1. Introduction
Cancer stem cells (CSCs) are a subset of tumor cells that mediate the initiation, resistance and metastasis of tumors [
1]. Although the origin and definition of CSCs is still not clear, the undifferentiated stem cell-like characteristics of CSCs may provide an advantage in the acquisition of new mutations to survive and migrate/adapt to another tissue environment [
2]. Previously, we screened novel therapeutic targets that eliminated CSCs efficiently in a glioblastoma multiform (GBM) cell line and found that several lipid metabolism enzymes are essential for CSC survival but are not essential in bulk cultured cells (BCCs) [
3]. In the same study, TONSL (NFκBIL2, Tonsoku-like protein) was found to be a candidate selective target for CSC elimination.
TONSL forms a complex with MMS22L (MMS22-like) that plays an important role in maintaining genome integrity [
4]. The complex mediates homologous recombination repair of double-strand breaks (DSBs) at stalled or collapsed replication forks [
5]. At the damaged site of a replication fork, TONSL–MMS22L loads Rad51 to replace RPA [
6] with the help of histone chaperones [
7] and the epigenetic modification of histones [
8]. Biallelic mutation of
TONSL causes spondyloepimetaphyseal dysplasia (SEMDSP) [
9,
10], probably due to the delay of replication, which is associated with several malignancies [
11].
Genomic integrity is maintained by many DNA damage repair (DDR) molecules. As cancer is a disease caused by accumulated DNA damage, loss of the DDR function is an important determinant of carcinogenesis [
12]. Therefore, both germline and somatic mutational defects in DDR genes can act as strong drivers of carcinogenesis [
13]. In particular, two major homologous recombination repair (HRR) genes,
BRCA1 and
BRCA2, become nonfunctional in cancer through loss of function (LOF) mutations or hypermutations in the promoter region [
14]. This suggests that LOF mutations of other HRR genes may contribute to tumorigenesis as well. Indeed, other core HRR-associated genes (
BARD1,
PALB2,
FANCC,
RAD51C and
RAD51D) are frequently lost in many cancers [
15]. Interestingly, it has been noted that the
TONSL gene is amplified in several cancers [
16], suggesting an oncogenic role different from those of other well-studied tumor-suppressive HRR genes in cancer or carcinogenesis.
In this study, we investigated whether the TONSL gene plays an important role in cancer and CSCs using RNAi and CSC enrichment cultures in addition to bioinformatics analyses of various public databases. We demonstrate that TONSL is enriched in cancer tissues versus normal tissues and that higher expression is associated with worse prognosis. The previous finding that TONSL may be essential in CSCs was demonstrated again in several cancers. The dependency of TONSL and other HRR genes suggests that HRR in the replication fork may be important in CSCs and could be a target for CSC therapy.
2. Results
Because we previously noted that GBM CSCs are more vulnerable to the loss of
TONSL than bulk cultured cells (BCCs), we investigated whether
TONSL plays an important role in cancer prognosis in other cancers (
Figure 1). The association of
TONSL mRNA expression with the survival of cancer patients was analyzed via the KM plotter online tool [
17]. Higher expression of
TONSL mRNA was significantly associated with worse overall survival (OS) in lung adenocarcinoma, gastric cancer (intestinal and diffuse), breast cancer (luminal A, luminal B and HER2-positive) and ovarian cancer (endometrial and serous) (hazard ratios were greater than 1.2). Lung squamous cell carcinoma (SQCC) showed a similar pattern, but without statistical significance. Interestingly, the Hazardous Ratio was less than 0.5 for the basal type of breast cancer, suggesting that higher expression of
TONSL mRNA is associated with better prognosis. These data showed that
TONSL expression may be a prognostic marker for worse survival in some subsets of tumors from the lung, breast, ovary and stomach, suggesting an oncogenic role in them. We also examined recurrence-free survival (RFS) in the same manner, and the pattern of RFS according to the expression of TONSL mRNA was similar to that of OS, except for luminal B breast cancer, which had the opposite result (
Supplementary Figure S1).
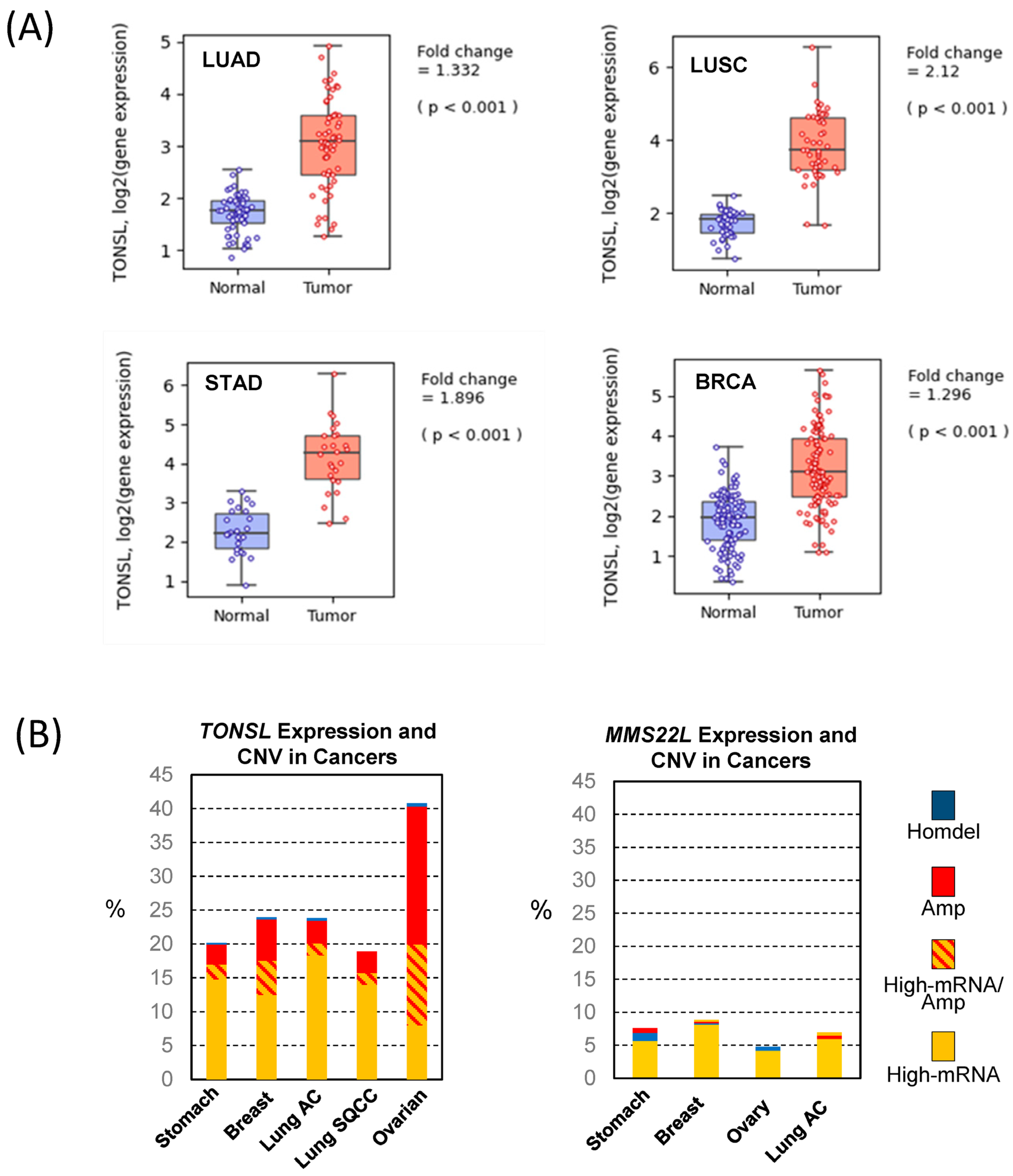
Because this finding suggests pro-cancer roles of
TONSL expression, we questioned whether the expression of
TONSL increases in cancer. We extracted mRNA expression data in cancers and corresponding normal tissues from the Pan-Cancer Atlas Study [
18] in The Cancer Genome Atlas (TCGA) and analyzed them using Qomics (
https://qomics.sookmyung.ac.kr/, accessed on 1 December 2022) [
19] or cBioportal (
https://www.cbioportal.org/, accessed on 1 December 2022) [
20,
21]. The expression of
TONSL was significantly higher in all tumors examined: lung, stomach and breast (
Figure 2A and
Figure S2). Because no normal tissue data were available for ovarian tumors, we could not analyze the relative expression between tumors and normal tissues for this cancer. Several oncogenes were found to be amplified, leading to an increase in mRNA expression. Therefore, we assessed the copy number variation (CNV) of
TONSL in the cancers indicated in
Figure 1. Interestingly, more than 5% of the tumors from all of the examined cancer tissues showed greater
TONSL gene amplification (
Supplementary Figure S3). Among those, more than 25% of ovarian cancers harbored gene amplification of
TONSL. As TONSL requires MMS22L to mediate repair in the replication fork, we also examined gene amplification of
MMS22L.
MMS22L gene amplification was very rare in all cancers assessed, and point mutations were the most common variations in stomach and lung adenocarcinomas. We then examined whether increased expression of
TONSL/MMS22L mRNA is associated with
TONSL and
MMS22L CNV (
Figure 2B). In ovarian cancers, 1/3 of the
TONSL-amplified tissues expressed higher
TONSL mRNA (of 32% of tumors with
TONSL gene amplification, only 12% highly expressed
TONSL mRNA). Approximately 8% of total tumors without gene amplification expressed TONSL mRNA at high levels. In other organ tumors (stomach, breast and lung adenocarcinoma and squamouscarcinoma), the majority of tumors that highly expressed
TONSL mRNA did not exhibit
TONSL gene amplification (14.7%, 12.5%, 18.3% and 14.0% respectively), and only a small portion of those tumors were accompanied with
TONSL gene amplification (2.2%, 5.0%, 1.8% and 1.8%, respectively). We also evaluated
MMS22L mRNA expression, point mutations and CNV in the same tumors. Only 4–8% of tumors had higher
MMS22L expression without CNV or mutation. This is somewhat surprising because TONSL functions as a complex with MMS22L. This suggests that
TONSL amplification may not be directly caused by cooperative function with
MMS22L, or that the amplification itself is not an oncogenic driver.
Accordingly, we sought to determine why amplification of the
TONSL gene frequently occurs in ovarian cancer, and its impact. One of the main mechanisms by which proto-oncogenes become oncogenes for carcinogenesis is amplification. The most frequently amplified genes in all cancers are
MYC,
EGFR and
ERBB2 (HER2). We explored the portion of cancers carrying these gene amplifications in the same dataset in TCGA. TCGA Pan-Cancer Atlas Study [
18] data were analyzed in cBioportal. The
MYC gene was amplified in 9% of total tumors and most amplified (32%) in ovarian cancer (serous).
ERBB2 amplification was found in 6% of total tumors and most amplified in gastric and breast cancers (11% and 10%, respectively).
EGFR amplification was detected in 7% of total tumors; glioblastoma presented an amplification frequency that was greater than 40%.
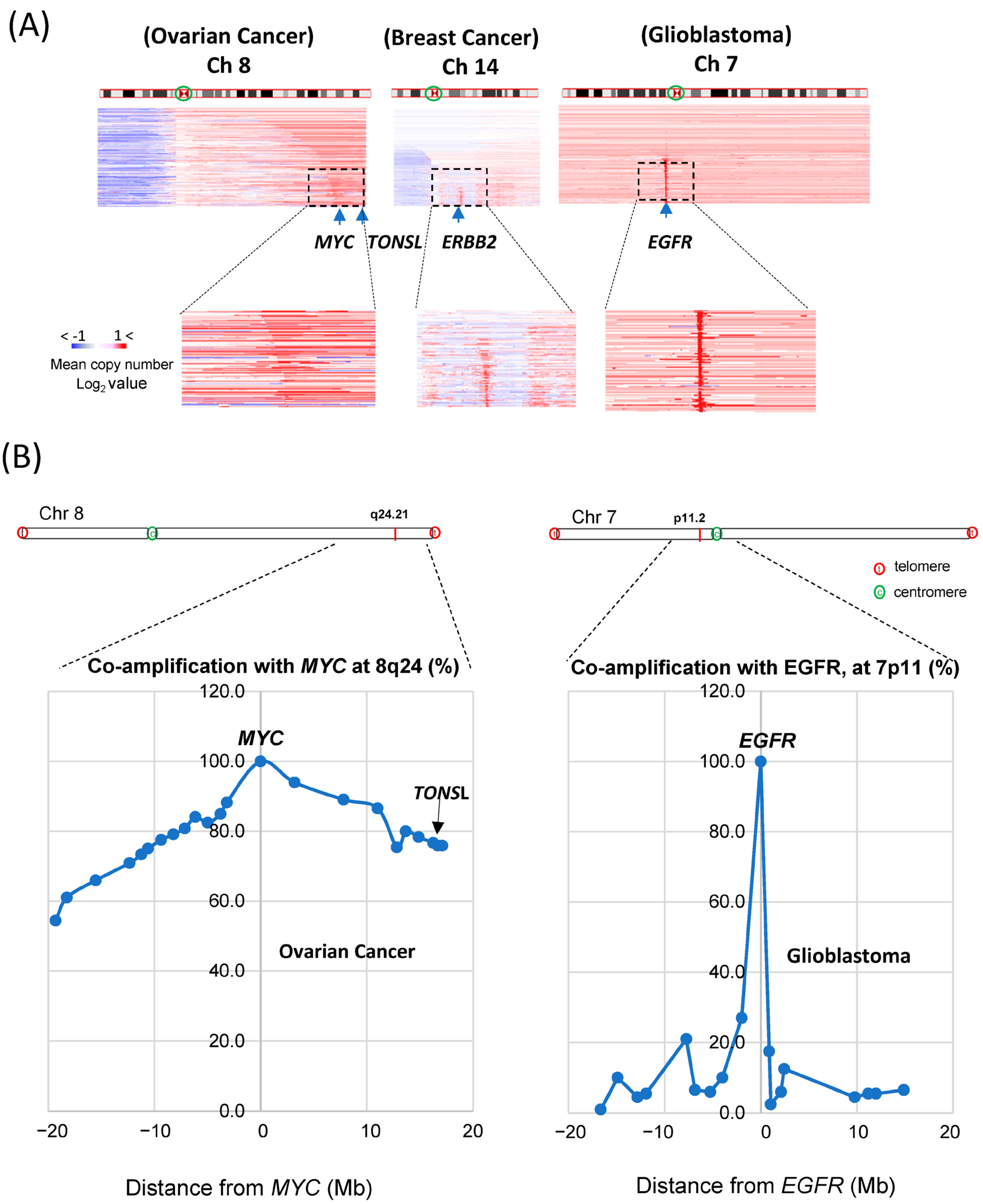
TONSL is located at the end of the long arm of chromosome 8 (Ch 8q24.3), close to where the
MYC gene is located (Ch 8q24.21). It has been reported that the long arm of chromosome 8 (Ch 8q) is the most highly amplified [
22] in the cancer genome. As
MYC is one of the most potent oncogenes, genes near
MYC might be coamplified as passenger genes, and
TONSL may be one of them. We then compared amplifications around
MYC,
ERBB2 and
EGFR in the whole chromosome view in corresponding highly amplified cancer tissues (
Figure 3A). For
MYC amplification, the whole region of Ch 8q was assessed as being largely amplified; the
TONSL gene, 17 Mb from the
MYC locus and near the telomere, was also amplified (the amplified area is shown in red throughout the arm in the figure). This may be caused by some positive or supportive effect derived from the coamplification of these genes with
MYC. In contrast,
ERBB2 and
EGFR amplifications were exclusively focal, suggesting that neighboring genes may not have a meaningful positive impact on the oncogenic roles of
EGFR and
HER2. However, it is still possible that coamplification at Ch 8q simply occurs because these regions are close in proximity to the potent oncogene
MYC. We then explored the correlation between the distance from
MYC and the coamplification rate. We collected CNV data for many genes on Ch 8q and around the MYC gene. Amplification of genes between 18 Mb up- and downstream of the
MYC gene (Ch 8q24.21) in ovarian cancer was examined (
Figure 3B, left). In total, 78% of
MYC amplification cases also showed amplification of
TONSL, which is approximately 18 Mb from
MYC and near the telomere. Interestingly, amplification of
PKHD1L1 and other genes at a similar distance from
MYC but in the opposite direction from
TONSL was observed in only 60% of
MYC-amplified cancers. Similar analysis was conducted with genes close to the
EGFR in the glioblastoma (
Figure 3B, right). The list of genes, the distance from
MYC or
EGFR and the coamplification rate are shown in
Supplementary Figure S4 and Supplementary Table S1. These data support the hypothesis that amplification of genes between
MYC and the telomere (including
TONSL, right side of
MYC in the chromosome shown in
Figure 3B) may play a supportive and/or additive role in
MYC-driven carcinogenesis. Several genes in this region mediate replication and repair, which may contribute to the oncogenic role of
MYC.
Based on the results, we hypothesized that the higher
TONSL expression in tumors compared to normal tissues, and the associated poor prognosis, is not caused by gene coamplification with
MYC alone. However, we sought to determine the effect of highly amplified
TONSL in ovarian cancer. Because ovarian cancer showed the highest gene amplification frequency of
TONSL, and the gene expression was associated with poor prognosis, we examined whether loss of
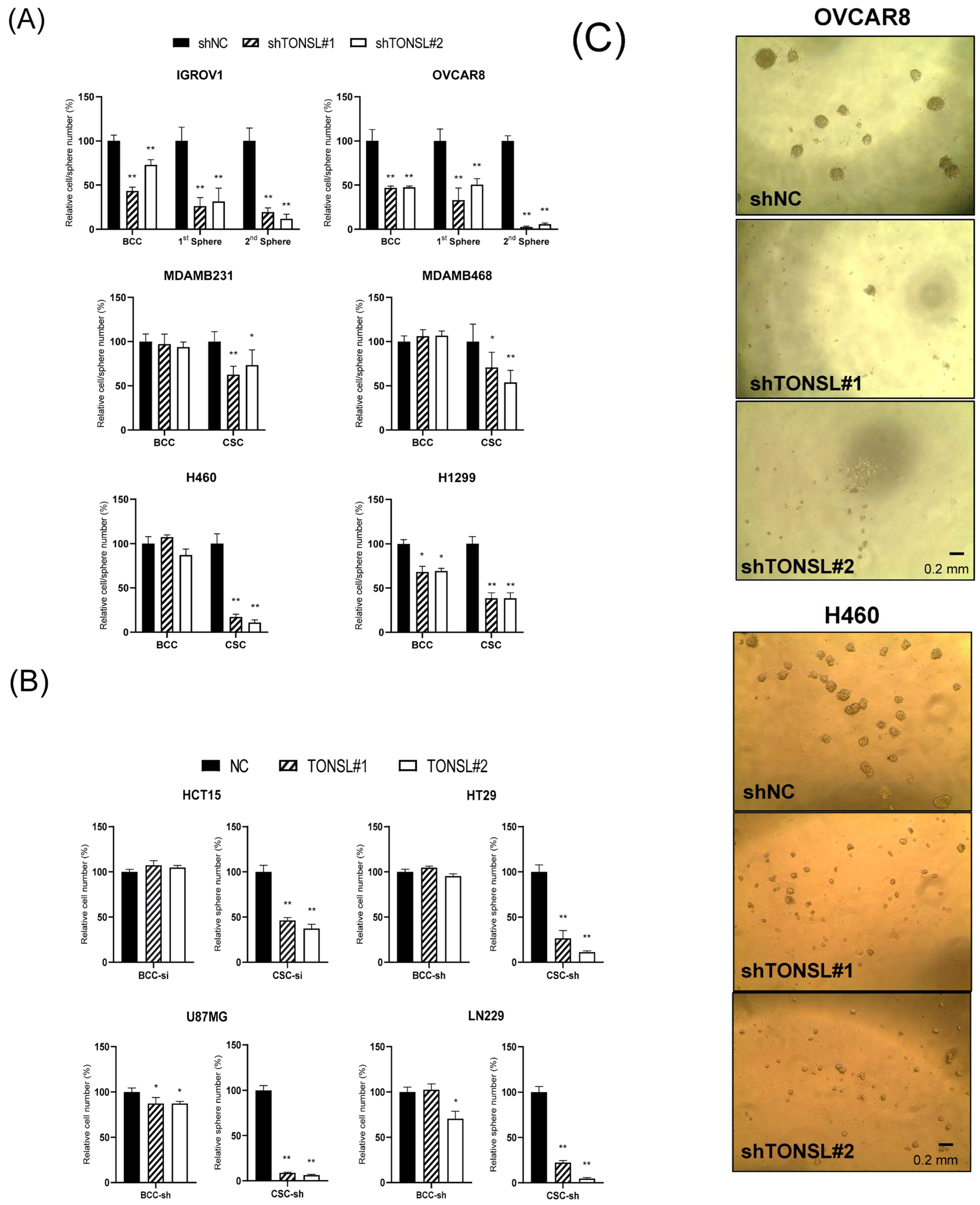
TONSL changes cancer cell behavior using ovarian serous adenocarcinoma cell lines (
Figure 4A) transduced by sh
TONSL- or shNC-expressing lentivirus. Based on the results of monolayer bulk cultured cells (BCCs) and CSC-enriched sphere cultures,
TONSL expression is required for growth in both cell populations. Notably, when
TONSL was suppressed, secondary CSC sphere formation was completely disrupted in the OVCAR8 cell line. This suggests a critical role in CSC maintenance or survival. We also confirmed the requirement of
TONSL in CSCs derived from lung (H1299 and H460) and breast (MDAMB468 and MDAMB231) cancer cell lines, of which we observed the clinical importance of in
Figure 1. Interestingly, TONSL was dispensable for BCC survival; the requirement of TONSL was always stricter in CSC than that in BCC. We also found that TONSL is required for CSCs in glioblastoma (U87MG and LN229) and colon cancer (HCT15 and HT29). (
Figure 4B). Representative images of CSCs are shown in
Figure 4C.
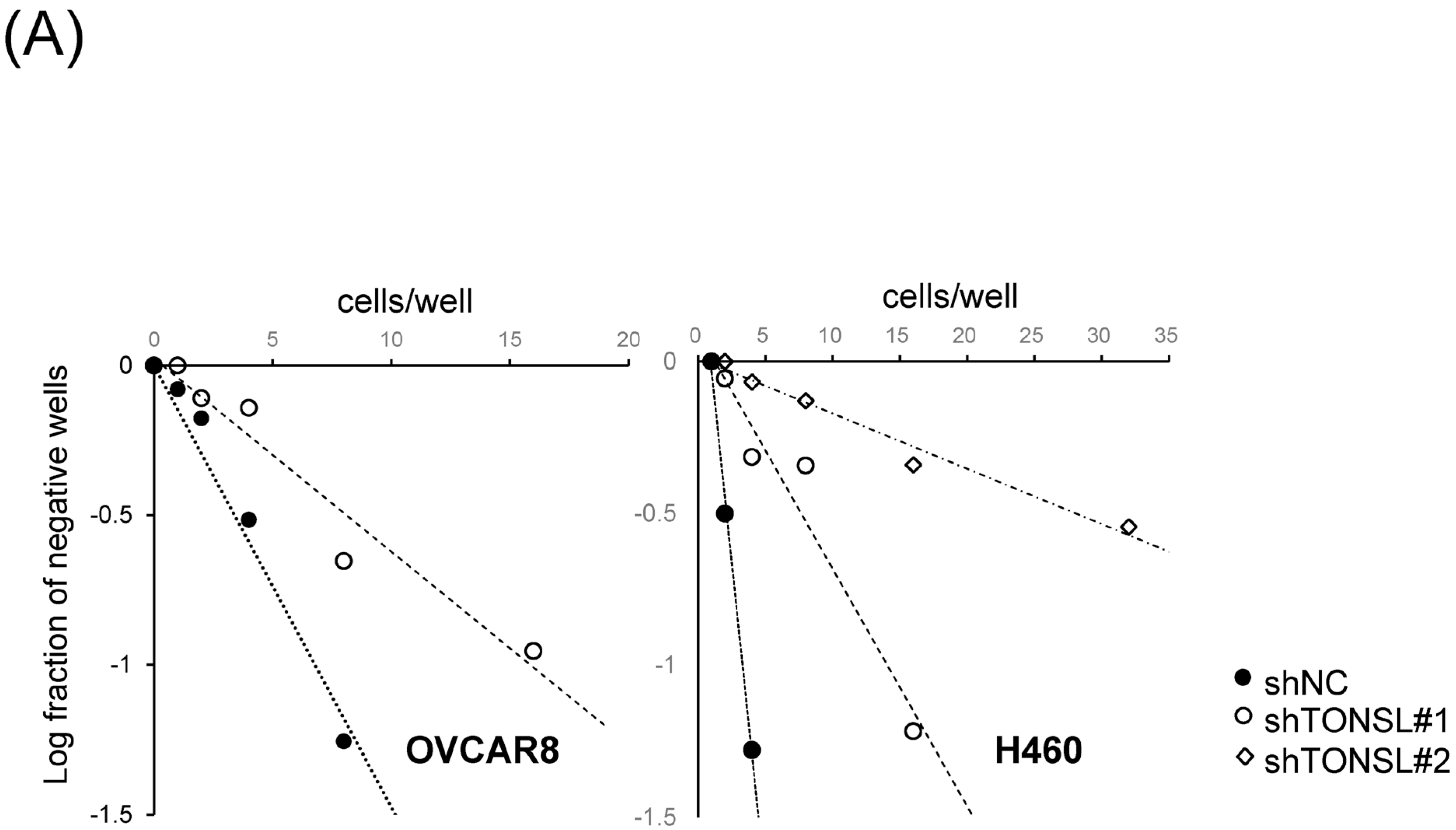
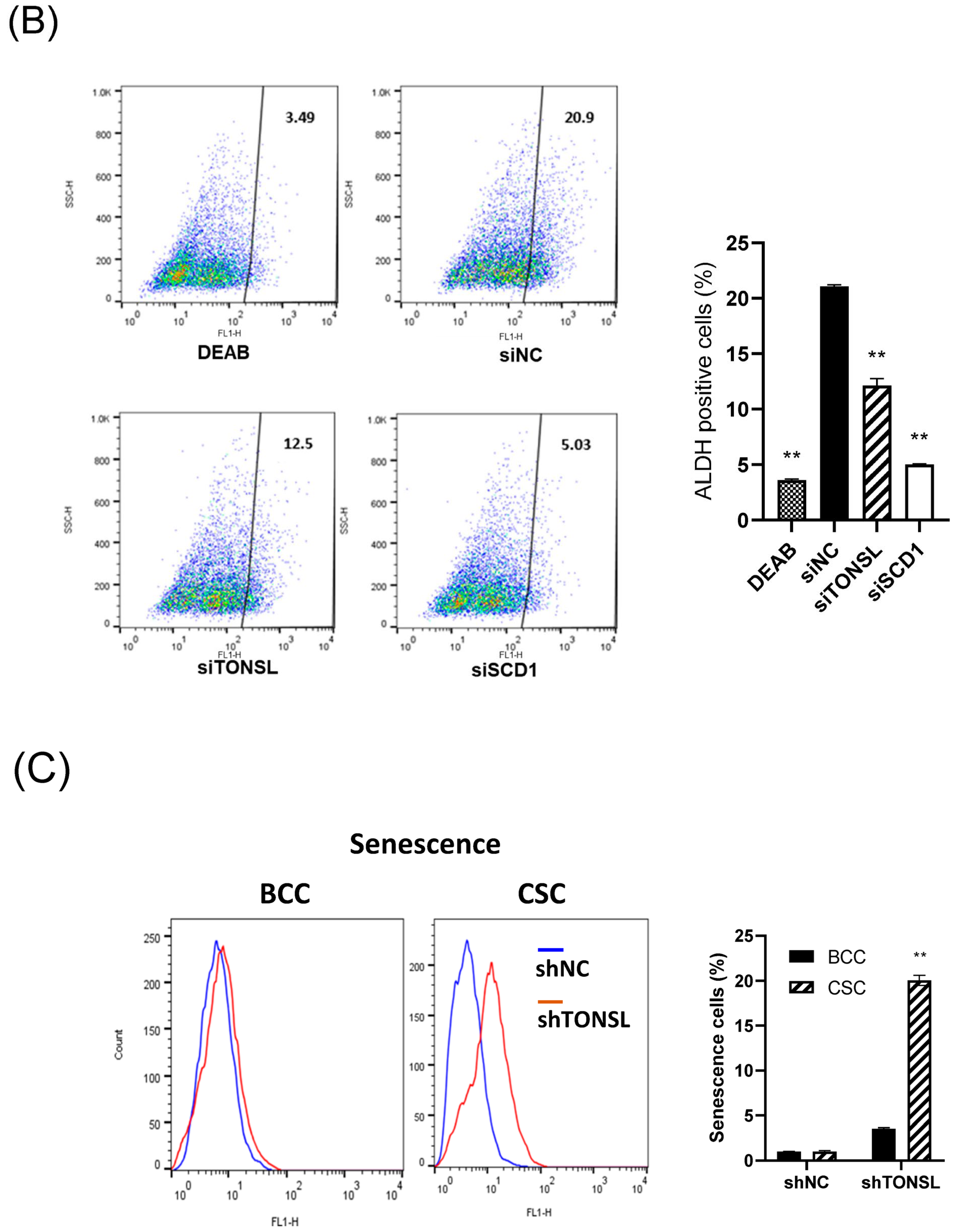
We also confirmed that the lost cells in CSC spheres due to TONSL depletion included the CSC population, using the limited dilution assay [
23] and a specific marker of CSCs, ALDH1 (
Figure 5A,B) [
23]. SCD1, which is essential for CSC survival, was used as the positive control [
24,
25]. We next investigated the mechanism by which CSCs are eliminated when TONSL is absent. Interestingly, TONSL-depleted OVCAR8 CSCs did not exhibit clear apoptosis in FACS analysis, but that of BCCs did. However, senescence increased dramatically in TONSL-depleted CSCs (
Figure 5C)
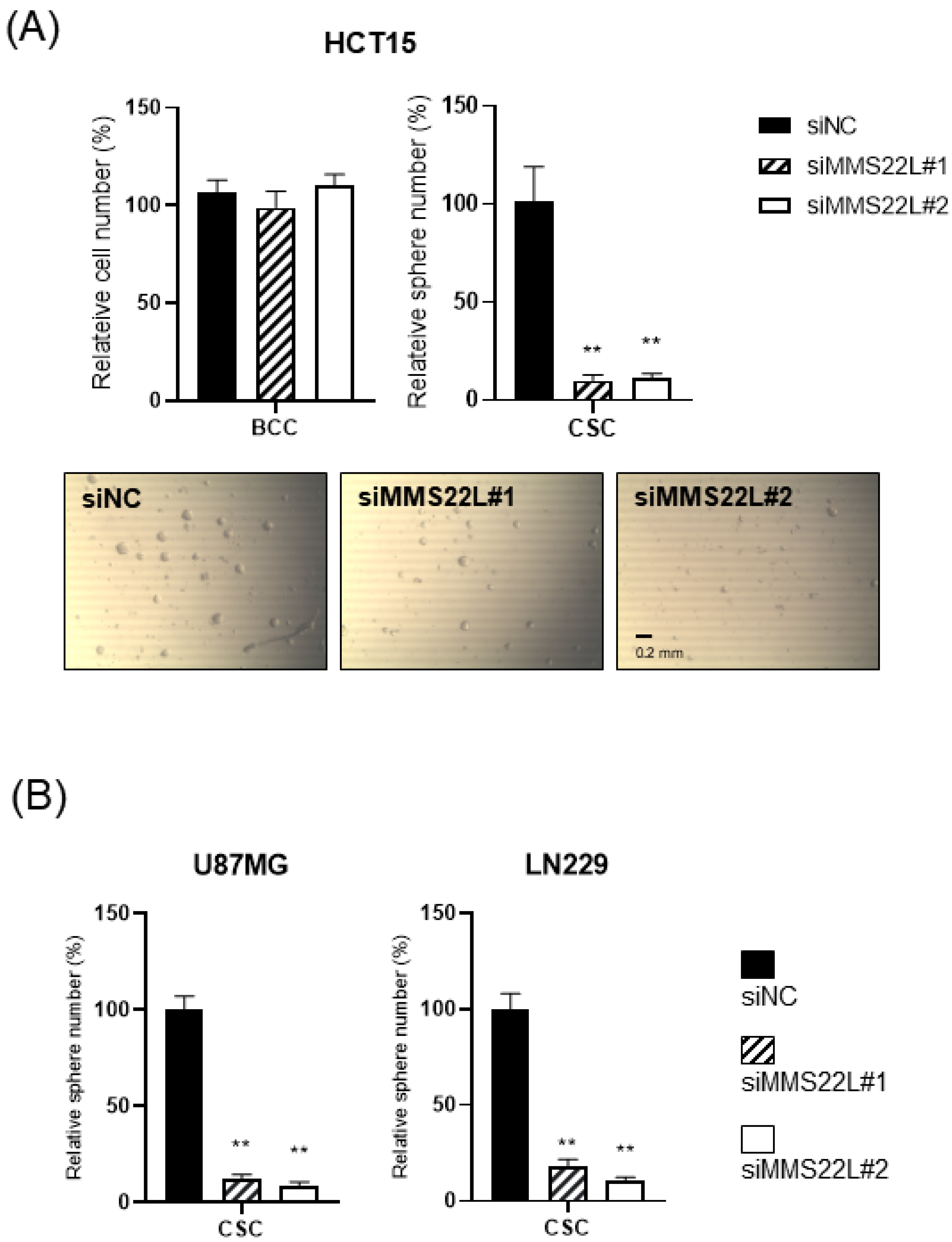
, suggesting that senescence is the main mechanism eliminating the CSC population in these ovarian cancer cells and can be different from the mechanism of BCC loss. We then tested whether the importance of TONSL in CSCs is related to its well-known role in the replication fork, which is mediated by its partner MMS22L (
Figure 6). siRNAs targeting MMS22L suppressed growth of HCT15 CSCs but had no effect on that of monolayer-cultured BCCs. This result was the same as the results acquired with
TONSL knockdown in
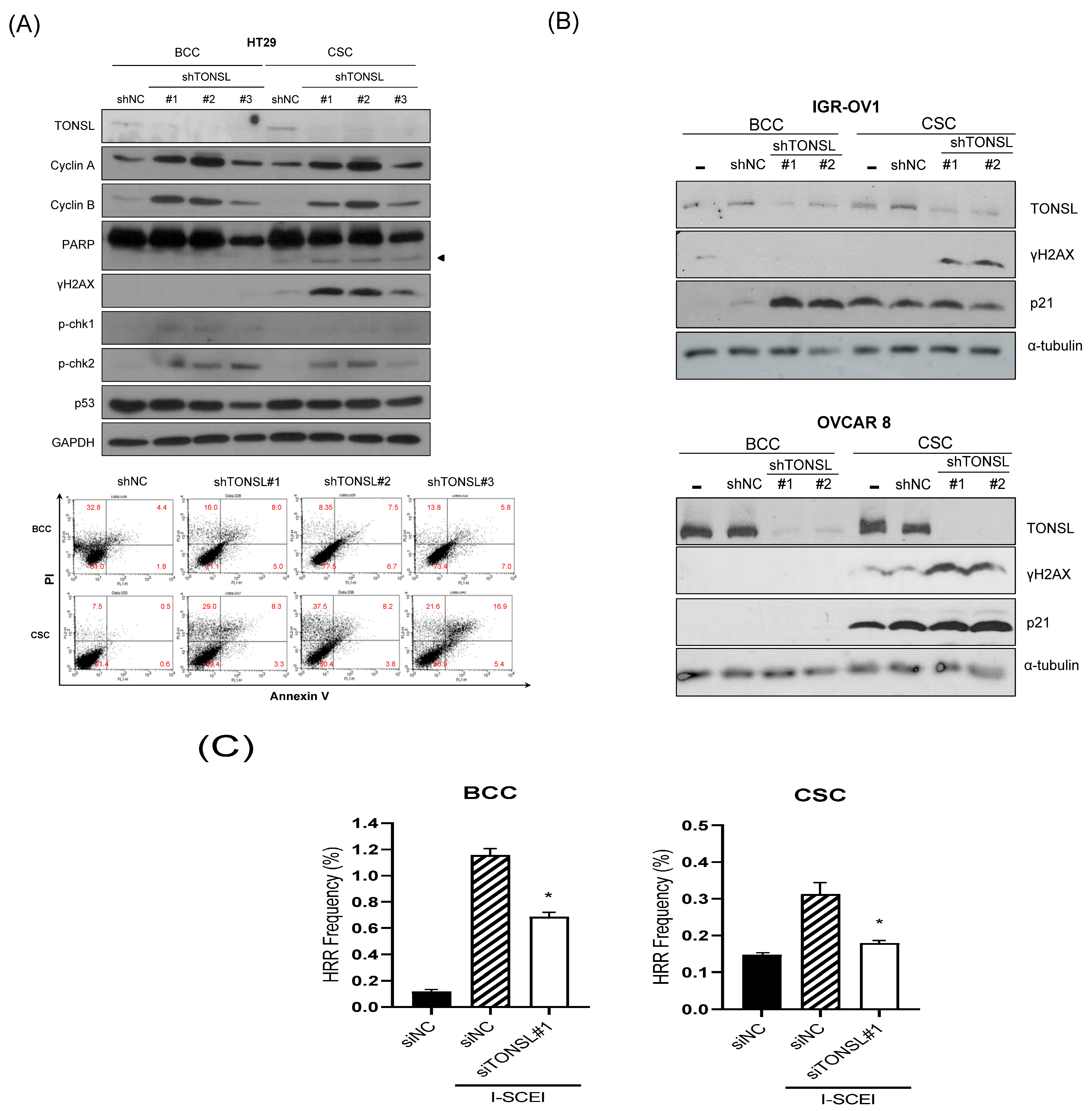
Figure 4 with HCT15. We also confirmed that CSCs in glioblastomas required MMS22L. These results strongly suggest that the complex composed of TONSL and MMS22L, which mediates HRR in stalled replication forks, plays the same pivotal role in CSC maintenance in colon cancer cells. Surprisingly, BCCs may circumvent the loss of these factors in some cancer cell lines. We explored the mechanism that resulted in differential responses in BCCs and CSCs. In the colon cancer cell HT29 (
Figure 7A), the siRNA targeting
TONSL increased the expression of G2/M cyclins (Cyclin B and Cyclin A) in both cell groups (BCC and CSC). γH2AX, the surrogate marker for dsDNA breaks, was also increased in CSCs with
TONSL knockdown, whereas the downstream signaling molecules p-chk1 and p-chk2 were increased in both BCCs and CSCs. We also detected PARP-1 cleavage in CSCs, which was not detectable in BCCs. After the loss of TONSL, more apoptotic cells were detected among CSCs by flow cytometry analysis. We also confirmed that γH2AX was increased by the suppression of
TONSL in ovarian cancers (
Figure 7B). We generated an HRR GFP reporter system in U87MG using pHPRT-DRGFP [
26]. The reporter-HRR efficiency was diminished by knockdown of
TONSL in both BCCs and CSCs of the U87MG cell line, suggesting that the HRR mechanism itself was damaged in both BCCs and CSCs (
Figure 7C).
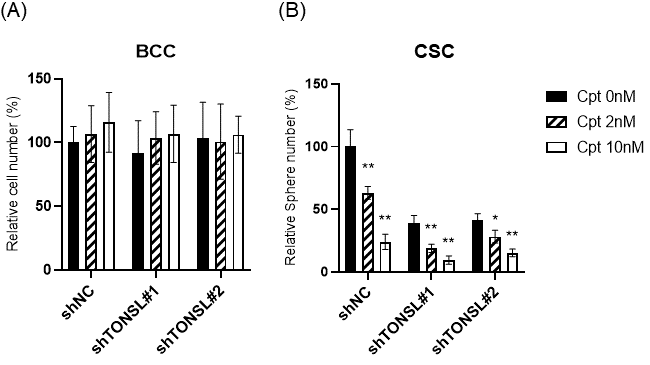
We hypothesized that TONSL loss-driven accumulation of dsDNA breaks is solved in the BCC population somehow, through error-prone nonhomologous end joining (NHEJ) or microhomology-mediated end joining (MMEJ), but accumulates in CSCs, preventing their survival. If this is true, more dsDNA breaks caused by treatment with DNA-damaging anticancer drugs should render CSCs more dependent on the TONSL/MMS22L complex. We tested this hypothesis with camptothecin (Cpt), which induces dsDNA breaks by binding to topoisomerase I [
27]. Treatment of U87MG cells with Cpt showed that Cpt suppressed CSC sphere growth (
Figure 8). The Cpt cytotoxicity (at 2 nM and 10 nM) was further enhanced by
TONSL knockdown in CSCs. However, the cytotoxic effect of Cpt on BCCs was not increased by
TONSL knockdown at the same concentration or at a much higher concentration. Therefore, it seems that TONSL plays a more critical role in genome integrity maintenance for survival in CSCs than in BCCs (
Figure 8).
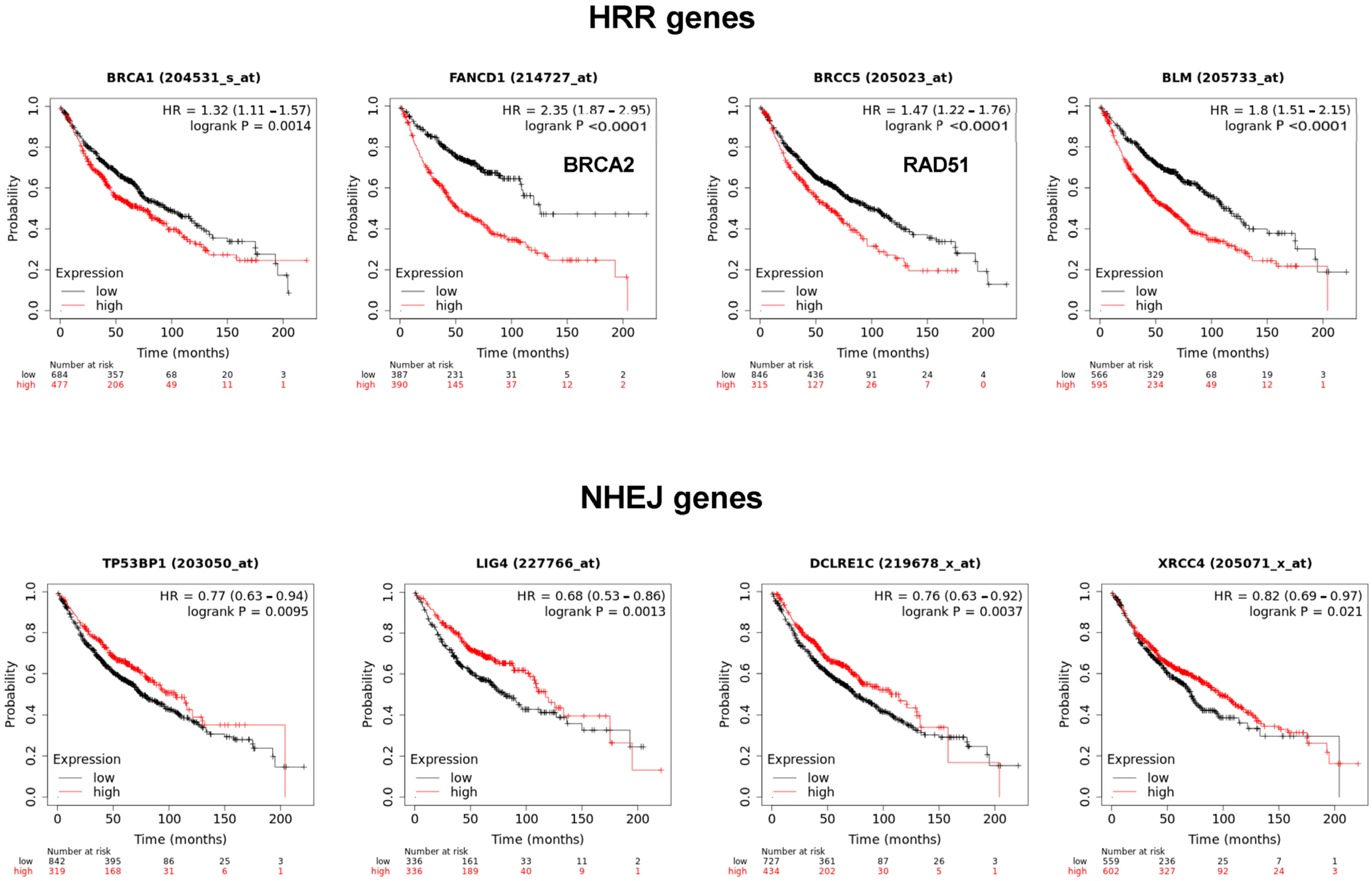
The MMS22L–TONSL complex resolves the stalled replication fork through homologous recombination repair (HRR). Many HRR mediators, such as BRCA1, BRCA2, BLM and Rad51, are directly involved in the replication fork resolution process. Accordingly, we determined whether the expression of these factors is also associated with poor prognosis of cancer, as observed with
TONSL in
Figure 1. Interestingly, the expression of
BRCA1,
BRCA2,
BLM and
RAD51 was significantly associated with poor prognosis in lung adenocarcinoma (
Figure 9). Strikingly, the factors that exclusively mediate NHEJ,
53BP1,
LIG4,
ARTEMIS and
XRCC4 were inversely associated with poor prognosis. Overall, higher expression in tumor tissue was associated with longer survival in lung adenocarcinomas, which was the opposite effect to that of
TONSL.
3. Discussion
In this study, we identified that the gene of a replication fork HRR mediator, TONSL, is amplified and transcriptionally upregulated in several cancers. Higher expression was associated with a worse survival rate in several cancer types. In addition, the loss of TONSL resulted in selective depletion of CSCs in the tested cell lines, including colon cancer, ovarian cancer and glioblastoma lines. CSC loss occurs through senescence and/or apoptosis, probably derived from accumulated DNA damage in the CSC population; in contrast, BCCs can manage the situation and survive in a subset of cell lines. The selective toxicity of TONSL loss to CSCs versus BCCs was more evident when dsDNA breakage was induced by anticancer therapeutic agents. As with TONSL, several HRR mediators at the fork are also associated with poor prognosis in lung adenocarcinoma. Conversely, NHEJ mediators are mostly associated with better survival. Because normal stem cells may invoke error-free HRR over error-prone NHEJ, CSCs may still maintain the characteristics of normal stem cells [
28].
CNVs are one of the most important classes of genomic mutations related to carcinogenesis [
29]. CNVs involve deletions or amplifications of large contiguous segments of the genome, including tumor-suppressive genes and oncogenic genes, respectively [
29]. Amplification of genes is occasionally associated with transcriptional upregulation of the amplified gene [
30]. Chromosome 8q is one of the most amplified segments in cancer [
31], and the gene
TONSL, which is located on the same arm, is also amplified in many cancers (
Supplementary Figure S3). However, as demonstrated in
Figure 2 and
Figure 3, enhanced transcription of
TONSL was detected in cancers regardless of
TONSL amplification.
Unrepaired lesions of DNA base adducts, mismatched bases and single-strand breaks can generate dsDNA breaks (DSBs) at the DNA replication fork. Unrepaired DSBs cause multiple chromosome instability (CIN) [
32], which is associated with poor prognosis, metastasis and therapeutic resistance. When DSB lesions occur, HRR, which preserves genomic integrity without errors, is invoked. However, when HRR is not available, cells activate other pathways despite junction site errors. The HRR gene group is one of the most frequently defective genes in cancer [
33]. Loss-of-function (LOF) germline mutations in HRR genes, such as
BRCA1 and
BRCA2, significantly contribute to the elevated risk of cancer in heterozygous carriers [
14,
34]. Sporadic mutations of these genes and hypermethylation of their promoters, leading to lowered expression, also contribute significantly to carcinogenesis. In addition to
BRCA1 and
BRCA2, many other HRR mediators (
ATM,
BRIP1,
CHEK2,
NBS1 or
RAD51C) are frequently defective in cancer [
14,
34]. In other words, loss of the DDR in cancer cells may contribute to the efficacy of DNA-damaging therapeutics [
35]. Specifically, homologous recombination repair (HRR) gene defects contribute to the efficacy of some DNA-damaging reagents, such as cisplatin and PARP inhibitors [
36,
37]. Therefore, HRR gene loss can contribute to both oncogenic processes and therapeutic biomarkers. Conversely, elevated expression of the essential HRR gene
TONSL in cancer versus normal tissues and its association with poor prognosis in many major cancer types is distinguished from the characteristics of many other HRR gene alterations.
TONSL is located on chromosome 8q24.3, in the region most often amplified in human cancers (up to 40% in breast cancer) [
38] and adjacent to the strong oncogenic driver
MYC (c-myc), which is located at 8q24.2. Recurrent amplification of this large region of chromosome 8q suggests that multiple genes in this segment, in addition to
MYC, support the “driver” role in oncogenesis [
19].
Figure 3 also shows that many genes (spanning over 30 Mb around
MYC) were coamplified with
MYC. Interestingly, the genes closer to telomeres (including
TONSL) than to centromeres were more frequently coamplified with
MYC (80% vs. 60%, respectively) (
Figure 3). Although we do not have sufficient data to support the idea that
TONSL itself is indeed a “driver” in carcinogenesis, it is possible that multiple genes in these regions together contribute to
MYC-driven oncogenesis. Several reported pro-proliferation genes, such as the potassium channel gene
KCNK [
39], the DNA helicase
RECQL4 [
40] and the chromatin modulator
PARP10, are also located at 8q24.3 and become coamplified. Collectively, it may be a reasonable hypothesis that some of these genes cooperate with
MYC to enhance its oncogenic “driver” effect.
Based on this hypothesis, we questioned the impact of
TONSL loss on ovarian cancer, lung cancer, breast cancer, colon cancer and glioblastoma.
TONSL loss resulted in depletion of the CSC population in many cell lines, whereas this loss had a less severe effect in the BCC population of several cell lines. In those several cell lines, CSCs required
TONSL for survival, showing that
TONSL is not dispensable for CSC survival. As TONSL-mediated HRR in the replication fork is essential, to preserve genome integrity in the repair process, the requirement of
TONSL should be the same in all cancer cells. Nevertheless, the necessity of HRR over other repair mechanisms may be more important in normal stem cells, and CSCs may be reminiscent of these stem cell characteristics. However, when HRR is not available, non-CSC cancer cells may survive through error-prone non-HRR repair processes. These processes at the stalled replication fork include MMEJ, which is mediated by DNA polymerase theta [
41].
The preference for HRR in normal germ/stem cells has been suggested multiple times. For example, stem cells barely express NHEJ genes but they do express HRR genes. Stem cells have a longer S phase to help the HRR process [
42]. In addition, the CSC preference for HRR has also been suggested in breast and gastric CSCs [
43,
44]. We also recently suggested that HRR may be indispensable for colon CSC survival [
45]. The current study shows that TONSL is indispensable to many tissue-originated CSCs, which further supports the idea that HRR can be a weak point for CSC survival.
In this study, consistently, the replication fork HRR mediator TONSL was shown to be essential for CSC survival. The elimination of CSCs is clinically important, and HRR may be a therapeutic target for this approach. Many HRR-targeting drugs that are being developed may be tested in this regard. However, the discrepancy in mutation patterns between many HRR genes and
TONSL suggests that the situation is more complicated. In addition, the
MMS22L mutation pattern, which is rarely amplified in cancer, should be considered when evaluating the significance of the oncogenic role of
TONSL. It is also necessary to consider the possibility that MMS22L plays an independent role from TONSL [
46].
In summary, we demonstrated the oncogenic role of the replication fork HRR gene TONSL, which is frequently amplified along with MYC, in the maintenance of CSC. Our findings suggest that this gene is a potential target in CSC elimination therapy. The results also indicate that CSC-preferred HRR is a vulnerable target in CSC, which is reminiscent of the normal stem cell characteristics that protect the genome integrity of stem cells. Further investigation regarding the clinical use of these potential target processes is warranted.
While this manuscript was being revised for publication, a paper was published demonstrating that TONSL is an immortalizing oncogene in breast cancer oncogenesis [
47]. The results of the present study will further increase the understanding of the role of TONSL in cancer stem cells specifically and provide a rationale for targeting TONSL to treat cancer.
4. Materials and Methods
4.1. Cell Lines
The HEK293T cell line was obtained from the Korean Cell Bank (Seoul, Republic of Korea). The human ovarian cancer cell lines IGR-OV1 and OVCAR8, the human breast cancer cell lines MDAMB231 and MDAMB468, the human lung cancer cell lines H1299 and H460, and the human colon cancer cell lines HT29 and HCT15 were obtained from the National Cancer Institute. The human glioblastoma cell lines U87MG and LN229 were obtained from ATCC (Manassas, VA, USA). All cells were cultured in RPMI-1640 or DMEM (high glucose) medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (all from Thermo) at 37 °C in a humid atmosphere containing 5% CO
2. To enrich the cancer stem cells, cells were cultured in suspension in DMEM/F12 medium supplemented with B27 supplement (Thermo), 20 ng/mL EGF and 40 ng/mL FGF (all from Thermo) on poly-HEMA-coated culture dishes (Millipore-Sigma, Burlington, MA, USA), as previously described [
39].
4.2. RNAi
siRNAs for
TONSL and
MMS22L were purchased from IDT (Coralville, IA, USA) and Genolution (Seoul, Republic of Korea), respectively. The sequences of each siRNA are listed in
Supplementary Table S2. siRNA transfections were performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Cells were assayed 3–5 days after transfection.
TONSL-specific shRNAs in the lentiviral pLKO.1 vector (TRCN0000424634, #1; TRCN0000424077, #2; and TRCN0000424443, #3) were obtained from Sigma-Aldrich (St. Louis, MO, USA), and the pLKO.1 vector was used as a control. Lentiviruses were produced in HEK293T cells by co-transfecting the shRNA-expressing vector, pMD2.G (Addgene, Watertown, MA, USA, #12259), and psPAX2 (Addgene) using jetPRIME (Polyplus-transfection, Illkirch, France) according to the manufacturer’s protocol. Cells were transduced with 5 μg/mL hexadimethrine bromide (Sigma). Two days later, the cell extract or RNA was used for western blotting or quantitative reverse transcription PCR to confirm the knockdown effect (
Supplementary Figure S5).
4.3. Western Blotting
Cells were lysed with RIPA buffer supplemented with a protease inhibitor cocktail (Sigma-Aldrich, Burlington, MA, USA), and protein concentrations in the extracts were measured using the BCA assay (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of proteins were separated by SDS–polyacrylamide gels and transferred onto PVDF membranes (Millipore-Sigma). The membrane was blocked with 5% non-fat dry milk in Tris-buffered saline containing 0.04% Tween-20 (TBST) and incubated with primary antibodies overnight at 4 °C. Antibodies against Cyclin A (#4656), Cyclin B (#4138), γH2AX (#9718), p-Chk1 (#2348), and p-Chk2 (#2197) were purchased from Cell Signaling Technology (Danvers, MA, USA), while PARP (sc-7150), p53 (SC-126), GAPDH (SC-32233), β-actin (SC-130657) and α-Tubulin (SC-23948) were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). The antibody against TONSL (ab101898) was purchased from Abcam (Cambridge, UK). The membranes were then washed with TBST and incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson Immuno Research, Philadelphia, PA, USA) for 1 h at room temperature. After washing with TBST, the immunoreactive bands were detected using ECL and visualized on X-ray films or with an Image 680 LAS (GE healthcare, Amersham, UK) imaging system.
4.4. Apoptosis Detection
For apoptosis detection, cells were transduced with shRNA, and after 72 h they were trypsinized and stained with FITC Annexin V and propidium iodide (PI) using the FITC Annexin V apoptosis detection kit from BD Biosciences (Franklin Lakes, NJ, USA). The stained cells were then analyzed using a FACSCalibur flow cytometer from BD Bioscience.
4.5. Senescence Measurement
Senescent cells were measured using the CellEvent™ Senescence Green Flow Cytometry Assay Kit (Thermo). Briefly, after shRNA transduction for 72 h, the cells were treated with 100 nM bafilomycin A1 for 1 h at 37 °C. Subsequently, the cells were incubated with C12FDG to a final concentration of 33 μM for 1 h. After harvesting, the cells were analyzed for senescence-associated β-galactosidase activity, which was indicated by raised green fluorescence, using a FACSCalibur flow cytometer.
4.6. MTS Assay for BCC and Sphere Counting for CSC
The cell viability of BCC was assessed using the CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI, USA). SiRNA-transfected cells were seeded at a density of 5 × 103 cells/200 μL into each well of a 96-well plate and cultured for 4 days. For drug treatment, the cells were then exposed to the indicated concentrations of Camptothecin or vehicle control (DMSO, final concentration below 0.1%). After 4 days of treatment, 10 μL of the MTS reagent was added to each well, and the plate was incubated at 37 °C in a humid atmosphere containing 5% CO2 for 90 min. The absorbance was measured at 450 nm using a 96-well microplate reader. Both media were supplemented with 1% penicillin and streptomycin (Welgene, Gyeongsan, Republic of Korea). CSC was cultured in 96 wells coated with POLYHEMA at a density between 250/well and 2000/well depending on cell lines. After 5–7 days, when the control cells had made 20–100 spheres with diameters greater than 100 µmeter, the samples were fixed with formaldehyde and the spheres (>100 µm) were counted under a microscope.
4.7. Quantitative Reverse Transcription PCR (qRT-PCR)
The total RNA was extracted from cells using TRIsure (Bioline, London, UK), and cDNA synthesis was performed using 2 µg of total RNA and Superscript II reverse transcriptase (Thermo). The reaction conditions involved incubation at 45 °C for 10 min, followed by 95 °C for 10 min, and then 40 cycles of amplification at 95 °C for 15 s and 60 °C for 1 min. SYBR Green (SensiFAST SYBR Hi-ROX, Bioline, London, UK) was used to quantify the PCR product, and the StepOnePlusTM Real-Time PCR system (Applied Biosystems, Waltham, MA, USA) was employed for detection. The primer sequences used are listed in
Supplementary Table S2. Relative mRNA expression levels were determined using the 2
−ΔΔCT method, with normalization to the expression levels of the housekeeping gene GAPDH.
4.8. Limiting Dilution Assay
The cells were adapted to siTONSL and plated in 96-well poly-HEMA-coated plates at various seeding densities (1–128 cells per well) containing sphere culture medium. After 7 days, the fraction of wells not containing spheres for each plating density was counted under a phase-contrast microscope. The data were then plotted against the number of cells per well.
4.9. ALDEFLUOR Assay
For the ALDEFLUOR assay, we utilized the Aldefluor kit (Stem cell Technologies, Vancouver, BC, Canada) to detect ALDH activity in cancer stem cell populations. Dissociated cells from CSC were incubated in assay buffer containing 1 μM of ALDH substrate for 30 min at 37 °C. As a negative control, a portion of ALDH substrate-treated cells were resuspended in buffer containing 15 μM of the specific ALDH enzyme inhibitor DEAB. ALDH-positive cells were quantified using FACS Calibur cytometry. The desired ALDH-positive cell population was determined based on the ALDH-positive regions set by DEAB-treated cells, which served as the control.
4.10. HRR Reporter Assay
The pHPRT-DRGFP reporter plasmid (#26476, AddGene, Cambridge, UK) was transfected into U87MG cells using Lipofectamine 2000. Single cells were seeded into 96-well plates, and Puromycin was added to establish stable clones at a final concentration of 1 μg/mL. The selected clone was then plated in a 6-well plate and transfected with 1 μg of I-SCEI plasmid (#26477, AddGene, Cambridge, UK) and 30 nM of siTONSL. After 72 h, cells were collected, and GFP-expressing cells were quantified using FACSCalibur cytometry.
4.11. Public Clinical Database Analyses
Publicly available clinical database analyses were conducted using data from The Cancer Genome Atlas (TCGA). mRNA expression in tumors and normal tissues was analyzed using Qomics (
http://qomics.sookmyung.ac.kr/). Additionally, genome copy number variations and mRNA expression data from the TCGA Pan-cancer Atlas Study were analyzed using cBioportal. To examine the association between mRNA expression and patient survival, Kaplan–Meier survival curves were generated using KM Plotter (
https://kmplot.com/analysis/). The “auto selection best cutoff” option was utilized to split patients into high- and low-expression groups based on the metadata of gene chips. The statistical significance was evaluated using the
p-value provided by the corresponding analysis program portal. A
p-value lower than 0.05 was considered statistically significant.

 and
and 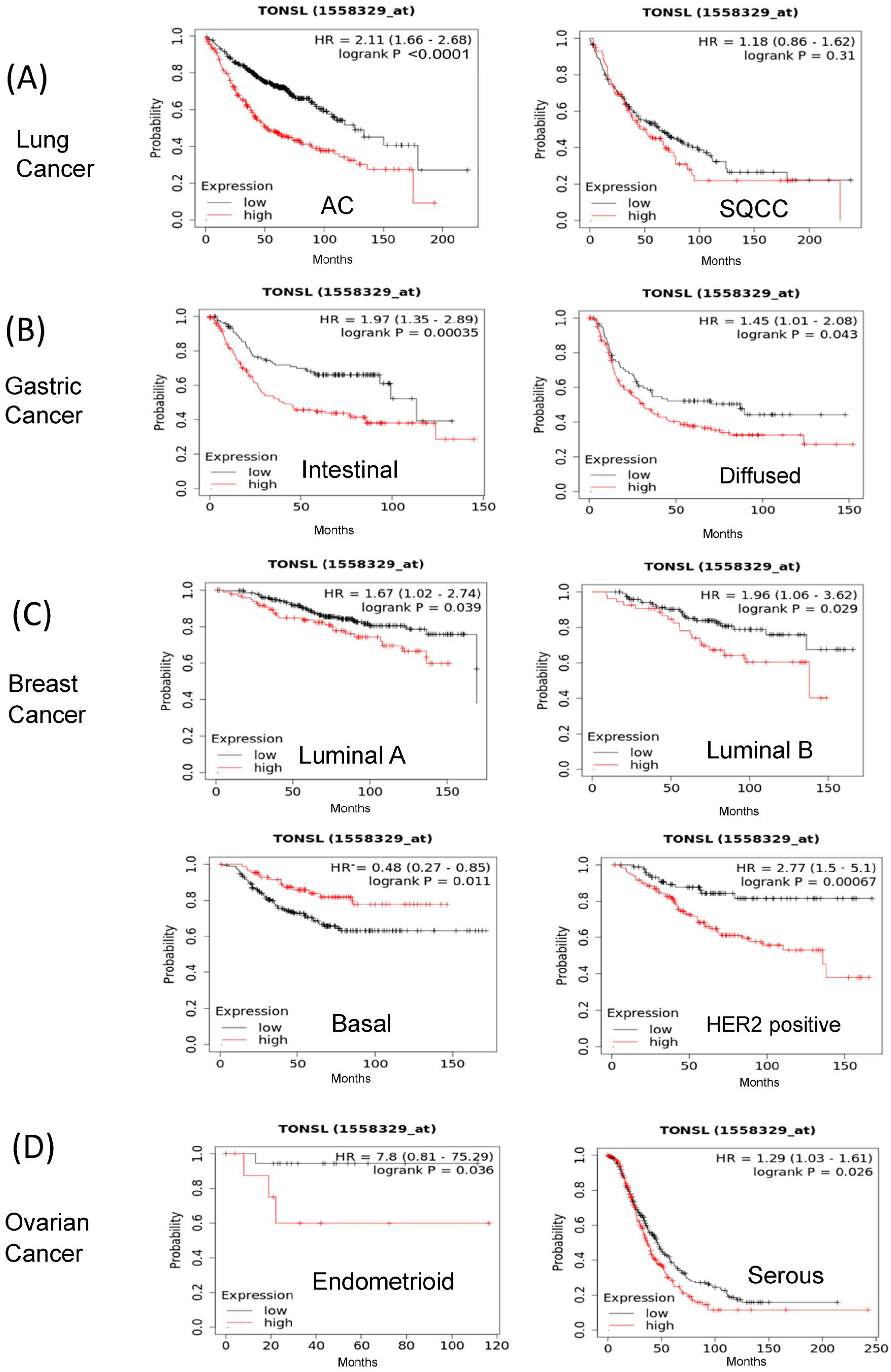{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









