
A New Culture Model for Enhancing Estrogen Responsiveness in HR+ Breast Cancer Cells through Medium Replacement: Presumed Involvement of Autocrine Factors in Estrogen Resistance


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Although CS-FBS Makes HR+ BC Cells Much More Responsive to 17β-Estradiol (E2) Than Normal FBS, It Has Some Limitations
2.2. Low Cell Density Increases E2 Responsiveness and Decreases Fulv Efficacy in T47D, and the Effectiveness of E2 and Fulv Decreases over Time
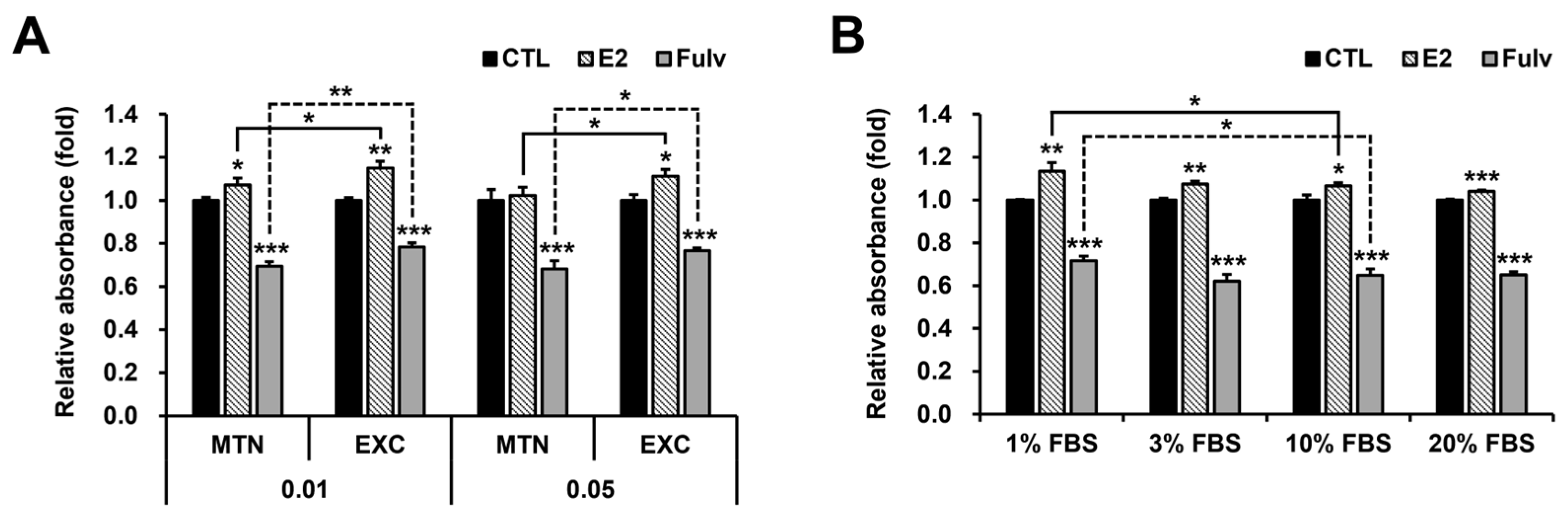
2.3. Medium Exchange (EXC) and Low FBS Concentration Independently Mimic Low Cell Density in Terms of E2 and Fulv Responses in T47D
2.4. BC Cell Culture Supernatants Mimic High Cell Density and MTN in Terms of E2 and Fulv Responses in T47D, Raising the Possibility of the Involvement of Autocrine Factors in Regulating Their Responses, Although Epidermal Growth Factor (EGF) Is Not Causative
2.5. The Results Previously Observed in the Parental T47D Cells Were Largely Reproduced in T47D Subclone and MCF-7 Cells, Albeit with Some Differences
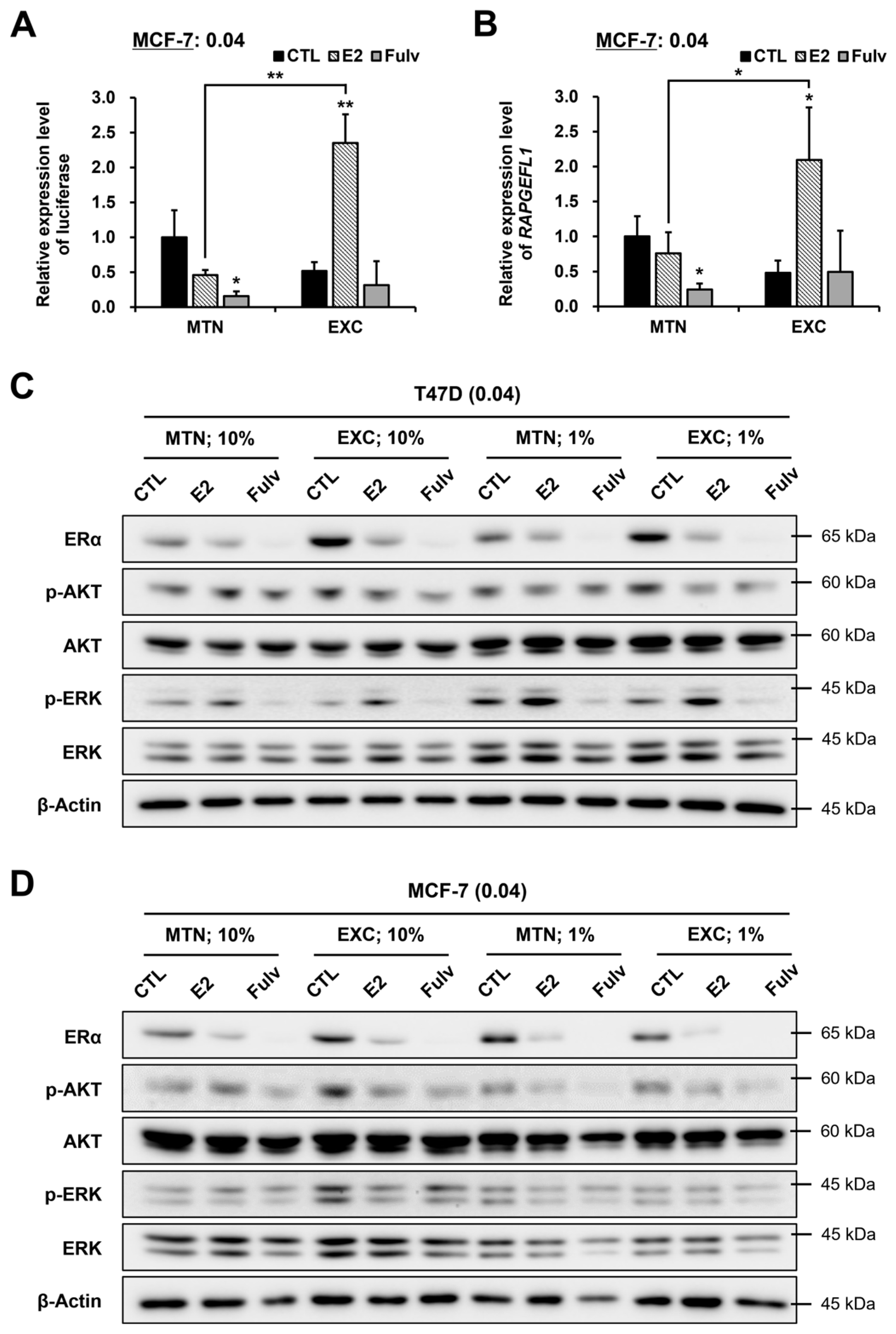
2.6. EXC Alters E2 and Fulv Responsiveness by Modulating ER Activity Rather Than the ERα Protein Level
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Reagents and Treatment
4.3. Cell Proliferation and Viability Assays
4.4. Western Blotting
4.5. Cell Cycle Analysis
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. Quantitative Reverse Transcription PCR (qRT-PCR) to Measure the Activity and Target Gene Expression of ER
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Cheang, M.C.; Chia, S.K.; Voduc, D.; Gao, D.; Leung, S.; Snider, J.; Watson, M.; Davies, S.; Bernard, P.S.; Parker, J.S.; et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst. 2009, 101, 736–750. [Google Scholar] [CrossRef]
- Porras, L.; Ismail, H.; Mader, S. Positive Regulation of Estrogen Receptor Alpha in Breast Tumorigenesis. Cells 2021, 10, 2966. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Russo, I.H. The role of estrogen in the initiation of breast cancer. J. Steroid Biochem. Mol. Biol. 2006, 102, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Augusto, T.V.; Correia-da-Silva, G.; Rodrigues, C.M.P.; Teixeira, N.; Amaral, C. Acquired resistance to aromatase inhibitors: Where we stand! Endocr. Relat. Cancer 2018, 25, R283–R301. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Johnston, S.R. Endocrine therapy—Current benefits and limitations. Breast Cancer Res. Treat. 2005, 93 (Suppl. 1), S3–S10. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative, G. Aromatase inhibitors versus tamoxifen in early breast cancer: Patient-level meta-analysis of the randomised trials. Lancet 2015, 386, 1341–1352. [Google Scholar] [CrossRef]
- Ma, C.X.; Reinert, T.; Chmielewska, I.; Ellis, M.J. Mechanisms of aromatase inhibitor resistance. Nat. Rev. Cancer 2015, 15, 261–275. [Google Scholar] [CrossRef]
- Jacquet, E.; Lardy-Cleaud, A.; Pistilli, B.; Franck, S.; Cottu, P.; Delaloge, S.; Debled, M.; Vanlemmens, L.; Leheurteur, M.; Guizard, A.V.; et al. Endocrine therapy or chemotherapy as first-line therapy in hormone receptor-positive HER2-negative metastatic breast cancer patients. Eur. J. Cancer 2018, 95, 93–101. [Google Scholar] [CrossRef]
- Li, L.; Chang, B.; Jiang, X.; Fan, X.; Li, Y.; Li, T.; Wu, S.; Zhang, J.; Kariminia, S.; Li, Q. Clinical outcomes comparison of 10 years versus 5 years of adjuvant endocrine therapy in patients with early breast cancer. BMC Cancer 2018, 18, 977. [Google Scholar] [CrossRef] [PubMed]
- Collin, L.J.; Cronin-Fenton, D.P.; Ahern, T.P.; Goodman, M.; McCullough, L.E.; Waller, L.A.; Kjaersgaard, A.; Damkier, P.; Christiansen, P.M.; Ejlertsen, B.; et al. Early Discontinuation of Endocrine Therapy and Recurrence of Breast Cancer among Premenopausal Women. Clin. Cancer Res. 2021, 27, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Tran, B.; Bedard, P.L. Luminal-B breast cancer and novel therapeutic targets. Breast Cancer Res. 2011, 13, 221. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Stebbing, J.; Giamas, G.; Murphy, J. Endocrine Resistance in Hormone Receptor Positive Breast Cancer-From Mechanism to Therapy. Front. Endocrinol. 2019, 10, 245. [Google Scholar] [CrossRef]
- Belachew, E.B.; Sewasew, D.T. Molecular Mechanisms of Endocrine Resistance in Estrogen-Positive Breast Cancer. Front. Endocrinol. 2021, 12, 599586. [Google Scholar] [CrossRef]
- Mills, J.N.; Rutkovsky, A.C.; Giordano, A. Mechanisms of resistance in estrogen receptor positive breast cancer: Overcoming resistance to tamoxifen/aromatase inhibitors. Curr. Opin. Pharmacol. 2018, 41, 59–65. [Google Scholar] [CrossRef]
- McAndrew, N.P.; Finn, R.S. Clinical Review on the Management of Hormone Receptor-Positive Metastatic Breast Cancer. JCO Oncol. Pract. 2022, 18, 319–327. [Google Scholar] [CrossRef]
- Berthois, Y.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Phenol red in tissue culture media is a weak estrogen: Implications concerning the study of estrogen-responsive cells in culture. Proc. Natl. Acad. Sci. USA 1986, 83, 2496–2500. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Ochi, Y.; Shiomi, K.; Hachiya, T.; Yoshimura, M.; Miyazaki, T. Dextran-coated charcoal technique to make the hormone-free serum as a diluent for standard curve of radioimmunoassay. Endocrinol. Jpn. 1973, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Vanetti, C.; Bifari, F.; Vicentini, L.M.; Cattaneo, M.G. Fatty acids rather than hormones restore in vitro angiogenesis in human male and female endothelial cells cultured in charcoal-stripped serum. PLoS ONE 2017, 12, e0189528. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Fiandalo, M.V.; Pop, E.; Stocking, J.J.; Azabdaftari, G.; Li, J.; Wei, H.; Ma, D.; Qu, J.; Mohler, J.L.; et al. Proteomic Analysis of Charcoal-Stripped Fetal Bovine Serum Reveals Changes in the Insulin-like Growth Factor Signaling Pathway. J. Proteome Res. 2018, 17, 2963–2977. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.J.; Johnson, M.D.; Lee, A.V.; Oesterreich, S. Endocrine Response Phenotypes Are Altered by Charcoal-Stripped Serum Variability. Endocrinology 2016, 157, 3760–3766. [Google Scholar] [CrossRef]
- Cao, Z.; West, C.; Norton-Wenzel, C.S.; Rej, R.; Davis, F.B.; Davis, P.J.; Rej, R. Effects of resin or charcoal treatment on fetal bovine serum and bovine calf serum. Endocr. Res. 2009, 34, 101–108. [Google Scholar] [CrossRef]
- Furuya, Y.; Kohno, N.; Fujiwara, Y.; Saitoh, Y. Mechanisms of estrogen action on the proliferation of MCF-7 human breast cancer cells in an improved culture medium. Cancer Res. 1989, 49, 6670–6674. [Google Scholar] [PubMed]
- Denver, N.; Khan, S.; Homer, N.Z.M.; MacLean, M.R.; Andrew, R. Current strategies for quantification of estrogens in clinical research. J. Steroid Biochem. Mol. Biol. 2019, 192, 105373. [Google Scholar] [CrossRef]
- Seneviratne, A.; Attia, E.; Williams, R.J.; Rodeo, S.A.; Hannafin, J.A. The effect of estrogen on ovine anterior cruciate ligament fibroblasts: Cell proliferation and collagen synthesis. Am. J. Sports Med. 2004, 32, 1613–1618. [Google Scholar] [CrossRef]
- Oh, A.S.; Lorant, L.A.; Holloway, J.N.; Miller, D.L.; Kern, F.G.; El-Ashry, D. Hyperactivation of MAPK induces loss of ERalpha expression in breast cancer cells. Mol. Endocrinol. 2001, 15, 1344–1359. [Google Scholar] [CrossRef]
- Stoica, A.; Saceda, M.; Doraiswamy, V.L.; Coleman, C.; Martin, M.B. Regulation of estrogen receptor-alpha gene expression by epidermal growth factor. J. Endocrinol. 2000, 165, 371–378. [Google Scholar] [CrossRef]
- Briand, P.; Lundholt, B.K.; Skouv, J.; Lykkesfeldt, A.E. Growth response of breast epithelial cells to estrogen is influenced by EGF. Mol. Cell. Endocrinol. 1999, 153, 1–9. [Google Scholar] [CrossRef]
- Zhu, J.; Meng, X.; Yan, F.; Qin, C.; Wang, M.; Ding, Q.; Li, P.; Yang, J.; Ju, X.; Zhang, Z.; et al. A functional epidermal growth factor (EGF) polymorphism, EGF serum levels and renal cell carcinoma risk in a Chinese population. J. Hum. Genet. 2010, 55, 236–240. [Google Scholar] [CrossRef]
- Reddel, R.R.; Alexander, I.E.; Koga, M.; Shine, J.; Sutherland, R.L. Genetic instability and the development of steroid hormone insensitivity in cultured T 47D human breast cancer cells. Cancer Res. 1988, 48, 4340–4347. [Google Scholar] [PubMed]
- Graham, M.L., 2nd; Krett, N.L.; Miller, L.A.; Leslie, K.K.; Gordon, D.F.; Wood, W.M.; Wei, L.L.; Horwitz, K.B. T47DCO cells, genetically unstable and containing estrogen receptor mutations, are a model for the progression of breast cancers to hormone resistance. Cancer Res. 1990, 50, 6208–6217. [Google Scholar] [PubMed]
- Nishi, K.; Fu, W.; Kiyama, R. Novel estrogen-responsive genes (ERGs) for the evaluation of estrogenic activity. PLoS ONE 2022, 17, e0273164. [Google Scholar] [CrossRef] [PubMed]
- Le Romancer, M.; Poulard, C.; Cohen, P.; Sentis, S.; Renoir, J.M.; Corbo, L. Cracking the estrogen receptor’s posttranslational code in breast tumors. Endocr. Rev. 2011, 32, 597–622. [Google Scholar] [CrossRef]
- Piggott, L.; Silva, A.; Robinson, T.; Santiago-Gomez, A.; Simoes, B.M.; Becker, M.; Fichtner, I.; Andera, L.; Young, P.; Morris, C.; et al. Acquired Resistance of ER-Positive Breast Cancer to Endocrine Treatment Confers an Adaptive Sensitivity to TRAIL through Posttranslational Downregulation of c-FLIP. Clin. Cancer Res. 2018, 24, 2452–2463. [Google Scholar] [CrossRef]
- Briukhovetska, D.; Dorr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in cancer: From biology to therapy. Nat. Rev. Cancer 2021, 21, 481–499. [Google Scholar] [CrossRef]
- Esquivel-Velazquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Morales-Montor, J. The role of cytokines in breast cancer development and progression. J. Interferon Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef]
- Jones, V.S.; Huang, R.Y.; Chen, L.P.; Chen, Z.S.; Fu, L.; Huang, R.P. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Biochim. Biophys. Acta 2016, 1865, 255–265. [Google Scholar] [CrossRef]
- Papadimitropoulou, A.; Vellon, L.; Atlas, E.; Steen, T.V.; Cuyas, E.; Verdura, S.; Espinoza, I.; Menendez, J.A.; Lupu, R. Heregulin Drives Endocrine Resistance by Altering IL-8 Expression in ER-Positive Breast Cancer. Int. J. Mol. Sci. 2020, 21, 7737. [Google Scholar] [CrossRef] [PubMed]
- Sasser, A.K.; Sullivan, N.J.; Studebaker, A.W.; Hendey, L.F.; Axel, A.E.; Hall, B.M. Interleukin-6 is a potent growth factor for ER-alpha-positive human breast cancer. FASEB J. 2007, 21, 3763–3770. [Google Scholar] [CrossRef]
- Yi, E.H.; Lee, C.S.; Lee, J.K.; Lee, Y.J.; Shin, M.K.; Cho, C.H.; Kang, K.W.; Lee, J.W.; Han, W.; Noh, D.Y.; et al. STAT3-RANTES autocrine signaling is essential for tamoxifen resistance in human breast cancer cells. Mol. Cancer Res. 2013, 11, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF receptor ligands. Exp. Cell Res. 2003, 284, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.J.; Gilmore, J.L.; Foley, J.; Lemmon, M.A.; Riese, D.J., 2nd. Functional selectivity of EGF family peptide growth factors: Implications for cancer. Pharmacol. Ther. 2009, 122, 1–8. [Google Scholar] [CrossRef]
- Pink, J.J.; Bilimoria, M.M.; Assikis, J.; Jordan, V.C. Irreversible loss of the oestrogen receptor in T47D breast cancer cells following prolonged oestrogen deprivation. Br. J. Cancer 1996, 74, 1227–1236. [Google Scholar] [CrossRef]
- Wang, P.; Henning, S.M.; Heber, D. Limitations of MTT and MTS-based assays for measurement of antiproliferative activity of green tea polyphenols. PLoS ONE 2010, 5, e10202. [Google Scholar] [CrossRef]
- Hall, J.M.; McDonnell, D.P. The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 1999, 140, 5566–5578. [Google Scholar] [CrossRef]
- Wilson, V.S.; Bobseine, K.; Gray, L.E., Jr. Development and characterization of a cell line that stably expresses an estrogen-responsive luciferase reporter for the detection of estrogen receptor agonist and antagonists. Toxicol. Sci. 2004, 81, 69–77. [Google Scholar] [CrossRef]
- Kutner, R.H.; Zhang, X.Y.; Reiser, J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc. 2009, 4, 495–505. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, S.-H.; Paek, S.H.; Kim, J.-K.; Seong, J.K.; Lim, W. A New Culture Model for Enhancing Estrogen Responsiveness in HR+ Breast Cancer Cells through Medium Replacement: Presumed Involvement of Autocrine Factors in Estrogen Resistance. Int. J. Mol. Sci. 2023, 24, 9474. https://doi.org/10.3390/ijms24119474
Jang S-H, Paek SH, Kim J-K, Seong JK, Lim W. A New Culture Model for Enhancing Estrogen Responsiveness in HR+ Breast Cancer Cells through Medium Replacement: Presumed Involvement of Autocrine Factors in Estrogen Resistance. International Journal of Molecular Sciences. 2023; 24(11):9474. https://doi.org/10.3390/ijms24119474
Chicago/Turabian StyleJang, Seok-Hoon, Se Hyun Paek, Jong-Kyu Kim, Je Kyung Seong, and Woosung Lim. 2023. "A New Culture Model for Enhancing Estrogen Responsiveness in HR+ Breast Cancer Cells through Medium Replacement: Presumed Involvement of Autocrine Factors in Estrogen Resistance" International Journal of Molecular Sciences 24, no. 11: 9474. https://doi.org/10.3390/ijms24119474
APA StyleJang, S. -H., Paek, S. H., Kim, J. -K., Seong, J. K., & Lim, W. (2023). A New Culture Model for Enhancing Estrogen Responsiveness in HR+ Breast Cancer Cells through Medium Replacement: Presumed Involvement of Autocrine Factors in Estrogen Resistance. International Journal of Molecular Sciences, 24(11), 9474. https://doi.org/10.3390/ijms24119474