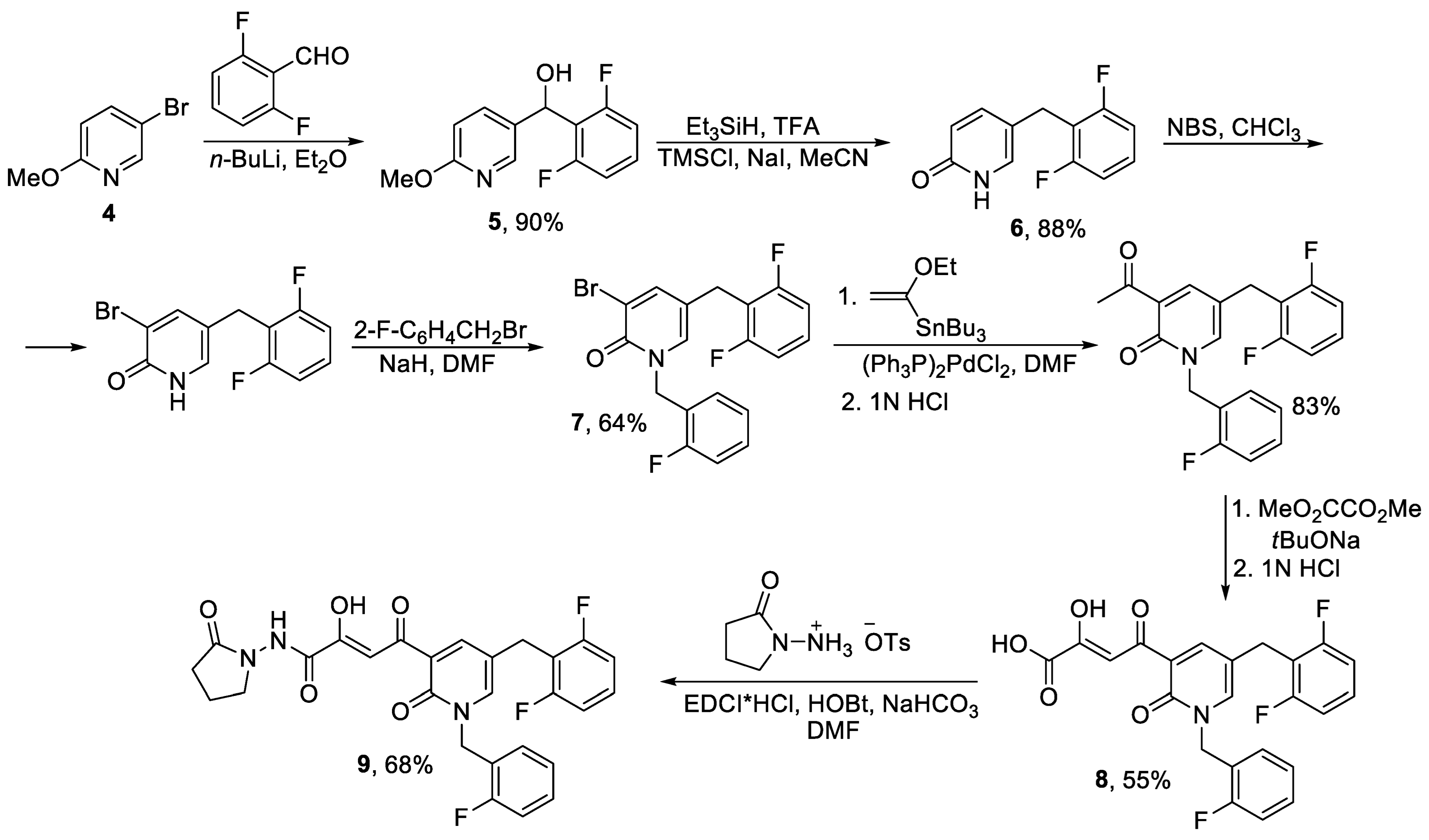
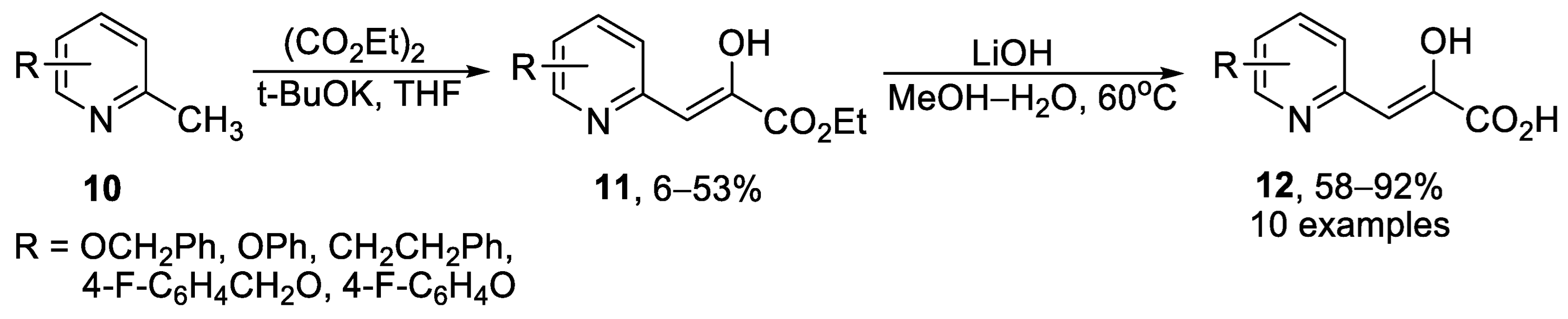
2.1. Monocyclic Pyridines
2.2. Quinolines and Isoquinolines
2.3. Pyridines Fused with Heterocycles
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AIDS | Acquired immunodeficiency syndrome |
| ARVT | Antiretroviral therapy |
| BIC | Bictegravir |
| BSA | Benzenesulfonic acid |
| CDI | 1,1′-Carbonyldiimidazole |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| DCE | 1,2-Dichloroethane |
| DCM | Dichloromethane |
| DEEMM | Diethyl ethoxymethylenemalonate |
| DFBA | 2,4-Difluorobenzyl amine |
| DIAD | Diisopropyl azodicarboxylate |
| DIPEA | N,N-Diisopropylethylamine |
| DMA | Dimethylacetamide |
| DMAD | Dimethyl acetylenedicarboxylate |
| DMADMF | Dimethylformamide dimethyl acetal |
| DMF | Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| DPPP | 1,3-Bis(diphenylphosphino)propane |
| DTG | Dolutegravir |
| EC50 | Half maximal effective concentration |
| EDC (EDCI) | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| EMME | Ethoxymethylenemalonic ester |
| EVG | Elvitegravir |
| FDA | Food and Drug Administration |
| FGI | Functional group interconversion |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate |
| HIV | Human immunodeficiency virus |
| HOBt | Hydroxybenzotriazole |
| IC50 | Half maximal inhibitory concentration |
| INI | Integrase inhibitor |
| MCPBA | Meta-chloroperoxybenzoic acid |
| NBS | N-Bromosuccinimide |
| NIS | N-Iodosuccinimide |
| NMM | N-Methylmorpholine |
| PIFA | Phenyliodine bis(trifluoroacetate) |
| PPA | Polyphosphoric acid |
| RAL | Raltegravir |
| TBHP | Tert-butyl hydroperoxide |
| TEA | Triethylamine |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
References
- FDA approves raltegravir tablets. AIDS Patient Care STDS 2007, 21, 889. [CrossRef]
- Cooper, D.A.; Steigbigel, R.T.; Gatell, J.M.; Rockstroh, J.K.; Katlama, C.; Yeni, P.; Lazzarin, A.; Clotet, B.; Kumar, P.N.; Eron, J.E.; et al. Subgroup and Resistance Analyses of Raltegravir for Resistant HIV-1 Infection. N. Engl. J. Med. 2008, 359, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Scarci, K.K.; Havens, J.P.; Podany, A.T.; Avedissian, S.N.; Fletcher, C.V. HIV-1 Integrase Inhibitors: A Comparative Review of Efficacy and Safety. Drugs 2020, 80, 1649–1676. [Google Scholar] [CrossRef] [PubMed]
- Jozwik, I.K.; Passos, D.O.; Lyumkis, D. Structural Biology of HIV Integrase Strand Transfer Inhibitors. Trends Pharmacol. Sci. 2020, 41, 611–626. [Google Scholar] [CrossRef] [PubMed]
- Hajimahdi, Z.; Zarghi, A. Progress in HIV-a Integrase Inhibitros: A review of their Chemical Structure Diversity. Iran J. Pharm. Res. 2016, 15, 595–628. [Google Scholar]
- Ingale, K.B.; Bhatia, M.S. HIV-1 Integrase Inhibitors: A Review of Their Chemical Development. Antivir. Chem. Chemother. 2011, 22, 95–105. [Google Scholar] [CrossRef]
- Trivedi, J.; Mahajan, D.; Jaffe, R.J.; Acharya, A.; Mitra, D.; Byrareddy, S.N. Recent Advances in the Development of Integrase Inhibitors for HIV Treatment. Curr. HIV/AIDS Rep. 2020, 17, 63–75. [Google Scholar] [CrossRef]
- Liman, W.; Ait Lahcen, N.; Oubahmane, M.; Hdoufane, I.; Cherqaoui, D.; Daoud, R.; El Allali, A. Hybrid Molecules as Potential Drugs for the Treatment of HIV: Design and Applications. Pharmaceuticals 2022, 15, 1092. [Google Scholar] [CrossRef]
- Zhou, J.; Hao, J.; Peng, L.; Duan, H.; Luo, Q.; Yan, H.; Wan, H.; Hu, Y.; Liang, L.; Xie, Z.; et al. Classification and Design of HIV-1 Integrase Inhibitors Based on Machine Learning. Comput. Math. Methods Med. 2021, 2021, 5559338. [Google Scholar] [CrossRef]
- Sala, M.; Spensiero, A.; Esposito, F.; Scala, M.C.; Vernieri, E.; Bertamino, A.; Manfra, M.; Carotenuto, A.; Grieco, P.; Novellino, E.; et al. Development and Identification of a Novel Anti-HIV-1 Peptide Derived by Modification of the N-Terminal Domain of HIV-1 Integrase. Front. Microbiol. 2016, 7, 845. [Google Scholar] [CrossRef]
- Nair, V.; Okello, M. Integrase Inhibitor Prodrugs: Approaches to Enhancing the Anti-HIV Activity of β-Diketo Acids. Molecules 2015, 20, 12623–12651. [Google Scholar] [CrossRef] [PubMed]
- Sawant, A.A.; Jadav, S.S.; Nayani, K.; Mainkar, P.S. Development of Synthetic Approaches Towards HIV Integrase Strand Transfer Inhibitors (INSTIs). Chemistryselect 2022, 7, e202201915. [Google Scholar] [CrossRef]
- Stranix, B.R.; Wu, J.J.; Milot, G.; Beaulieu, F.; Bouchard, J.-E.; Gouveia, K.; Forte, A.; Garde, S.; Wang, Z.; Mouscadet, J.-F.; et al. Pyridoxine hydroxamic acids as novel HIV-integrase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1233–1236. [Google Scholar] [CrossRef]
- Okello, M.; Nishonov, M.; Singh, P.; Mishra, S.; Mangu, N.; Seo, B.; Gund, M.; Nair, V. Approaches to the synthesis of a novel, anti-HIV active integrase inhibitor. Org. Biomol. Chem. 2013, 11, 7852–7858. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Okello, M.O.; Nishonov, A.A.; Mishra, S. Pyridinone Hydroxycyclopentyl Carboxamides: HIV Integrase Inhibitors with Therapeutic Applications. WO Patent 2011071849 A2, 16 June 2011. [Google Scholar]
- Okello, M.; Mishra, S.; Nishonov, M.; Nair, V. Notable difference in anti-HIV activity of integrase inhibitors as a consequence of geometric and enantiomeric configurations. Bioorg. Med. Chem. Lett. 2013, 23, 4112–4116. [Google Scholar] [CrossRef]
- Seo, B.I.; Uchil, V.R.; Okello, M.; Mishra, S.; Ma, X.-H.; Nishonov, M.; Shu, Q.; Chi, G.; Nair, V. Discovery of a Potent HIV Integrase Inhibitor That Leads to a Prodrug with Significant anti-HIV Activity. ACS Med. Chem. Lett. 2011, 2, 877–881. [Google Scholar] [CrossRef]
- Kawasuji, T.; Yoshinaga, T.; Sato, A.; Yodo, M.; Fujiwara, T.; Kiyama, R. A platform for designing HIV integrase inhibitors. Part 1: 2-Hydroxy-3-heteroaryl acrylic acid derivatives as novel HIV integrase inhibitor and modeling of hydrophilic and hydrophobic pharmacophores. Bioorg. Med. Chem. 2006, 14, 8430–8445. [Google Scholar] [CrossRef]
- Wang, Z.; Vince, R. Design and synthesis of dual inhibitors of HIV reverse transcriptase and integrase: Introducing a diketoacid functionality into delavirdine. Bioorg. Med. Chem. 2008, 16, 3587–3595. [Google Scholar] [CrossRef]
- Cavalluzzo, C.; Voet, A.; Christ, F.; Singh, B.K.; Sharma, A.; Debyser, Z.; De Maeyer, M.; Van der Eycken, E. De novo design of small molecule inhibitors targeting the LEDGF/p75-HIVintegrase interaction. RSC Adv. 2012, 2, 974–984. [Google Scholar] [CrossRef]
- Sugiyama, S.; Akiyama, T.; Taoda, Y.; Iwaki, T.; Matsuoka, E.; Akihisa, E.; Seki, T.; Yoshinaga, T.; Kawasuji, T. Discovery of novel HIV-1 integrase-LEDGF/p75 allosteric inhibitors based on a pyridine scaffold forming an intramolecular hydrogen bond. Bioorg. Med. Chem. Lett. 2021, 33, 127742. [Google Scholar] [CrossRef]
- Naidu, B.N.; Patel, M.; McAuliffe, B.; Ding, B.; Cianci, C.; Simmermacher, J.; Jenkins, S.; Parker, D.D.; Sivaprakasam, P.; Khan, J.A.; et al. Design, Synthesis, and Preclinical Profiling of GSK3739936 (BMS-986180), an Allosteric Inhibitor of HIV-1 Integrase with Broad-Spectrum Activity toward 124/125 Polymorphs. J. Med. Chem. 2022, 65, 4949–4971. [Google Scholar] [CrossRef] [PubMed]
- Kawasuji, T.; Johns, B.A.; Yoshida, H.; Taishi, T.; Taoda, Y.; Murai, H.; Kiyama, R.; Fuji, M.; Yoshinaga, T.; Seki, T.; et al. Carbamoyl Pyridone HIV-1 Integrase Inhibitors. 1. Molecular Design and Establishment of an Advanced Two-Metal Binding Pharmacophore. J. Med. Chem. 2012, 55, 8735–8744. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H. Carbamoylpyridone Derivative Having HIV Integrase Activity. EP Patent 1790638 A1, 30 May 2007. [Google Scholar]
- Rostami, M.; Sirous, H.; Zabihollahi, R.; Aghasadeghi, M.R.; Sadat, S.M.; Namazi, R.; Saghaie, L.; Memarian, H.R.; Fassihi, A. Design, synthesis and anti-HIV-1 evaluation of a series of 5-hydroxypyridine-4-one derivatives as possible integrase inhibitors. Med. Chem. Res. 2015, 24, 4113–4127. [Google Scholar] [CrossRef]
- Sirous, H.; Fassihi, A.; Brogi, S.; Campiani, G.; Christ, F.; Debyser, Z.; Gemma, S.; Butini, S.; Chemi, G.; Grillo, A.; et al. Synthesis, Molecular Modelling and Biological Studies of 3-hydroxypyrane-4-one and 3-hydroxy-pyridine-4-one Derivatives as HIV-1 Integrase Inhibitors. Med. Chem. 2019, 15, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Aramaki, H.; Yamashita, M.; Inoue, M.; Kawakami, H.; Shinkai, H.; Nakamura, H.; Matsuzaki, Y.; Wamaki, S. 6-(Heterocycle-substitited benzyl)-4-oxoquinoline Compound and Use of the Same as HIV Integrase Inhibitor. WO Patent 2007148780 A1, 27 December 2007. [Google Scholar]
- Dowdy, E.; Chen, X.; Pfeiffer, S. Process and Intermediates for Preparing Integrase Inhibitors. WO Patent 2008033836 A2, 20 March 2008. [Google Scholar]
- Hu, L.; Yan, S.; Luo, Z.; Han, X.; Wang, Y.; Wang, Z.; Zeng, C. Design, Practical Synthesis, and Biological Evaluation of Novel 6-(Pyrazolylmethyl)-4-quinoline-3-carboxylic Acid Derivatives as HIV-1 Integrase Inhibitors. Molecules 2012, 17, 10652–10666. [Google Scholar] [CrossRef]
- Deo, K.D.; Singhvi, I.J.; Patil, S.R.; Patil, A.V. Docking, Synthesis and Biological Evaluation of Novel Diketoquinoline Analogues as HIV-1 Integrase Inhibitor. Asian J. Chem. 2019, 31, 2000–2008. [Google Scholar] [CrossRef]
- Deo, K.D.; Singhvi, I.J.; Murugesan, S.; Vadnere, G.P.; Patil, A.V. Design, synthesis and biological evaluation of novel quinoline analogues as HIV-1 integrase inhibitor. Int. J. Pharm. Sci. Res. 2020, 11, 1210–1223. [Google Scholar] [CrossRef]
- He, Q.-Q.; Zhang, X.; Wu, H.-Q.; Gu, S.-X.; Ma, X.-D.; Yang, L.-M.; Zheng, Y.-T.; Chen, F.-E. Synthesis and biological evaluation of HQCAs with aryl or benzyl substituents on N-1 position as potential HIV-1 integrase inhibitors. Bioorg. Med. Chem. 2011, 19, 5553–5558. [Google Scholar] [CrossRef]
- He, Q.-Q.; Zhang, X.; Yang, L.-M.; Zheng, Y.-T.; Chen, F. Synthesis and biological evaluation of 5-fluoroquinolone-3-carboxylic acids as potential HIV-1 integrase inhibitors. J. Enzym. Inhib. Med. Chem. 2013, 28, 671–676. [Google Scholar] [CrossRef]
- Sato, M.; Kawakami, H.; Motomura, T.; Aramaki, H.; Matsuda, T.; Yamashita, M.; Ito, Y.; Matsuzaki, Y.; Yamataka, K.; Ikeda, S.; et al. Quinolone Carboxylic Acids as a Novel Monoketo Acid Class of Human Immunodeficiency Virus Type 1 Integrase Inhibitors. J. Med. Chem. 2009, 52, 4869–4882. [Google Scholar] [CrossRef]
- Velthuisen, E.J.; Johns, B.A.; Temelkoff, D.P.; Brown, K.W.; Danehower, S.C. The Design of 8-Hydroxyquinoline Tetracyclic Lactams as HIV-1 Integrase Strand Transfer Inhibitors. Eur. J. Med. Chem. 2016, 117, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Rong, J.; Mao, Z.; Wang, Y.; Zeng, C. 1-N-Substituted Benzyl-6-N′-substituent-2,3,6,9-tetralin-1H-[1,4] Benzoxazine [3,2-g] Quinolone-9-ketone-8-formic Acid Compound and Preparation Method and Application Thereof. CN Patent 104693216 A, 10 June 2015. [Google Scholar]
- Carcelli, M.; Bacchi, A.; Pelagatti, P.; Rispoli, G.; Rogolino, D.; Sanchez, T.W.; Sechi, M.; Neamati, N. Ruthenium arene complexes as HIV-1 integrase strand transfer inhibitors. J. Inorg. Biochem. 2013, 118, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, A.; Carcelli, M.; Compari, C.; Fisicaro, E.; Pala, N.; Rispoli, G.; Rogolino, D.; Sanchez, T.W.; Sechi, M.; Sinisi, V.; et al. Investigating the Role of Metal Chelation in HIV-1 Integrase Strand Transfer Inhibitors. J. Med. Chem. 2011, 54, 8407–8420. [Google Scholar] [CrossRef]
- Fandrick, K.R.; Li, W.; Zhang, Y.; Tang, W.; Gao, J.; Rodriguez, S.; Patel, N.D.; Reeves, D.C.; Wu, J.-P.; Sanyal, S.; et al. Concise and Practical Asymmetric Synthesis of a Challenging Atropisomeric HIV Integrase Inhibitor. Angew. Chem. Int. Ed. 2015, 54, 7144–7148. [Google Scholar] [CrossRef]
- Haddad, N.; Mangunuru, H.P.R.; Fandrick, K.R.; Qu, B.; Sieber, J.D.; Rodriguez, S.; Desrosiers, J.-N.; Patel, N.; Lee, H.; Kurouski, D.; et al. Reengineered BI-DIME Ligand Core Based on Computer Modeling to Increase Selectivity in Asymmetric Suzuki-Miyaura Coupling for the Challenging Axially Chiral HIV Integrase Inhibitor. Adv. Synth. Catal. 2016, 358, 3522–3527. [Google Scholar] [CrossRef]
- Brown, B.H.; Wang, X.; Fandrick, K.R.; Gao, J.J.; Haddad, N.; Landry, S.R.; Li, W.; Lu, Z.H.; Qu, B.; Reeves, D.C.; et al. Process for the Preparation of an HIV Integrase Inhibitor. U.S. Patent 2014094609 A1, 3 April 2014. [Google Scholar]
- Jentsch, N.G.; Hart, A.P.; Hume, J.D.; Sun, J.; McNeely, K.A.; Lama, C.; Pigza, J.A.; Donahue, M.J.; Kessl, J.J. Synthesis and Evaluation of Aryl Quinolines as HIV-1 Integrase Multimerization Inhibitors. ACS Med. Chem. Lett. 2018, 9, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Sekgota, K.C.; Majumder, S.; Isaacs, M.; Mnkandhla, D.; Hoppe, H.C.; Khanye, S.D.; Kriel, F.H.; Coates, J.; Kaye, P.T. Application of the Morita-Baylis-Hillman Reaction in the Synthesis of 3-[(N-Cycloalkylbenzamido)methyl]-2-quinolones as potential HIV-1 Integrase Inhibitors. Bioorg. Chem. 2017, 75, 310–316. [Google Scholar] [CrossRef]
- Corvaglia, V.; Carbajo, D.; Prabhakaran, P.; Ziach, K.; Mandal, P.K.; Dos Santos, V.; Legeay, C.; Vogel, R.; Parissi, V.; Pourquier, P.; et al. Carboxylate-functionalized foldamer inhibitors of HIV-1 integrase and Topoisomerase 1: Artificial analogues of DNA mimic proteins. Nucleic Acids Res. 2019, 47, 5511–5521. [Google Scholar] [CrossRef]
- Adams, M.M.; Bats, J.B.; Nikolaus, N.V.; Witvrouw, M.; Debyser, Z.; Engels, J.W. Microwave-assisted synthesis of fluoroquinolones and their nucleosides as inhibitors of HIV integrase. Collect. Czechoslov. Chem. Commun. 2006, 71, 978–990. [Google Scholar] [CrossRef]
- Polanski, J.; Niedbala, H.; Musiol, R.; Podeszwa, B.; Tabak, D.; Palka, A.; Mencel, A.; Finster, J.; Mouscadet, J.-F.; Le Bret, M. 5-Hydroxy-6-Quinaldic Acid as a Novel Molecular Scaffold for HIV-1 Integrase Inhibitors. Lett. Drug Des. Discov. 2006, 3, 175–178. [Google Scholar] [CrossRef]
- Mouscadet, J.-F.; Desmaële, D. Chemistry and Structure-Activity Relationship of the Styrylquinoline-Type HIV Integrase Inhibitors. Molecules 2010, 15, 3048–3078. [Google Scholar] [CrossRef] [PubMed]
- Sechi, M.; Rizzi, G.; Bacchi, A.; Carcelli, M.; Rogolino, D.; Pala, N.; Sanchez, T.W.; Taheri, L.; Dayam, R.; Neamati, N. Design and synthesis of novel dihydroquinoline-3-carboxylic acids as HIV-1 integrase inhibitors. Bioorg. Med. Chem. 2009, 17, 2925–2935. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Vince, R. Synthesis of pyrimidine and quinolone conjugates as a scaffold for dual inhibitors of HIV reverse transcriptase and integrase. Bioorg. Med. Chem. Lett. 2008, 18, 1293–1296. [Google Scholar] [CrossRef] [PubMed]
- Metobo, S.E.; Jin, H.; Tsiang, M.; Kim, C.U. Design, synthesis, and biological evaluation of novel tricyclic HIV-1 integrase inhibitors by modification of its pyridine ring. Bioorg. Med. Chem. Lett. 2006, 16, 3985–3988. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Cai, R.Z.; Schacherer, L.; Jabri, S.; Tsiang, M.; Fardis, M.; Chen, X.; Chen, J.M.; Kim, C.U. Design, synthesis, and SAR studies of novel and highly active tri-cyclic HIV integrase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 3989–3992. [Google Scholar] [CrossRef]
- Jin, H.; Wright, M.; Pastor, R.; Mish, M.; Metobo, S.; Jabri, S.; Lansdown, R.; Cai, R.; Pyun, P.; Tsiang, M.; et al. Tricyclic HIV integrase inhibitors: Potent and orally bioavailable C5-aza analogs. Bioorg. Med. Chem. Lett. 2008, 18, 1388–1391. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Fukuda, T.; Ishibashi, F.; Iwao, M. The first total synthesis of lamellarin a 20-sulfate, a selective inhibitor of HIV-1 integrase. Tetrahedron Lett. 2006, 47, 3755–3757. [Google Scholar] [CrossRef]
- Di Santo, R.; Costi, R.; Roux, A.; Miele, G.; Crucitti, G.C.; Iacovo, A.; Rosi, F.; Lavecchia, A.; Marinelli, L.; Giovanni, C.D.; et al. Novel Quinolinonyl Diketo Acid Derivatives as HIV-1 Integrase Inhibitors: Design, Synthesis, and Biological Activities. J. Med. Chem. 2008, 51, 4744–4750. [Google Scholar] [CrossRef]
- Tandon, V.; Urvashi; Yadav, P.; Sur, S.; Abbat, S.; Tiwari, V.; Hewer, R.; Papathanasopoulos, M.A.; Raja, R.; Banerjea, A.C.; et al. Design, Synthesis, and Biological Evaluation of 1,2-Dihydroisoquinolines as HIV-1 Integrase Inhibitors. ACS Med. Chem. Lett. 2015, 6, 1065–1070. [Google Scholar] [CrossRef]
- Wilson, T.A.; Koneru, P.C.; Rebensburg, S.V.; Lindenberger, J.J.; Kobe, M.J.; Cockroft, N.T.; Adu-Ampratwum, D.; Larue, R.C.; Kvaratskhelia, M.; Fuchs, J.R. An Isoquinoline Scaffold as a Novel Class of Allosteric HIV-1 Integrase Inhibitors. ACS Med. Chem. Lett. 2019, 10, 215–220. [Google Scholar] [CrossRef]
- Reddy, A.G.K.; Satyanarayana, G. A simple efficient sequential one-pot intermolecular aza-Michael addition and intramolecular Buchwald–Hartwig arylation of amines: Synthesis of functionalized tetrahydroisoquinolines. Tetrahedron 2012, 68, 8003–8010. [Google Scholar] [CrossRef]
- George, A.; Reddy, A.G.K.; Satyanarayana, G.; Raghavendra, N.K. 1,2,3,4-Tetrahydroisoquinolines as inhibitors of HIV-1 integrase and human LEDGF/p75 interaction. Chem. Biol. Drug Des. 2018, 91, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Billamboz, M.; Bailly, F.; Barreca, M.L.; De Luca, L.; Mouscadet, J.-F.; Calmels, C.; Andréola, M.-L.; Witvrouw, M.; Christ, F.; Debyser, Z.; et al. Design, Synthesis, and Biological Evaluation of a Series of 2-Hydroxyisoquinoline-1,3(2H,4H)-diones as Dual Inhibitors of Human Immunodeficiency Virus Type 1 Integrase and the Reverse Transcriptase RNase H Domain. J. Med. Chem. 2008, 51, 7717–7730. [Google Scholar] [CrossRef] [PubMed]
- Pankaj, W.; Priti, J.; Jadhav, H.R.; Santosh, R. 4-Substituted Benzylideneisoquinoline-1,3(2H,4H)-dione Derivatives: Synthesis and Biological Evaluation as Potential HIV-1 Integrase Inhibitors. Der Pharm. Lett. 2018, 10, 18–31. [Google Scholar]
- Billamboz, M.; Bailly, F.; Lion, C.; Calmels, C.; Andréola, M.-L.; Witvrouw, M.; Christ, F.; Debyser, Z.; De Luca, L.; Chimirri, A.; et al. 2-Hydroxyisoquinoline-1,3(2H,4H)-diones as inhibitors of HIV-1 integrase and reverse transcriptase RNase H domain: Influence of the alkylation of position 4. Eur. J. Med. Chem. 2011, 46, 535–546. [Google Scholar] [CrossRef]
- Hughes, D.L. Review of Synthetic Routes and Final Forms of Integrase Inhibitors Dolutegravir, Cabotegravir, and Bictegravir. Org. Process Res. Dev. 2019, 23, 716–729. [Google Scholar] [CrossRef]
- Vellanki, S.P.; Nadella, M.; Bhalme, M.; Ramabhotla, R.S.; Arumalla, V.S.R.; Kilaru, R.B. Process for the Preparation of Dolutegravir. WO Patent 2016/125192 A2, 11 August 2016. [Google Scholar]
- Ziegler, R.E.; Desai, B.K.; Jee, J.-A.; Gupton, B.F.; Roper, T.D.; Jamison, T.F. 7-Step Flow Synthesis of the HIV Integrase Inhibitor Dolutegravir. Angew. Chem. Int. Ed. 2018, 57, 7181–7185. [Google Scholar] [CrossRef]
- Yasukata, T.; Masui, M.; Ikarashi, F.; Okamoto, K.; Kurita, T.; Nagai, M.; Sugata, Y.; Miyake, N.; Hara, S.; Adachi, Y.; et al. Practical Synthetic Method for the Preparation of Pyrone Diesters: An Efficient Synthetic Route for the Synthesis of Dolutegravir Sodium. Org. Process Res. Dev. 2019, 23, 565–570. [Google Scholar] [CrossRef]
- Aoyama, Y.; Hakogi, T.; Fukui, Y.; Yamada, D.; Ooyama, T.; Nishino, Y.; Shinomoto, S.; Nagai, M.; Miyake, N.; Taoda, Y.; et al. Practical and Scalable Synthetic Method for Preparation of Dolutegravir Sodium: Improvement of a Synthetic Route for Large-Scale Synthesis. Org. Process Res. Dev. 2019, 23, 558–564. [Google Scholar] [CrossRef]
- Johns, B.A.; Kawasuji, T.; Weatherhead, J.G.; Taishi, T.; Temelkoff, D.P.; Yoshida, H.; Akiyama, T.; Taoda, Y.; Murai, H.; Kiyama, R.; et al. Carbamoyl Pyridone HIV-1 Integrase Inhibitors 3. A Diastereomeric Approach to Chiral Nonracemic Tricyclic Ring Systems and the Discovery of Dolutegravir (S/GSK1349572) and (S/GSK1265744). J. Med. Chem. 2015, 56, 5901–5916. [Google Scholar] [CrossRef]
- Ramanathan, S.; Mathias, A.A.; German, P.; Kearney, B.P. Clinical pharmacokinetic and pharmacodynamic profile of the HIV integrase inhibitor elvitegravir. Clin. Pharmacokinet. 2011, 50, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Dietz, J.-P.; Lucas, T.; Groß, J.; Seitel, S.; Brauer, J.; Ferenc, D.; Gupton, B.F.; Opatz, T. Six-Step Gram-Scale Synthesis of the Human Immunodeficiency Virus Integrase Inhibitor Dolutegravir Sodium. Org. Process Res. Dev. 2021, 25, 1898–1910. [Google Scholar] [CrossRef]
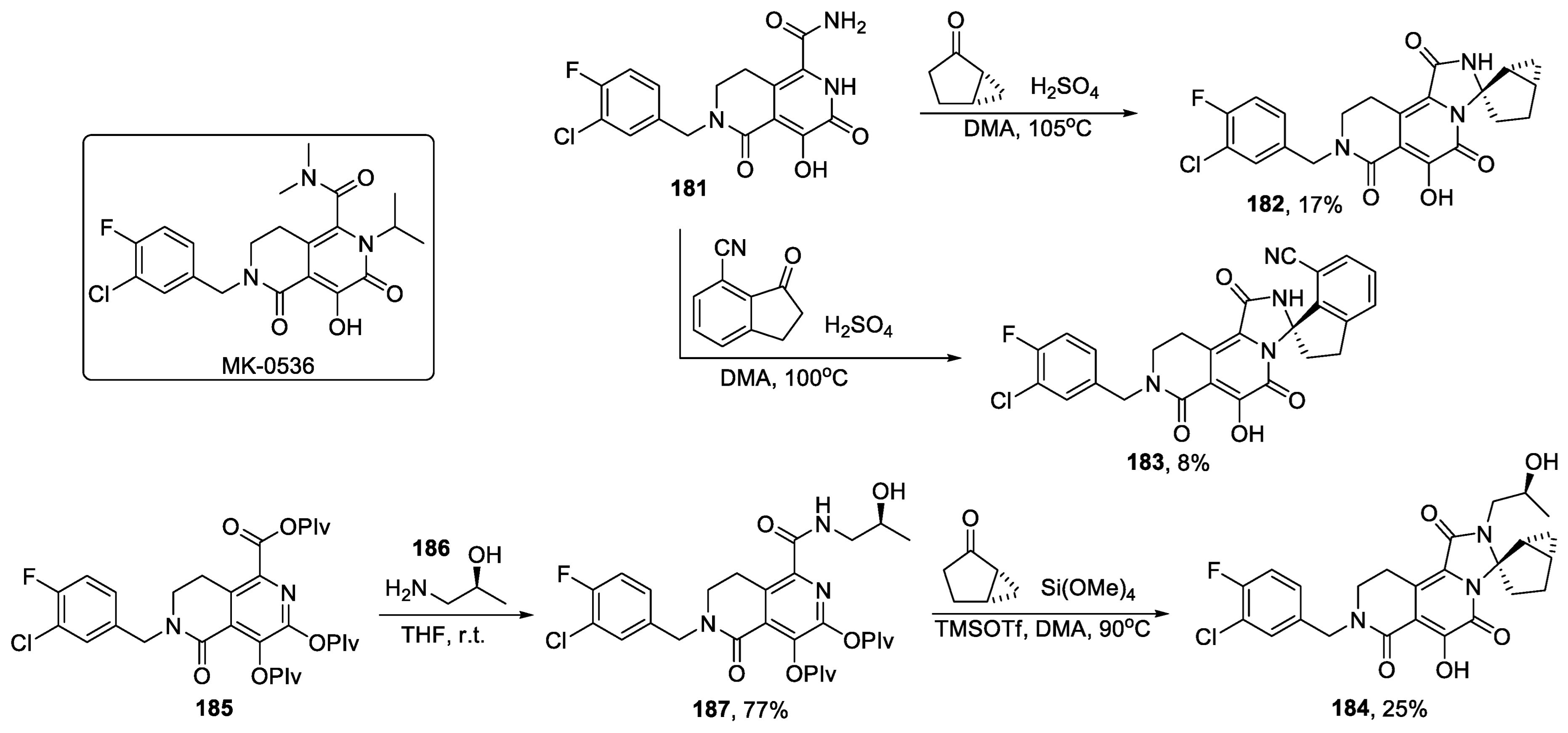
- Schreier, J.D.; Embrey, M.W.; Raheem, I.T.; Barbe, G.; Campeau, L.-C.; Dubost, D.; McCabe Dunn, J.; Grobler, J.; Hartingh, T.J.; Hazuda, D.J.; et al. Discovery and optimization of 2-pyridinone aminal integrase strand transfer inhibitors for the treatment of HIV. Bioorg. Med. Chem. Lett. 2017, 27, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Gao, P.; Dong, G.; Zhang, X.; Cheng, X.; Ding, X.; Wang, X.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; et al. 5-Hydroxypyrido[2,3-b]pyrazin-6(5H)-One Derivatives as Novel Dual Inhibitors of HIV-1 Reverse Transcriptase-Associated Ribonuclease H and Integrase. Eur. J. Med. Chem. 2018, 155, 714–724. [Google Scholar] [CrossRef]
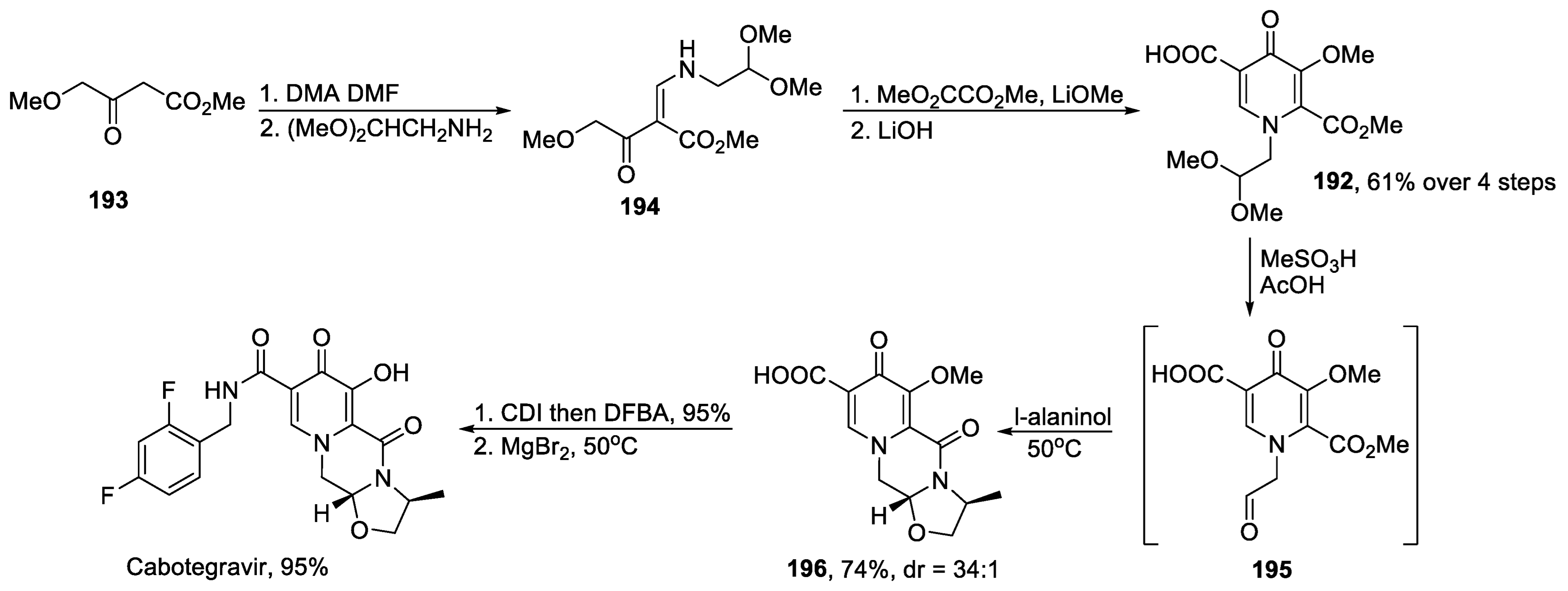
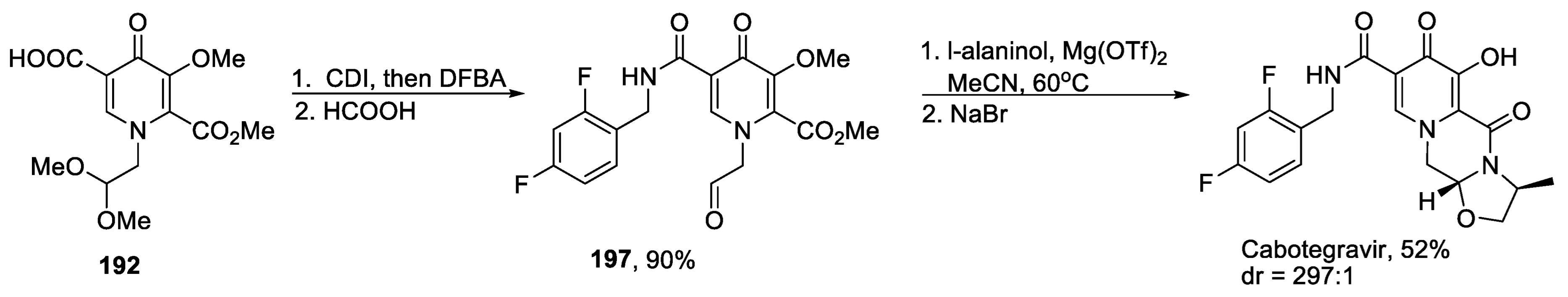
- Wang, H.; Kowalski, M.D.; Lakdawala, A.S.; Vogt, F.G.; Wu, L. An Efficient and Highly Diastereoselective Synthesis of GSK1265744, a Potent HIV Integrase Inhibitor. Org. Lett. 2015, 17, 564–567. [Google Scholar] [CrossRef]
- Cao, Y.; Xie, Y.; Nie, W.; Fu, C.; Hong, Y.; Lei, H. Alkynyl-Coupled Difluorophenylamino Pyridone HIV Integrase Inhibitor as Well as Preparation Method and Application Thereof. CN Patent 114149449 A, 8 March 2022. [Google Scholar]
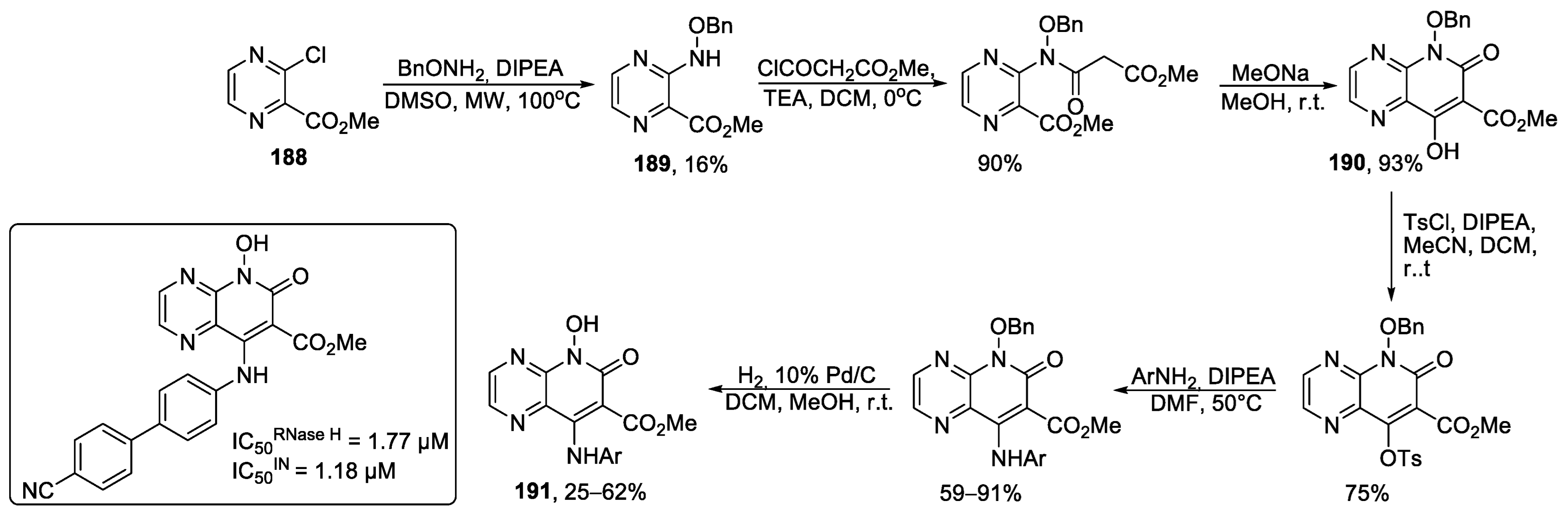
- Kuethe, J.T.; Humphrey, G.R.; Journet, M.; Peng, Z.; Childers, K.G. Asymmetric Synthesis of a Potent HIV-1 Integrase Inhibitor. J. Org. Chem. 2016, 81, 10256–10265. [Google Scholar] [CrossRef]
- Vacca, J.P.; Wai, J.S.; Payne, L.S.; Isaacs, R.C.A.; Han, W.; Egbertson, M.; Pracitto, R. HIV Integrase Inhibitors. PCT International Application. WO Patent 2006121831 A2, 16 November 2006. [Google Scholar]
- Zhao, X.Z.; Smith, S.J.; Métifiot, M.; Johnson, B.C.; Marchand, C.; Pommier, Y.; Hughes, S.H.; Burke, T.R., Jr. Bicyclic 1-Hydroxy-2-oxo-1,2-dihydropyridine-3-carboxamide-Containing HIV-1 Integrase Inhibitors Having High Antiviral Potency against Cells Harboring Raltegravir-Resistant Integrase Mutants. J. Med. Chem. 2014, 57, 1573–1582. [Google Scholar] [CrossRef]
- Zhao, X.Z.; Smith, S.J.; Maskell, D.P.; Métifiot, M.; Pye, V.E.; Fesen, K.; Marchand, C.; Pommier, Y.; Cherepanov, P.; Hughes, S.H.; et al. Structure-Guided Optimization of HIV Integrase Strand Transfer Inhibitors. J. Med. Chem. 2017, 60, 7315–7332. [Google Scholar] [CrossRef]
- Hu, L.; Ju, L.; Mao, Z.; Li, Z.; Zeng, C. 6-Chlorine-N-(substituted benzyl)-1H-pyrrolo[2,3-b]pyridine-2-amide Compound and Preparation Method as Well as Application Thereof. CN Patent 105294688 A, 3 February 2016. [Google Scholar]
- Hu, L.; Ju, L.; Mao, Z.; Li, Z.; Zeng, C. HIV-1 Integrase Inhibitor Compound as Well as Preparation Method and Application Thereof. CN Patent 105348282 A, 24 February 2016. [Google Scholar]
- Lee, S.U.; Park, J.H.; Kwon, T.H.; Yoo, Y.J.; Lee, J.Y.; Shin, C.G.; Yoo, K.H.; Lee, Y.S. Synthesis and HIV-1 Integrase Inhibitory Activities of 4-Hydroxy-5-azacoumarin 3-Carboxamides. Bull. Korean Chem. Soc. 2007, 28, 1510–1514. [Google Scholar] [CrossRef]
- Zhao, X.Z.; Maddali, K.; Metifiot, M.; Smith, S.J.; Vu, B.C.; Marchand, C.; Hughes, S.H.; Pommier, Y.; Burke, T.R., Jr. Bicyclic Hydroxy-1H-pyrrolopyridine-trione Containing HIV-1 Integrase Inhibitors. Chem. Biol. Drug Des. 2011, 79, 157–165. [Google Scholar] [CrossRef]
- Plewe, M.B.; Butler, S.L.; Dress, K.R.; Hu, O.; Johnson, T.W.; Kuehler, J.E.; Kuki, A.; Lam, H.; Liu, W.; Nowlin, D.; et al. Azaindole Hydroxamic Acids are Potent HIV-1 Integrase Inhibitors. J. Med. Chem. 2009, 52, 7211–7219. [Google Scholar] [CrossRef] [PubMed]
- Boros, E.E.; Edwards, C.E.; Foster, S.A.; Fuji, M.; Fujiwara, T.; Garvey, E.P.; Golden, P.L.; Hazen, R.J.; Jeffrey, J.L.; Johns, B.A.; et al. Synthesis and Antiviral Activity of 7-Benzyl-4-hydroxy-1,5-naphthyridin-2(1H)-one HIV Integrase Inhibitors. J. Med. Chem. 2009, 52, 2754–2761. [Google Scholar] [CrossRef] [PubMed]
- Johns, B.A.; Kawasuji, T.; Weatherhead, J.G.; Boros, E.E.; Thompson, J.B.; Garvey, E.P.; Foster, S.A.; Jeffrey, J.L.; Miller, W.H.; Kurose, N.; et al. Combining symmetry elements results in potent naphthyridinone (NTD) HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 6461–6464. [Google Scholar] [CrossRef] [PubMed]
- Korolev, S.P.; Pustovarova, M.A.; Starosotnikov, A.M.; Bastrakov, M.A.; Agapkina, Y.Y.; Shevelev, S.A.; Gottikh, M.B. Nitrobenzofuroxane Derivatives as Dual Action HIV-1 Inhibitors. Biochem. Mosc. Suppl. Ser. B Biomed. Chem. 2017, 11, 286–290. [Google Scholar] [CrossRef]
- Starosotnikov, A.M.; Shkaev, D.V.; Bastrakov, M.A.; Fedyanin, I.V.; Shevelev, S.A.; Dalinger, I.L. Nucleophilic dearomatization of 4-aza-6-nitrobenzofuroxan by CH acids in the synthesis of pharmacology-oriented compounds. Beilstein J. Org. Chem. 2017, 13, 2854–2861. [Google Scholar] [CrossRef]
- Bastrakov, M.A.; Starosotnikov, A.M.; Fedyanin, I.V.; Kachala, V.V.; Shevelev, S.A. 5-Nitro-7,8-furoxanoquinoline: A new type of fused nitroarenes possessing Diels–Alder reactivity. Mendeleev Commun. 2014, 24, 203–205. [Google Scholar] [CrossRef]
- Starosotnikov, A.M.; Nikol’skiy, V.V.; Borodulya, A.N.; Kachala, V.V.; Bastrakov, M.A.; Solkan, V.N.; Shevelev, S.A. Synthesis and Functionalization of 5,7-Dinitroquinoline and Its N-Oxide. Asian J. Org. Chem. 2016, 5, 685–690. [Google Scholar] [CrossRef]
- Platts, M.Y.; Barber, C.G.; Chiva, G.-Y.; Eastwood, R.L.; Fenwick, D.R.; Paradowski, K.A.; Blakemore, D.C. A concise synthesis of HIV integrase inhibitors bearing the dipyridone acid motif. Tetrahedron Lett. 2011, 52, 512–514. [Google Scholar] [CrossRef]
- Kinzel, O.D.; Monteagudo, E.; Muraglia, E.; Orvieto, F.; Pescatore, G.; Rosario Rico Ferreira, M.D.; Rowley, M.; Summa, V. The synthesis of tetrahydropyridopyrimidones as a new scaffold for HIV-1 integrase inhibitors. Tetrahedron Lett. 2007, 48, 6552–6555. [Google Scholar] [CrossRef]
- Kinzel, O.D.; Ball, R.G.; Donghi, M.; Maguire, C.K.; Muraglia, E.; Pesci, S.; Rowley, M.; Summa, V. 3-Hydroxy-4-oxo-4H-pyrido[1,2-a]pyrimidine-2-carboxylates—Fast access to a heterocyclic scaffold for HIV-1 integrase inhibitors. Tetrahedron Lett. 2008, 49, 6556–6558. [Google Scholar] [CrossRef]
- Johns, B.A.; Weatherhead, J.G.; Allen, S.H.; Thompson, J.B.; Garvey, E.P.; Foster, S.A.; Jeffrey, J.L.; Miller, W.H. The use of oxadiazole and triazole substituted naphthyridines as HIV-1 integrase inhibitors. Part 1: Establishing the pharmacophore. Bioorg. Med. Chem. Lett. 2009, 19, 1802–1806. [Google Scholar] [CrossRef] [PubMed]
- Le, G.; Vandegraaff, N.; Rhodes, D.I.; Jones, E.D.; Coates, J.A.V.; Thienthong, N.; Winfield, L.J.; Lu, L.; Li, X.; Yu, C.; et al. Design of a series of bicyclic HIV-1 integrase inhibitors. Part 2: Azoles: Effective metal chelators. Bioorg. Med. Chem. Lett. 2010, 20, 5909–5912. [Google Scholar] [CrossRef] [PubMed]


























































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Starosotnikov, A.M.; Bastrakov, M.A. Recent Developments in the Synthesis of HIV-1 Integrase Strand Transfer Inhibitors Incorporating Pyridine Moiety. Int. J. Mol. Sci. 2023, 24, 9314. https://doi.org/10.3390/ijms24119314
Starosotnikov AM, Bastrakov MA. Recent Developments in the Synthesis of HIV-1 Integrase Strand Transfer Inhibitors Incorporating Pyridine Moiety. International Journal of Molecular Sciences. 2023; 24(11):9314. https://doi.org/10.3390/ijms24119314
Chicago/Turabian StyleStarosotnikov, Alexey M., and Maxim A. Bastrakov. 2023. "Recent Developments in the Synthesis of HIV-1 Integrase Strand Transfer Inhibitors Incorporating Pyridine Moiety" International Journal of Molecular Sciences 24, no. 11: 9314. https://doi.org/10.3390/ijms24119314
APA StyleStarosotnikov, A. M., & Bastrakov, M. A. (2023). Recent Developments in the Synthesis of HIV-1 Integrase Strand Transfer Inhibitors Incorporating Pyridine Moiety. International Journal of Molecular Sciences, 24(11), 9314. https://doi.org/10.3390/ijms24119314