
VE-Cadherin in Cancer-Associated Angiogenesis: A Deceptive Strategy of Blood Vessel Formation
 , , and
, , and 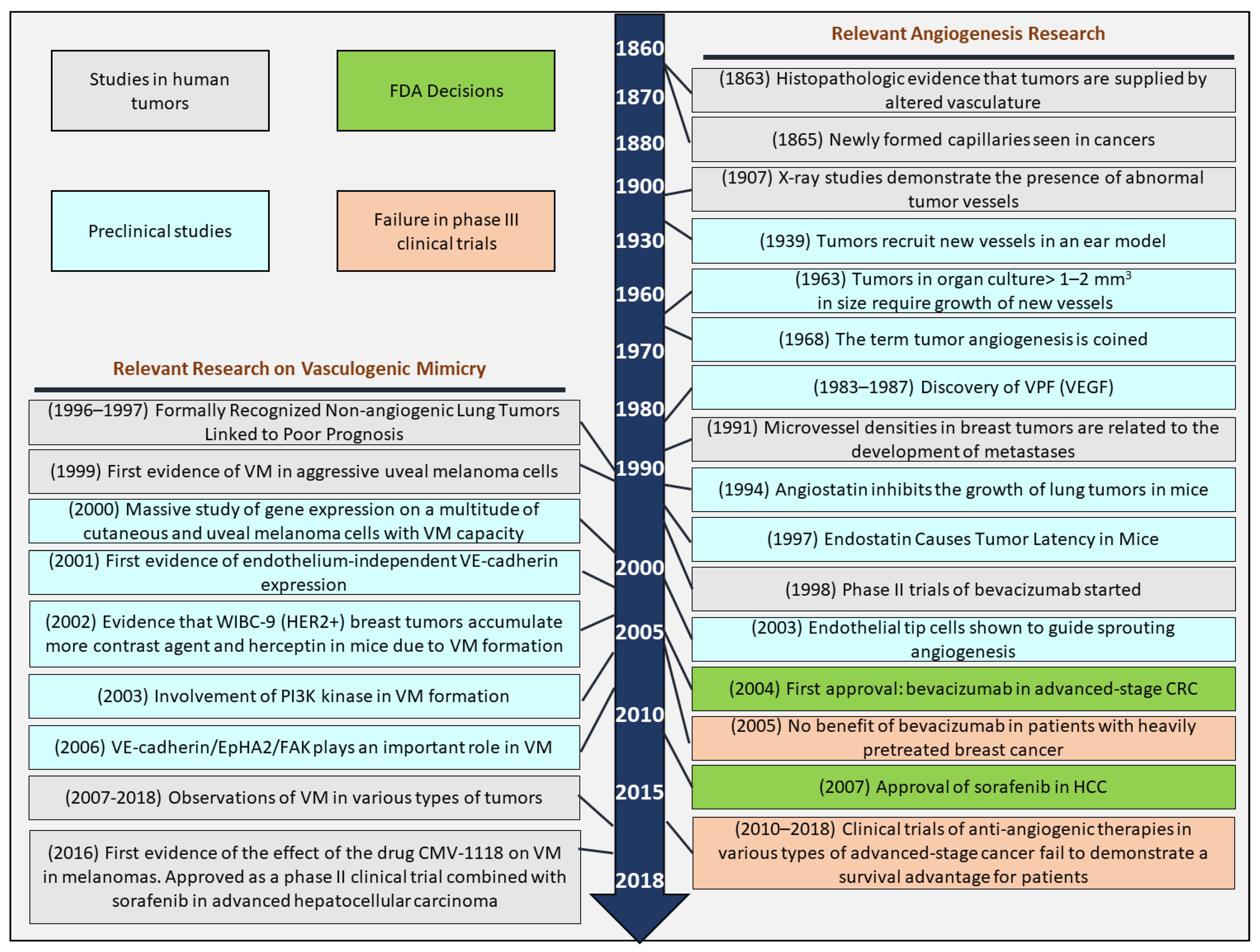{kind=link}
{kind=link}
{kind=link}
Abstract
:1. A Glance at Angiogenesis Past
2. Facts
- There is a correlation between VM and high tumor grade, cancer cell invasion, cancer cell metastasis, and reduced survival of cancer patients;
- VE-cadherin, in conjunction with focal adhesion kinase (FAK) activity and tethered β-catenin, serves as a regulator of the tumor microenvironment, promoting the formation of VM;
- Anti-angiogenic therapy fails in several types of cancer in overall survival;
- FAK plays a pivotal role in the dynamics of cell permeabilization in the metastatic tumor endothelium.
3. Mechanism of Vascularization in Cancer: Endothelial Sprouting
- Angiogenic stimuli can cause the local degradation of the basement membrane on the side of the post-capillary venule that is dilated and located closer to the tumor. This can weaken the inter-endothelial contacts, and the endothelial cells (ECs) may migrate into the surrounding connective tissue;
- A solid bead is then formed by the ECs that accomplish each other in a bipolar manner;
- The formation of blood vessels occurs through the curvature of one or more ECs in conjunction with forming a new basement membrane and recruiting the pericytic or mural cells.
- Structural alteration of the basement membrane due to a loss of electron density in almost the entire area of the dilated “original vessel”. However, immunohistochemical techniques can still locate basement membrane components such as laminin and collagen IV. The impaired basement membrane’s partially regulated degradation occurs only at sites where endothelial cell processes, connected by gap junctions, project into the surrounding connective tissue;
- The migration of ECs, which maintain their basal-luminal polarity and form a slit-like lumen in parallel with the lumen of the main vessel, takes place continuously and is stamped by intact inter-endothelial junctions. The low electron density basement membrane is continuously deposited by polarized ECs, while only the tip of the growing capillary is free of basement membrane material;
- Pericytes [27] from the mother vessel relocate along the basement membrane of the capillary sprout, resulting in complete coverage of the new vessel. Similarly, the appearance of an electron-dense basement membrane can be observed around the mature capillary buds. In contrast to the previous model, this model suggests that the loss of endothelial cell polarity is not a necessary stimulus for the induction of lumen formation.
4. Mechanism of Vascularization in Cancer: Vessel Co-Option
5. Mechanism of Vascularization in Cancer: Vasculogenic Mimicry
6. Vasculogenic Mimicry (VM): Functionality of Tumor-Mimicked Vessels in Cancer
7. Vasculogenic Mimicry (VM): Intracellular Signaling
8. Vasculogenic Mimicry (VM): Intracellular Signaling, Hypoxia, and Tumor Microenvironments
9. Vasculogenic Mimicry (VM): Intracellular Signaling, Focus on Non-Endothelial VE-Cadherin
10. Concluding Remarks
11. Open Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lenzi, P.; Bocci, G.; Natale, G. John Hunter and the origin of the term “angiogenesis”. Angiogenesis 2016, 19, 255–256. [Google Scholar] [CrossRef]
- Natale, G.; Bocci, G.; Lenzi, P. Looking for the Word “Angiogenesis” in the History of Health Sciences: From Ancient Times to the First Decades of the Twentieth Century. World J. Surg. 2017, 41, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef]
- Döme, B.; Hendrix, M.J.C.; Paku, S.; Tóvári, J.; Tímár, J. Alternative vascularization mechanisms in cancer: Pathology and therapeutic implications. Am. J. Pathol. 2007, 170, 1–15. [Google Scholar] [CrossRef]
- Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Bilecz, A.; Daley, F.; Kostaras, E.; Nathan, M.R.; Wan, E.; Frentzas, S.; Schweiger, T.; et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J. Pathol. 2017, 241, 362–374. [Google Scholar] [CrossRef]
- Delgado-Bellido, D.; Serrano-Saenz, S.; Fernández-Cortés, M.; Oliver, F.J. Vasculogenic mimicry signaling revisited: Focus on non-vascular VE-cadherin. Mol. Cancer 2017, 16, 65. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007, 6, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, L.M.; Parris, E.E.; Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Pezzella, F.; Di Bacco, A.D.; Andreola, S.; Nicholson, A.G.; Pastorino, U.; Harris, A.L. Angiogenesis in primary lung cancer and lung secondaries. Eur. J. Cancer Part A 1996, 32, 2494–2500. [Google Scholar] [CrossRef]
- Pezzella, F.; Pastorino, U.; Tagliabue, E.; Andreola, S.; Sozzi, G.; Gasparini, G.; Menard, S.; Gatter, K.C.; Harris, A.L.; Fox, S.; et al. Non-small-cell lung carcinoma tumor growth without morphological evidence of neo-angiogenesis. Am. J. Pathol. 1997, 151, 1417. [Google Scholar]
- Ebos, J.M.L.; Kerbel, R.S. Antiangiogenic therapy: Impact on invasion, disease progression, and metastasis. Nat. Rev. Clin. Oncol. 2011, 8, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Sánchez-Gastaldo, A.; Kempf, E.; González del Alba, A.; Duran, I. Systemic treatment of renal cell cancer: A comprehensive review. Cancer Treat. Rev. 2017, 60, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Tabernero, J.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.E.; Portnoy, D.C.; Van Cutsem, E.; Grothey, A.; et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): A randomised, double-blin. Lancet Oncol. 2015, 16, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Kindler, H.L.; Niedzwiecki, D.; Hollis, D.; Sutherland, S.; Schrag, D.; Hurwitz, H.; Innocenti, F.; Mulcahy, M.F.; O’Reilly, E.; Wozniak, T.F.; et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: Phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J. Clin. Oncol. 2010, 28, 3617–3622. [Google Scholar] [CrossRef]
- Miller, K.D.; Chap, L.I.; Holmes, F.A.; Cobleigh, M.A.; Marcom, P.K.; Fehrenbacher, L.; Dickler, M.; Overmoyer, B.A.; Reimann, J.D.; Sing, A.P.; et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J. Clin. Oncol. 2005, 23, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Lee, S.J.; Zhao, F.; Schuchter, L.M.; Flaherty, L.; Kefford, R.; Atkins, M.B.; Leming, P.; Kirkwood, J.M. Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma. J. Clin. Oncol. 2013, 31, 373–379. [Google Scholar] [CrossRef]
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 2007, 357, 2666–2676. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.W.; Chan, A.; Dirix, L.Y.; Cortés, J.; Pivot, X.; Tomczak, P.; Delozier, T.; Sohn, J.H.; Provencher, L.; Puglisi, F.; et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J. Clin. Oncol. 2010, 28, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- Ausprunk, D.H.; Folkman, J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc. Res. 1977, 14, 53–65. [Google Scholar] [CrossRef]
- Paku, S.; Paweletz, N. First steps of tumor-related angiogenesis. Lab. Investig. 1991, 65, 334–346. [Google Scholar]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Brown, L.F.; Detmar, M.; Dvorak, A.M. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 1995, 146, 1029–1039. [Google Scholar]
- Saharinen, P.; Eklund, L.; Alitalo, K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat. Rev. Drug Discov. 2017, 16, 635–661. [Google Scholar] [CrossRef]
- van Meeteren, L.A.; Goumans, M.-J.; ten Dijke, P. TGF-β Receptor Signaling Pathways in Angiogenesis; Emerging Targets for Anti-Angiogenesis Therapy. Curr. Pharm. Biotechnol. 2011, 12, 2108–2120. [Google Scholar] [CrossRef]
- Serini, G.; Valdembri, D.; Bussolino, F. Integrins and angiogenesis: A sticky business. Exp. Cell Res. 2006, 312, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Latacz, E.; Caspani, E.; Barnhill, R.; Lugassy, C.; Verhoef, C.; Grünhagen, D.; Van Laere, S.; Fernández Moro, C.; Gerling, M.; Dirix, M.; et al. Pathological features of vessel co-option versus sprouting angiogenesis. Angiogenesis 2020, 23, 43–54. [Google Scholar] [CrossRef]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef]
- Dme, B.; Paku, S.; Somlai, B.; Tmr, J. Vascularization of cutaneous melanoma involves vessel co-option and has clinical significance. J. Pathol. 2002, 197, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Vasudev, N.S.; Reynolds, A.R. Anti-angiogenic therapy for cancer: Current progress, unresolved questions and future directions. Angiogenesis 2014, 17, 471–494. [Google Scholar] [CrossRef]
- Donnem, T.; Hu, J.; Ferguson, M.; Adighibe, O.; Snell, C.; Harris, A.L.; Gatter, K.C.; Pezzella, F. Vessel co-option in primary human tumors and metastases: An obstacle to effective anti-angiogenic treatment? Cancer Med. 2013, 2, 427–436. [Google Scholar] [CrossRef]
- Leenders, W.P.J.; Küsters, B.; De Waal, R.M.W. Vessel co-option: How tumors obtain blood supply in the absence of sprouting angiogenesis. Endothel. J. Endothel. Cell Res. 2002, 9, 83–87. [Google Scholar] [CrossRef]
- Pezzella, F.; Gatter, K.C. Evidence showing that tumors can grow without angiogenesis and can switch between angiogenic and nonangiogenic phenotypes. J. Natl. Cancer. Inst. 2016, 108, djw032. [Google Scholar] [CrossRef]
- Winkler, F. Hostile takeover: How tumours hijack pre-existing vascular environments to thrive. J. Pathol. 2017, 242, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Nierodzik, M.L.; Karpatkin, S. Thrombin induces tumor growth, metastasis, and angiogenesis: Evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell 2006, 10, 355–362. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Teuwen, L.A.; De Rooij, L.P.M.H.; Cuypers, A.; Rohlenova, K.; Dumas, S.J.; García-Caballero, M.; Meta, E.; Amersfoort, J.; Taverna, F.; Becker, L.M.; et al. Tumor vessel co-option probed by single-cell analysis. Cell Rep. 2021, 35, 109253. [Google Scholar] [CrossRef]
- Rosińska, S.; Gavard, J. Tumor vessels fuel the fire in glioblastoma. Int. J. Mol. Sci. 2021, 22, 6514. [Google Scholar] [CrossRef]
- Qiao, L.; Liang, N.; Zhang, J.; Xie, J.; Liu, F.; Xu, D.; Yu, X.; Tian, Y. Advanced research on vasculogenic mimicry in cancer. J. Cell. Mol. Med. 2015, 19, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Fidler, I.J. Finding the tumor copycat: Therapy fails, patients don’t. Nat. Med. 2010, 16, 974–975. [Google Scholar] [CrossRef]
- Folberg, R.; Hendrix, M.J.C.; Maniotis, A.J. Vasculogenic mimicry and tumor angiogenesis. Am. J. Pathol. 2000, 156, 361–381. [Google Scholar] [CrossRef]
- Paulis, Y.W.J.; Soetekouw, P.M.M.B.; Verheul, H.M.W.; Tjan-Heijnen, V.C.G.; Griffioen, A.W. Signalling pathways in vasculogenic mimicry. Biochim. Biophys. Acta-Rev. Cancer 2010, 1806, 18–28. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.G.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J.C. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Folberg, R.; Maniotis, A.J. Vasculogenic mimicry. APMIS 2004, 112, 508–525. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, N.V.; Nadeev, A.D.; Jenkins, R.O.; Avdonin, P.V. Markers and Biomarkers of Endothelium: When Something Is Rotten in the State. Oxid. Med. Cell. Longev. 2017, 2017, 9759735. [Google Scholar] [CrossRef]
- Hendrix, M.J.C.; Seftor, E.A.; Hess, A.R.; Seftor, R.E.B. Vasculogenic mimicry and tumour-cell plasticity: Lessons from melanoma. Nat. Rev. Cancer 2003, 3, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Barnett, F.H.; Rosenfeld, M.; Wood, M.; Kiosses, W.B.; Usui, Y.; Marchetti, V.; Aguilar, E.; Friedlander, M. Macrophages form functional vascular mimicry channels in vivo. Sci. Rep. 2016, 6, 36659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, D.; Sun, B. Vasculogenic mimicry: Current status and future prospects. Cancer Lett. 2007, 254, 157–164. [Google Scholar] [CrossRef]
- Cao, Z.; Bao, M.; Miele, L.; Sarkar, F.H.; Wang, Z.; Zhou, Q. Tumour vasculogenic mimicry is associated with poor prognosis of human cancer patients: A systemic review and meta-analysis. Eur. J. Cancer 2013, 49, 3914–3923. [Google Scholar] [CrossRef]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef]
- Vartanian, A.; Stepanova, E.; Grigorieva, I.; Solomko, E.; Belkin, V.; Baryshnikov, A.; Lichinitser, M. Melanoma Vasculogenic Mimicry Capillary-Like Structure Formation Depends on Integrin and Calcium Signaling. Microcirculation 2011, 18, 390–399. [Google Scholar] [CrossRef]
- Rezaei, M.; Martins Cavaco, A.C.; Stehling, M.; Nottebaum, A.; Brockhaus, K.; Caliandro, M.F.; Schelhaas, S.; Schmalbein, F.; Vestweber, D.; Eble, J.A. Extracellular vesicle transfer from endothelial cells drives ve-cadherin expression in breast cancer cells, thereby causing heterotypic cell contacts. Cancers 2020, 12, 2138. [Google Scholar] [CrossRef]
- Martini, C.; DeNichilo, M.; King, D.P.; Cockshell, M.P.; Ebert, B.; Dale, B.; Ebert, L.M.; Woods, A.; Bonder, C.S. CD36 promotes vasculogenic mimicry in melanoma by mediating adhesion to the extracellular matrix. BMC Cancer 2021, 21, 765. [Google Scholar] [CrossRef]
- Tan, L.Y.; Cockshell, M.P.; Moore, E.; Myo Min, K.K.; Ortiz, M.; Johan, M.Z.; Ebert, B.; Ruszkiewicz, A.; Brown, M.P.; Ebert, L.M.; et al. Vasculogenic mimicry structures in melanoma support the recruitment of monocytes. Oncoimmunology 2022, 11, 2043673. [Google Scholar] [CrossRef]
- Benjakul, N.; Prakobphol, N.; Tangshewinsirikul, C.; Dulyaphat, W.; Svasti, J.; Charngkaew, K.; Kangsamaksin, T. Notch signaling regulates vasculogenic mimicry and promotes cell morphogenesis and the epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma. PLoS ONE 2022, 17, e0279001. [Google Scholar] [CrossRef]
- Wechman, S.L.; Emdad, L.; Sarkar, D.; Das, S.K.; Fisher, P.B. Vascular mimicry: Triggers, molecular interactions and in vivo models. Adv. Cancer Res. 2020, 148, 27–67. [Google Scholar]
- Shubik, P.; Warren, B.A. Additional literature on “vasculogenic mimicry” not cited. Am. J. Pathol. 2000, 156, 736. [Google Scholar] [CrossRef] [PubMed]
- Warren, B.A.; Shubik, P. The growth of the blood supply to melanoma transplants in the hamster cheek pouch. Lab. Investig. 1966, 4, 652. [Google Scholar] [CrossRef]
- Folberg, R.; Rummelt, V.; Parys-Van Ginderdeuren, R.; Hwang, T.; Woolson, R.F.; Pe’er, J.; Gruman, L.M. The Prognostic Value of Tumor Blood Vessel Morphology in Primary Uveal Melanoma. Ophthalmology 1993, 100, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Mäkitie, T.; Summanen, P.; Tarkkanen, A.; Kivelä, T. Microvascular loops and networks as prognostic indicators in choroidal and ciliary body melanomas. J. Natl. Cancer Inst. 1999, 91, 359–367. [Google Scholar] [CrossRef]
- Sakamoto, T.; Sakamoto, M.; Yoshikawa, H.; Hata, Y.; Ishibashi, T.; Ohnishi, Y.; Inomata, H. Histologic findings and prognosis of uveal malignant melanoma in Japanese patients. Am. J. Ophthalmol. 1996, 121, 276–283. [Google Scholar] [CrossRef]
- Seregard, S.; Spångberg, B.; Juul, C.; Oskarsson, M. Prognostic accuracy of the mean of the largest nucleoli, vascular patterns, and PC-10 in posterior uveal melanoma. Ophthalmology 1998, 105, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Thies, A.; Mangold, U.; Moll, I.; Schumacher, U. PAS-positive loops and networks as a prognostic indicator in cutaneous malignant melanoma. J. Pathol. 2001, 195, 537–542. [Google Scholar] [CrossRef]
- Warso, M.A.; Maniotis, A.J.; Chen, X.; Majumdar, D.; Patel, M.K.; Shilkaitis, A.; Das Gupta, T.K.; Folberg, R. Prognostic significance of periodic acid-schiff-positive patterns in primary cutaneous melanoma. Clin. Cancer Res. 2001, 7, 473–477. [Google Scholar]
- Rummelt, V.; Mehaffey, M.G.; Campbell, R.J.; Pe’Er, J.; Bentler, S.E.; Woolson, R.F.; Naumann, G.O.H.; Folberg, R.H. Microcirculation architecture of metastases from primary ciliary body and choroidal melanomas. Am. J. Ophthalmol. 1998, 126, 303–305. [Google Scholar] [CrossRef]
- Mueller, A.J.; Maniotis, A.J.; Freeman, W.R.; Bartsch, D.U.; Schaller, U.C.; Bergeron-Lynn, G.; Cheng, L.; Taskintuna, I.; Chen, X.; Kan-Mitchell, J.; et al. An orthotopic model for human uveal melanoma in SCID mice. Microvasc. Res. 2002, 64, 207–213. [Google Scholar] [CrossRef]
- Folberg, R.; Leach, L.; Valyi-Nagy, K.; Lin, A.Y.; Apushkin, M.A.; Ai, Z.; Barak, V.; Majumdar, D.; Pe’er, J.; Maniotis, A.J. Modeling the behavior of uveal melanoma in the liver. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2967–2974. [Google Scholar] [CrossRef] [PubMed]
- Clarijs, R.; Otte-Höller, I.; Ruiter, D.J.; De Waal, R.M.W. Presence of a fluid-conducting meshwork in xenografted cutaneous and primary human uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2002, 43, 912–918. [Google Scholar]
- Maniotis, A.J.; Chen, X.; Garcia, C.; DeChristopher, P.J.; Wu, D.; Pe’er, J.; Folberg, R. Control of melanoma morphogenesis, endothelial survival, and perfusion by extracellular matrix. Lab. Investig. 2002, 82, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Pötgens, A.J.G.; Van Altena, M.C.; Lubsen, N.H.; Ruiter, D.J.; De Waal, R.M.W. Analysis of the tumor vasculature and metastatic behavior of xenografts of human melanoma cell lines transfected with vascular permeability factor. Am. J. Pathol. 1996, 148, 1203–1217. [Google Scholar]
- Thijssen, V.L.J.L.; Paulis, Y.W.J.; Nowak-Sliwinska, P.; Deumelandt, K.L.; Hosaka, K.; Soetekouw, P.M.M.B.; Cimpean, A.M.; Raica, M.; Pauwels, P.; van den Oord, J.J.; et al. Targeting PDGF-mediated recruitment of pericytes blocks vascular mimicry and tumor growth. J. Pathol. 2018, 246, 447–458. [Google Scholar] [CrossRef]
- Rodríguez, M.I.; Peralta-Leal, A.; O’Valle, F.; Rodriguez-Vargas, J.M.; Gonzalez-Flores, A.; Majuelos-Melguizo, J.; López, L.; Serrano, S.; de Herreros, A.G.; Rodríguez-Manzaneque, J.C.; et al. PARP-1 Regulates Metastatic Melanoma through Modulation of Vimentin-induced Malignant Transformation. PLoS Genet. 2013, 9, e1003531. [Google Scholar] [CrossRef]
- Fernández-Cortés, M.; Delgado-Bellido, D.; Bermúdez-Jiménez, E.; Paramio, J.M.; O’Valle, F.; Vinckier, S.; Carmeliet, P.; Garcia-Diaz, A.; Oliver, F.J. PARP inhibition promotes endothelial-like traits in melanoma cells and modulates pericyte coverage dynamics during vasculogenic mimicry. J. Pathol. 2023, 259, 318–330. [Google Scholar] [CrossRef]
- Bittner, M.; Meltzer, P.; Chen, Y.; Jiang, Y.; Seftor, E.; Hendrix, M.; Radmacher, M.; Simon, R.; Yakhini, Z.; Ben-Dor, A.; et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000, 406, 536–540. [Google Scholar] [CrossRef]
- Epha, E.; Hess, A.R.; Seftor, E.A.; Gardner, L.M.G.; Role, P.; Kinase, C.; Carles-Kinch, K.; Schneider, G.B.; Seftor, R.E.B.; Kinch, M.S.; et al. Molecular Regulation of Tumor Cell Vasculogenic Mimicry by Tyrosine Phosphorylation: Role of Epithelial Cell Kinase Advances in Brief Molecular Regulation of Tumor Cell Vasculogenic Mimicry by Tyrosine. Cancer Res. 2001, 61, 3250–3255. [Google Scholar]
- Hess, A.R.; Seftor, E.A.; Gruman, L.M.; Kinch, M.S.; Seftor, R.E.B.; Hendrix, M.J.C. VE-cadherin regulates EphA2 in aggressive melanoma cells through a novel signaling pathway: Implications for vasculogenic mimicry. Cancer Biol. Ther. 2006, 5, 228–233. [Google Scholar] [CrossRef]
- Hess, A.R.; Margaryan, N.V.; Seftor, E.A.; Hendrix, M.J.C. Deciphering the signaling events that promote melanoma tumor cell vasculogenic mimicry and their link to embryonic vasculogenesis: Role of the Eph receptors. Dev. Dyn. 2007, 236, 3283–3296. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.S.; Sun, W.; Ge, C.Y.; Zhang, W.Z.; Fan, Y.Z. Contribution of the PI3K/MMPs/Ln-5γ2 and EphA2/FAK/Paxillin signaling pathways to tumor growth and vasculogenic mimicry of gallbladder carcinomas. Int. J. Oncol. 2013, 42, 2103–2115. [Google Scholar] [CrossRef] [PubMed]
- Seftor, R.E.B.; Seftor, E.A.; Koshikawa, N.; Meltzer, P.S.; Gardner, L.M.G.; Bilban, M.; Stetler-Stevenson, W.G.; Quaranta, V.; Hendrix, M.J.C. Cooperative interactions of laminin 5 γ2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 2001, 61, 6322–6327. [Google Scholar] [PubMed]
- Welch, D.R.; Bisi, J.E.; Miller, B.E.; Conaway, D.; Seftor, E.A.; Yohem, K.H.; Gilmore, L.B.; Seftor, R.E.B.; Nakajima, M.; Hendrix, M.J.C. Characterization of a highly invasive and spontaneously metastatic human malignant melanoma cell line. Int. J. Cancer 1991, 47, 227–237. [Google Scholar] [CrossRef]
- Peris-Torres, C.; Plaza-Calonge, M.D.C.; López-Domínguez, R.; Domínguez-García, S.; Barrientos-Durán, A.; Carmona-Sáez, P.; Rodríguez-Manzaneque, J.C. Extracellular protease adamts1 is required at early stages of human uveal melanoma development by inducing stemness and endothelial-like features on tumor cells. Cancers 2020, 12, 801. [Google Scholar] [CrossRef] [PubMed]
- Casal, C.; Torres-Collado, A.X.; Plaza-Calonge, M.D.C.; Martino-Echarri, E.; Ramon y Cajal, S.; Rojo, F.; Griffioen, A.W.; Rodríguez-Manzaneque, J.C. ADAMTS1 contributes to the acquisition of an endothelial-like phenotype in plastic tumor cells. Cancer Res. 2010, 70, 4676–4686. [Google Scholar] [CrossRef]
- Hendrix, M.J.C.; Seftor, E.A.; Meltzer, P.S.; Gardner, L.M.G.; Hess, A.R.; Kirschmann, D.A.; Schatteman, G.C.; Seftor, R.E.B. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: Role in vasculogenic mimicry. Proc. Natl. Acad. Sci. USA 2001, 98, 8018–8023. [Google Scholar] [CrossRef]
- Nobre, A.R.; Entenberg, D.; Wang, Y.; Condeelis, J.; Aguirre-Ghiso, J.A. The Different Routes to Metastasis via Hypoxia-Regulated Programs. Trends Cell Biol. 2018, 28, 941–956. [Google Scholar] [CrossRef]
- Sun, B.; Zhang, D.; Zhang, S.; Zhang, W.; Guo, H.; Zhao, X. Hypoxia influences vasculogenic mimicry channel formation and tumor invasion-related protein expression in melanoma. Cancer Lett. 2007, 249, 188–197. [Google Scholar] [CrossRef]
- Wei, X.; Chen, Y.; Jiang, X.; Peng, M.; Liu, Y.; Mo, Y.; Ren, D.; Hua, Y.; Yu, B.; Zhou, Y.; et al. Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Mol. Cancer 2021, 20, 7. [Google Scholar] [CrossRef]
- Andreucci, E.; Peppicelli, S.; Ruzzolini, J.; Bianchini, F.; Calorini, L. Physicochemical aspects of the tumour microenvironment as drivers of vasculogenic mimicry. Cancer Metastasis Rev. 2022, 41, 935–951. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Calvani, M.; Giannoni, E.; Bianchini, F.; Calorini, L.; Torre, E.; Migliore, C.; Giordano, S.; Chiarugi, P. HIF-1α stabilization by mitochondrial ROS promotes Met-dependent invasive growth and vasculogenic mimicry in melanoma cells. Free Radic. Biol. Med. 2011, 51, 893–904. [Google Scholar] [CrossRef]
- Spinella, F.; Caprara, V.; Di Castro, V.; Rosanò, L.; Cianfrocca, R.; Natali, P.G.; Bagnato, A. Endothelin-1 induces the transactivation of vascular endothelial growth factor receptor-3 and modulates cell migration and vasculogenic mimicry in melanoma cells. J. Mol. Med. 2013, 91, 395–405. [Google Scholar] [CrossRef]
- Fu, R.; Du, W.; Ding, Z.; Wang, Y.; Li, Y.; Zhu, J.; Zeng, Y.; Zheng, Y.; Liu, Z.; Huang, J. HIF-1α promoted vasculogenic mimicry formation in lung adenocarcinoma through NRP1 upregulation in the hypoxic tumor microenvironment. Cell Death Dis. 2021, 12, 394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, B.; Zhao, X.; Liu, Z.; Wang, X.; Yao, X.; Dong, X.; Chi, J. Clinical significances and prognostic value of cancer stem-like cells markers and vasculogenic mimicry in renal cell carcinoma. J. Surg. Oncol. 2013, 108, 414–419. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, X.; Zhu, D.; Liu, T.; Liang, X.; Liu, F.; Zhang, Y.; Dong, X.; Sun, B. HIF-1α promoted vasculogenic mimicry formation in hepatocellular carcinoma through LOXL2 up-regulation in hypoxic tumor microenvironment. J. Exp. Clin. Cancer Res. 2017, 36, 60. [Google Scholar] [CrossRef]
- Bedal, K.B.; Grässel, S.; Spanier, G.; Reichert, T.E.; Bauer, R.J. The NC11 domain of human collagen XVI induces vasculogenic mimicry in oral squamous cell carcinoma cells. Carcinogenesis 2015, 36, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Biondani, G.; Zeeberg, K.; Greco, M.R.; Cannone, S.; Dando, I.; Dalla Pozza, E.; Mastrodonato, M.; Forciniti, S.; Casavola, V.; Palmieri, M.; et al. Extracellular matrix composition modulates PDAC parenchymal and stem cell plasticity and behavior through the secretome. FEBS J. 2018, 285, 2104–2124. [Google Scholar] [CrossRef]
- Velez, D.O.; Tsui, B.; Goshia, T.; Chute, C.L.; Han, A.; Carter, H.; Fraley, S.I. 3D collagen architecture induces a conserved migratory and transcriptional response linked to vasculogenic mimicry. Nat. Commun. 2017, 8, 1651. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, R.; Niwa, Y.; Simizu, S. Integrin β1 is an essential factor in vasculogenic mimicry of human cancer cells. Cancer Sci. 2018, 109, 2490–2496. [Google Scholar] [CrossRef]
- Rong, X.; Huang, B.; Qiu, S.; Li, X.; He, L.; Peng, Y. Tumor-associated macrophages induce vasculogenic mimicry of glioblastoma multiforme through cyclooxygenase-2 activation. Oncotarget 2016, 7, 83976–83986. [Google Scholar] [CrossRef]
- Hutchenreuther, J.; Vincent, K.; Norley, C.; Racanelli, M.; Gruber, S.B.; Johnson, T.M.; Fullen, D.R.; Raskin, L.; Perbal, B.; Holdsworth, D.W.; et al. Activation of cancer-associated fibroblasts is required for tumor neovascularization in a murine model of melanoma. Matrix Biol. 2018, 74, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Van Hinsbergh, V.W.M. Endothelial permeability for macromolecules: Mechanistic aspects of pathophysiological modulation. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1018–1023. [Google Scholar] [CrossRef]
- Gavard, J.; Gutkind, S.J. VE-cadherin and claudin-5: It takes two to tango. Nat. Cell Biol. 2008, 10, 883–885. [Google Scholar] [CrossRef]
- Yuan, S.Y.; Rigor, R.R. Regulation of Endothelial Barrier Function; Colloquium Series on Integrated Systems Physiology: From Molecule to Function; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2011; Volume 3, 146p. [Google Scholar] [CrossRef]
- Esser, S.; Lampugnani, M.G.; Corada, M.; Dejana, E.; Risau, W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 1998, 111, 1853–1865. [Google Scholar] [CrossRef]
- Andriopoulou, P.; Navarro, P.; Zanetti, A.; Lampugnani, M.G.; Dejana, E. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2286–2297. [Google Scholar] [CrossRef]
- Chen, X.L.; Nam, J.O.; Jean, C.; Lawson, C.; Walsh, C.T.; Goka, E.; Lim, S.T.; Tomar, A.; Tancioni, I.; Uryu, S.; et al. VEGF-Induced Vascular Permeability Is Mediated by FAK. Dev. Cell 2012, 22, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Breier, G.; Grosser, M.; Rezaei, M. Endothelial cadherins in cancer. Cell Tissue Res. 2014, 355, 523–527. [Google Scholar] [CrossRef]
- Carmeliet, P.; Lampugnani, M.G.; Moons, L.; Breviario, F.; Compernolle, V.; Bono, F.; Balconi, G.; Spagnuolo, R.; Oosthuyse, B.; Dewerchin, M.; et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 1999, 98, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Jean, C.; Chen, X.L.; Nam, J.O.; Tancioni, I.; Uryu, S.; Lawson, C.; Ward, K.K.; Walsh, C.T.; Miller, N.L.G.; Ghassemian, M.; et al. Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J. Cell Biol. 2014, 204, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Orsenigo, F.; Giampietro, C.; Ferrari, A.; Corada, M.; Galaup, A.; Sigismund, S.; Ristagno, G.; Maddaluno, L.; Koh, G.Y.; Franco, D.; et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat. Commun. 2012, 3, 1208. [Google Scholar] [CrossRef]
- Wessel, F.; Winderlich, M.; Holm, M.; Frye, M.; Rivera-Galdos, R.; Vockel, M.; Linnepe, R.; Ipe, U.; Stadtmann, A.; Zarbock, A.; et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat. Immunol. 2014, 15, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Bellido, D.; Fernández-Cortés, M.; Rodríguez, M.I.; Serrano-Sáenz, S.; Carracedo, A.; Garcia-Diaz, A.; Oliver, F.J. VE-cadherin promotes vasculogenic mimicry by modulating kaiso-dependent gene expression. Cell Death Differ. 2019, 26, 348–361. [Google Scholar] [CrossRef]
- Lampugnani, M.G.; Corada, M.; Andriopoulou, P.; Esser, S.; Risau, W.; Dejana, E. Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J. Cell Sci. 1997, 110, 2065–2077. [Google Scholar] [CrossRef]
- Bäumer, S.; Keller, L.; Holtmann, A.; Funke, R.; August, B.; Gamp, A.; Wolburg, H.; Wolburg-Buchholz, K.; Deutsch, U.; Vestweber, D. Vascular endothelial cell-specific phosphotyrosine phosphatase (VE-PTP) activity is required for blood vessel development. Blood 2006, 107, 4754–4762. [Google Scholar] [CrossRef]
- Delgado-Bellido, D.; Bueno-Galera, C.; López-Jiménez, L.; Garcia-Diaz, A.; Oliver, F.J. Endothelial Phosphatase VE-PTP Participates in Vasculogenic Mimicry by Preventing Autophagic Degradation of VE-Cadherin. Front. Oncol. 2020, 10, 18. [Google Scholar] [CrossRef]
- Delgado-Bellido, D.; Zamudio-Martínez, E.; Fernández-Cortés, M.; Herrera-Campos, A.B.; Olmedo-Pelayo, J.; Perez, C.J.; Expósito, J.; de Álava, E.; Amaral, A.T.; Valle, F.O.; et al. VE-Cadherin modulates β-catenin/TCF-4 to enhance Vasculogenic Mimicry. Cell Death Dis. 2023, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Kalucka, J.; de Rooij, L.P.M.H.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779.e20. [Google Scholar] [CrossRef]
- Pasut, A.; Becker, L.M.; Cuypers, A.; Carmeliet, P. Endothelial cell plasticity at the single-cell level. Angiogenesis 2021, 24, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Paik, D.T.; Tian, L.; Williams, I.M.; Rhee, S.; Zhang, H.; Liu, C.; Mishra, R.; Wu, S.M.; Red-Horse, K.; Wu, J.C. Single-Cell RNA Sequencing Unveils Unique Transcriptomic Signatures of Organ-Specific Endothelial Cells. Circulation 2020, 142, 1848–1862. [Google Scholar] [CrossRef] [PubMed]
- Rada, M.; Lazaris, A.; Kapelanski-Lamoureux, A.; Mayer, T.Z.; Metrakos, P. Tumor microenvironment conditions that favor vessel co-option in colorectal cancer liver metastases: A theoretical model. Semin. Cancer Biol. 2021, 71, 52–64. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.; Dudley, A.C. Models and molecular mechanisms of blood vessel co-option by cancer cells. Angiogenesis 2020, 23, 17–25. [Google Scholar] [CrossRef]
- Haas, G.; Fan, S.; Ghadimi, M.; De Oliveira, T.; Conradi, L.C. Different Forms of Tumor Vascularization and Their Clinical Implications Focusing on Vessel Co-option in Colorectal Cancer Liver Metastases. Front. Cell Dev. Biol. 2021, 9, 612774. [Google Scholar] [CrossRef]
- Boire, A.; Coffelt, S.B.; Quezada, S.A.; Vander Heiden, M.G.; Weeraratna, A.T. Tumour Dormancy and Reawakening: Opportunities and Challenges. Trends Cancer 2019, 5, 762–765. [Google Scholar] [CrossRef]
- Recasens, A.; Munoz, L. Targeting Cancer Cell Dormancy. Trends Pharmacol. Sci. 2019, 40, 128–141. [Google Scholar] [CrossRef]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef]
- Belotti, D.; Pinessi, D.; Taraboletti, G. Alternative vascularization mechanisms in tumor resistance to therapy. Cancers 2021, 13, 1912. [Google Scholar] [CrossRef] [PubMed]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.R.; Wotherspoon, A.; Gao, Z.H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgado-Bellido, D.; Oliver, F.J.; Vargas Padilla, M.V.; Lobo-Selma, L.; Chacón-Barrado, A.; Díaz-Martin, J.; de Álava, E. VE-Cadherin in Cancer-Associated Angiogenesis: A Deceptive Strategy of Blood Vessel Formation. Int. J. Mol. Sci. 2023, 24, 9343. https://doi.org/10.3390/ijms24119343
Delgado-Bellido D, Oliver FJ, Vargas Padilla MV, Lobo-Selma L, Chacón-Barrado A, Díaz-Martin J, de Álava E. VE-Cadherin in Cancer-Associated Angiogenesis: A Deceptive Strategy of Blood Vessel Formation. International Journal of Molecular Sciences. 2023; 24(11):9343. https://doi.org/10.3390/ijms24119343
Chicago/Turabian StyleDelgado-Bellido, Daniel, F. J. Oliver, María Victoria Vargas Padilla, Laura Lobo-Selma, Antonio Chacón-Barrado, Juan Díaz-Martin, and Enrique de Álava. 2023. "VE-Cadherin in Cancer-Associated Angiogenesis: A Deceptive Strategy of Blood Vessel Formation" International Journal of Molecular Sciences 24, no. 11: 9343. https://doi.org/10.3390/ijms24119343
APA StyleDelgado-Bellido, D., Oliver, F. J., Vargas Padilla, M. V., Lobo-Selma, L., Chacón-Barrado, A., Díaz-Martin, J., & de Álava, E. (2023). VE-Cadherin in Cancer-Associated Angiogenesis: A Deceptive Strategy of Blood Vessel Formation. International Journal of Molecular Sciences, 24(11), 9343. https://doi.org/10.3390/ijms24119343