
Screening and Analysis of Possible Drugs Binding to PDGFRα: A Molecular Modeling Study

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
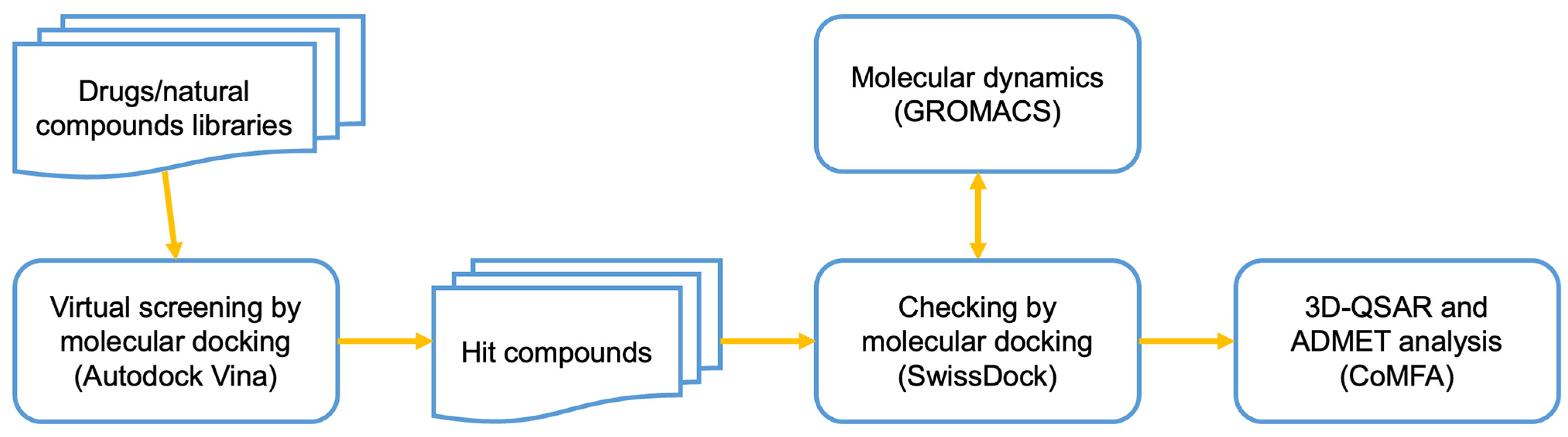
2.1. Virtual Screening of the Small Compounds Database
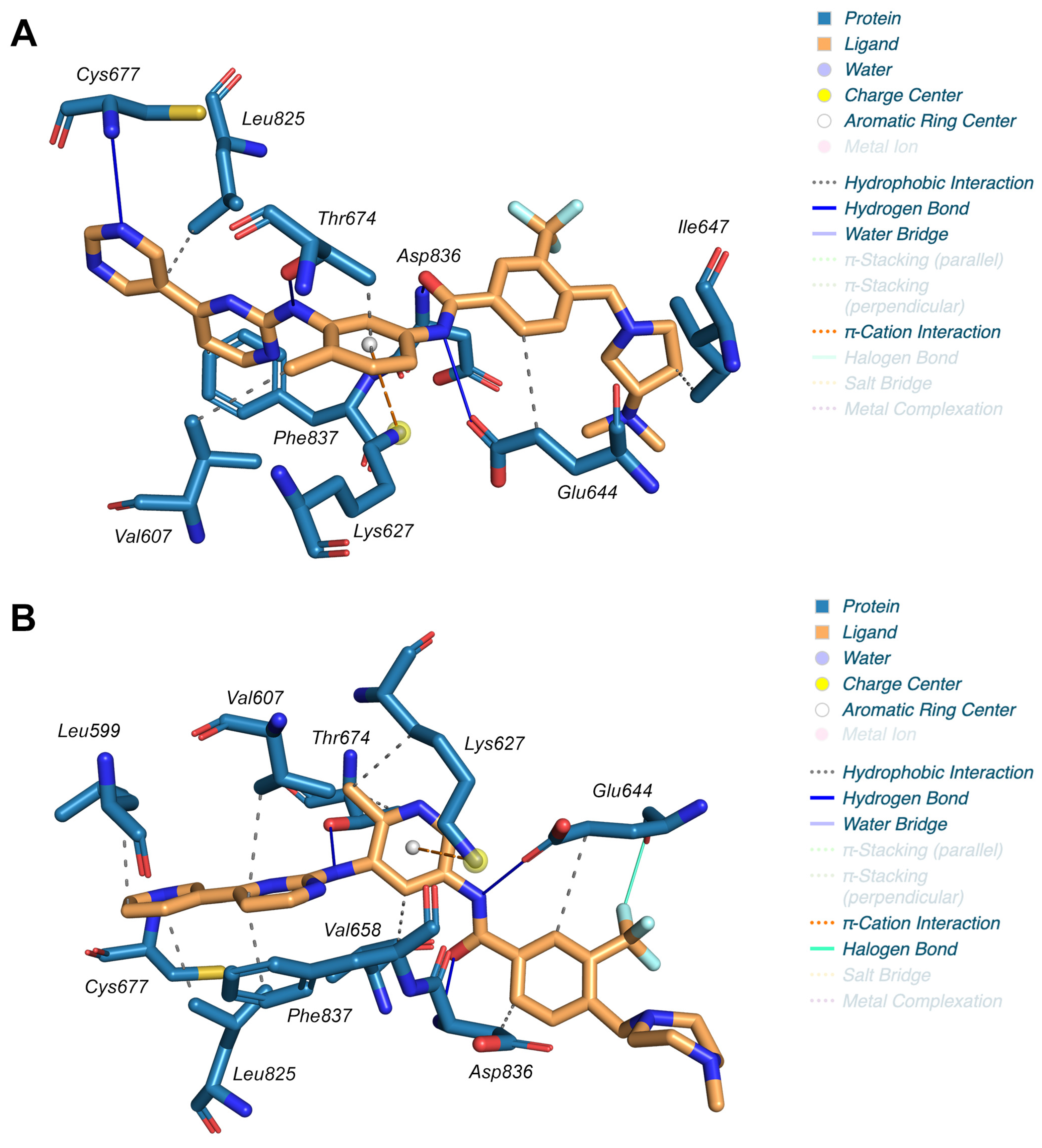
2.2. Molecular Docking
2.3. Molecular Dynamics
2.4. Three-Dimensional Quantitative Structure–Activity Relationship (3D-QSAR) and ADMET Analyses
3. Discussion
4. Materials and Methods
4.1. Virtual Screening of the Small Compounds Database
4.2. Molecular Docking
4.3. Molecular Dynamics
4.4. Three-Dimensional Quantitative Structure–Activity Relationship (3D-QSAR) and ADMET Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Winger, J.A.; Hantschel, O.; Superti-Furga, G.; Kuriyan, J. The structure of the leukemia drug imatinib bound to human quinone reductase 2 (NQO2). BMC Struct. Biol. 2009, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.S.H.; Abdel Aziz, Y.M.; Elgawish, M.S.; Said, M.M.; Abouzid, K.A.M. Design, synthesis, biological evaluation and molecular modeling study of new thieno[2,3-d]pyrimidines with anti-proliferative activity on pancreatic cancer cell lines. Bioorg. Chem. 2020, 94, 103472. [Google Scholar] [CrossRef] [PubMed]
- Keretsu, S.; Ghosh, S.; Cho, S.J. Molecular Modeling Study of c-KIT/PDGFRalpha Dual Inhibitors for the Treatment of Gastrointestinal Stromal Tumors. Int. J. Mol. Sci. 2020, 21, 8232. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhu, X.; Zhang, L.; Qin, J.J.; Feng, C.; Li, Q. In Silico Screening and Validation of PDGFRA Inhibitors Enhancing Radioiodine Sensitivity in Thyroid Cancer. Front. Pharmacol. 2022, 13, 883581. [Google Scholar] [CrossRef]
- Paolini, C.; Agarbati, S.; Benfaremo, D.; Mozzicafreddo, M.; Svegliati, S.; Moroncini, G. PDGF/PDGFR: A Possible Molecular Target in Scleroderma Fibrosis. Int. J. Mol. Sci. 2022, 23, 3904. [Google Scholar] [CrossRef] [PubMed]
- Moroncini, G.; Grieco, A.; Nacci, G.; Paolini, C.; Tonnini, C.; Pozniak, K.N.; Cuccioloni, M.; Mozzicafreddo, M.; Svegliati, S.; Angeletti, M.; et al. Epitope Specificity Determines Pathogenicity and Detectability of Anti-Platelet-Derived Growth Factor Receptor alpha Autoantibodies in Systemic Sclerosis. Arthritis Rheumatol. 2015, 67, 1891–1903. [Google Scholar] [CrossRef]
- Makino, K.; Makino, T.; Stawski, L.; Mantero, J.C.; Lafyatis, R.; Simms, R.; Trojanowska, M. Blockade of PDGF Receptors by Crenolanib Has Therapeutic Effect in Patient Fibroblasts and in Preclinical Models of Systemic Sclerosis. J. Invest. Dermatol. 2017, 137, 1671–1681. [Google Scholar] [CrossRef]
- Huang, J.; Beyer, C.; Palumbo-Zerr, K.; Zhang, Y.; Ramming, A.; Distler, A.; Gelse, K.; Distler, O.; Schett, G.; Wollin, L.; et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann. Rheum. Dis. 2016, 75, 883–890. [Google Scholar] [CrossRef]
- Benfaremo, D.; Svegliati, S.; Paolini, C.; Agarbati, S.; Moroncini, G. Systemic Sclerosis: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2022, 10, 163. [Google Scholar] [CrossRef]
- Grzesk, G.; Wozniak-Wisniewska, A.; Blazejewski, J.; Gorny, B.; Wolowiec, L.; Rogowicz, D.; Nowaczyk, A. The Interactions of Nintedanib and Oral Anticoagulants-Molecular Mechanisms and Clinical Implications. Int. J. Mol. Sci. 2020, 22, 282. [Google Scholar] [CrossRef]
- Liang, L.; Yan, X.E.; Yin, Y.; Yun, C.H. Structural and biochemical studies of the PDGFRA kinase domain. Biochem. Biophys. Res. Commun. 2016, 477, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Seeliger, M.A. Targeting conformational plasticity of protein kinases. ACS Chem. Biol. 2015, 10, 190–200. [Google Scholar] [CrossRef]
- Moroncini, G.; Maccaroni, E.; Fiordoliva, I.; Pellei, C.; Gabrielli, A.; Berardi, R. Developments in the management of advanced soft-tissue sarcoma—olaratumab in context. OncoTargets Ther. 2018, 11, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, F.A.; Piera-Velazquez, S.; Jimenez, S.A. Tyrosine kinases in the pathogenesis of tissue fibrosis in systemic sclerosis and potential therapeutic role of their inhibition. Transl. Res. 2021, 231, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Moroncini, G.; Cuccioloni, M.; Mozzicafreddo, M.; Pozniak, K.N.; Grieco, A.; Paolini, C.; Tonnini, C.; Spadoni, T.; Svegliati, S.; Funaro, A.; et al. Characterization of binding and quantification of human autoantibodies to PDGFRalpha using a biosensor-based approach. Anal. Biochem. 2017, 528, 26–33. [Google Scholar] [CrossRef]
- Svegliati, S.; Amico, D.; Spadoni, T.; Fischetti, C.; Finke, D.; Moroncini, G.; Paolini, C.; Tonnini, C.; Grieco, A.; Rovinelli, M.; et al. Agonistic Anti-PDGF Receptor Autoantibodies from Patients with Systemic Sclerosis Impact Human Pulmonary Artery Smooth Muscle Cells Function In Vitro. Front. Immunol. 2017, 8, 75. [Google Scholar] [CrossRef]
- Chu, J.S.; Ge, F.J.; Zhang, B.; Wang, Y.; Silvestris, N.; Liu, L.J.; Zhao, C.H.; Lin, L.; Brunetti, A.E.; Fu, Y.L.; et al. Expression and prognostic value of VEGFR-2, PDGFR-beta, and c-Met in advanced hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2013, 32, 16. [Google Scholar] [CrossRef]
- Huang, W.; Fridman, Y.; Bonfil, R.D.; Ustach, C.V.; Conley-LaComb, M.K.; Wiesner, C.; Saliganan, A.; Cher, M.L.; Kim, H.R. A novel function for platelet-derived growth factor D: Induction of osteoclastic differentiation for intraosseous tumor growth. Oncogene 2012, 31, 4527–4535. [Google Scholar] [CrossRef]
- Jechlinger, M.; Sommer, A.; Moriggl, R.; Seither, P.; Kraut, N.; Capodiecci, P.; Donovan, M.; Cordon-Cardo, C.; Beug, H.; Grunert, S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Invest. 2006, 116, 1561–1570. [Google Scholar] [CrossRef]
- Lin, C.L.; Tsai, M.L.; Chen, Y.H.; Liu, W.N.; Lin, C.Y.; Hsu, K.W.; Huang, C.Y.; Chang, Y.J.; Wei, P.L.; Chen, S.H.; et al. Platelet-Derived Growth Factor Receptor-alpha Subunit Targeting Suppresses Metastasis in Advanced Thyroid Cancer In Vitro and In Vivo. Biomol. Ther. 2021, 29, 551–561. [Google Scholar] [CrossRef]
- Lopez-Campistrous, A.; Adewuyi, E.E.; Benesch, M.G.K.; Ko, Y.M.; Lai, R.; Thiesen, A.; Dewald, J.; Wang, P.; Chu, K.; Ghosh, S.; et al. PDGFRalpha Regulates Follicular Cell Differentiation Driving Treatment Resistance and Disease Recurrence in Papillary Thyroid Cancer. EBioMedicine 2016, 12, 86–97. [Google Scholar] [CrossRef]
- Matei, D.; Kelich, S.; Cao, L.; Menning, N.; Emerson, R.E.; Rao, J.; Jeng, M.H.; Sledge, G.W. PDGF BB induces VEGF secretion in ovarian cancer. Cancer Biol. Ther. 2007, 6, 1951–1959. [Google Scholar] [CrossRef]
- Heldin, C.H.; Lennartsson, J. Structural and functional properties of platelet-derived growth factor and stem cell factor receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009100. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. The role of small molecule platelet-derived growth factor receptor (PDGFR) inhibitors in the treatment of neoplastic disorders. Pharmacol. Res. 2018, 129, 65–83. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef]
- Alvarez, R.H.; Kantarjian, H.M.; Cortes, J.E. Biology of platelet-derived growth factor and its involvement in disease. Mayo Clin. Proc. 2006, 81, 1241–1257. [Google Scholar] [CrossRef]
- Bonner, J.C. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor. Rev. 2004, 15, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Trojanowska, M. Role of PDGF in fibrotic diseases and systemic sclerosis. Rheumatology 2008, 47 (Suppl. 5), v2–v4. [Google Scholar] [CrossRef] [PubMed]
- Aarthy, M.; Panwar, U.; Selvaraj, C.; Singh, S.K. Advantages of Structure-Based Drug Design Approaches in Neurological Disorders. Curr. Neuropharmacol. 2017, 15, 1136–1155. [Google Scholar] [CrossRef]
- Huckleby, A.E.; Saul, J.G.; Shin, H.; Desmarais, S.; Bokka, A.; Jeon, J.; Kim, S.K. Development of Hydroxamic Acid Compounds for Inhibition of Metallo-beta-Lactamase from Bacillus anthracis. Int. J. Mol. Sci. 2022, 23, 9163. [Google Scholar] [CrossRef]
- Santos, F.P.; Kantarjian, H.; Cortes, J.; Quintas-Cardama, A. Bafetinib, a dual Bcr-Abl/Lyn tyrosine kinase inhibitor for the potential treatment of leukemia. Curr. Opin. Investig. Drugs 2010, 11, 1450–1465. [Google Scholar]
- Kantarjian, H.; le Coutre, P.; Cortes, J.; Pinilla-Ibarz, J.; Nagler, A.; Hochhaus, A.; Kimura, S.; Ottmann, O. Phase 1 study of INNO-406, a dual Abl/Lyn kinase inhibitor, in Philadelphia chromosome-positive leukemias after imatinib resistance or intolerance. Cancer 2010, 116, 2665–2672. [Google Scholar] [CrossRef]
- Campochiaro, C.; De Luca, G.; Lazzaroni, M.G.; Armentaro, G.; Spinella, A.; Vigone, B.; Ruaro, B.; Stanziola, A.; Benfaremo, D.; De Lorenzis, E.; et al. Real-life efficacy and safety of nintedanib in systemic sclerosis-interstitial lung disease: Data from an Italian multicentre study. RMD Open. 2023, 9, e002850. [Google Scholar] [CrossRef]
- Labbe, C.M.; Rey, J.; Lagorce, D.; Vavrusa, M.; Becot, J.; Sperandio, O.; Villoutreix, B.O.; Tuffery, P.; Miteva, M.A. MTiOpenScreen: A web server for structure-based virtual screening. Nucleic Acids Res. 2015, 43, W448–W454. [Google Scholar] [CrossRef]
- Lagarde, N.; Rey, J.; Gyulkhandanyan, A.; Tuffery, P.; Miteva, M.A.; Villoutreix, B.O. Online structure-based screening of purchasable approved drugs and natural compounds: Retrospective examples of drug repositioning on cancer targets. Oncotarget 2018, 9, 32346–32361. [Google Scholar] [CrossRef]
- Ferraro, G.; Mozzicafreddo, M.; Ettari, R.; Corsi, L.; Monti, M.C. A Proteomic Platform Unveils the Brain Glycogen Phosphorylase as a Potential Therapeutic Target for Glioblastoma Multiforme. Int. J. Mol. Sci. 2022, 23, 8200. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. Fast docking using the CHARMM force field with EADock DSS. J. Comput. Chem. 2011, 32, 2149–2159. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef]
- Ragno, R. www.3d-qsar.com: A web portal that brings 3-D QSAR to all electronic devices-the Py-CoMFA web application as tool to build models from pre-aligned datasets. J. Comput. Aided Mol. Des. 2019, 33, 855–864. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Database | ΔG (kcal/mol) | Kd,pred (M) |
|---|---|---|---|
| Bafetinib | Drugs-lib | −12.26 | 1.01 × 10−9 |
| Radotinib | Drugs-lib | −11.74 | 2.43 × 10−9 |
| Imatinib | Drugs-lib | −11.67 | 2.74 × 10−9 |
| Flumatinib | Drugs-lib | −11.64 | 2.88 × 10−9 |
| Nilotinib | Drugs-lib | −10.93 | 9.53 × 10−9 |
| Ag-13958 | Drugs-lib | −10.79 | 1.21 × 10−8 |
| Tg100-801 | Drugs-lib | −10.76 | 1.28 × 10−8 |
| Ditercalinium | Drugs-lib | −10.15 | 3.60 × 10−8 |
| Dasatinib | Drugs-lib | −10.06 | 4.14 × 10−8 |
| R428_Bemcentinib | Drugs-lib | −9.98 | 4.78 × 10−8 |
| Glisolamide | Drugs-lib | −9.91 | 5.35 × 10−8 |
| MolPort-002-524-598 | NP-lib | −9.83 | 6.19 × 10−8 |
| Bms-833923 | Drugs-lib | −9.53 | 1.01 × 10−7 |
| Benfluorex | Drugs-lib | −9.40 | 1.28 × 10−7 |
| MolPort-039-052-621 | NP-lib | −9.23 | 1.69 × 10−7 |
| MolPort-009-018-791_Curcumin | NP-lib | −9.10 | 2.12 × 10−7 |
| 47194043_iPPI | iPPI-lib | −9.08 | 2.18 × 10−7 |
| Crenolanib | Drugs-lib | −9.08 | 2.18 × 10−7 |
| MolPort-001-740-946_luteolin-7-O-glucuronide | NP-lib | −9.07 | 2.21 × 10−7 |
| Nintedanib | Drugs-lib | −8.80 | 3.48 × 10−7 |
| 24301892_iPPI | iPPI-lib | −8.78 | 3.60 × 10−7 |
| 24824231_div | Diverse-lib | −8.74 | 3.88 × 10−7 |
| MolPort-001-741-358_EGCG | NP-lib | −8.70 | 4.18 × 10−7 |
| 29215783_iPPI | iPPI-lib | −8.48 | 6.00 × 10−7 |
| MolPort-001-740-557_Quercetin | NP-lib | −8.40 | 6.88 × 10−7 |
| Hispaglabridin_B | Food-lib | −8.36 | 7.38 × 10−7 |
| MolPort-001-768-161_Binaphthalene | NP-lib | −8.06 | 1.23 × 10−6 |
| Complex | Coulombic Interaction Energy (kJ/mol) | Lennard–Jones Energy (kJ/mol) |
|---|---|---|
| Bafetinib/hiPDGFRα | −135.72 ± 0.96 | −257.94 ± 0.60 |
| Radotinib/hiPDGFRα | −142.66 ± 1.10 | −234.45 ± 1.50 |
| Imatinib/hiPDGFRα | −121.95 ± 1.20 | −234.49 ± 1.80 |
| Flumatinib/hiPDGFRα | −135.79 ± 2.10 | −244.41 ± 3.00 |
| Field | r2 | q2 | Optimal PC |
|---|---|---|---|
| STE | 1.000 | 0.525 | 5 |
| ELE | 0.761 | 0.574 | 4 |
| STE-ELE | 0.999 | 0.692 | 6 |
| Compound | Lipinski | Pfizer | GSK | Golden Triangle | MW | LogP | LogD | nHA | nHD | TPSA |
|---|---|---|---|---|---|---|---|---|---|---|
| Recommended Range | 100/600 | 0/3 | 1/3 | 0/12 | 0/7 | 0/140 | ||||
| Benfluorex | Accepted | Rejected | Rejected | Accepted | 351.14 | 4.229 | 4.039 | 3 | 1 | 38.33 |
| Glisolamide | Accepted | Accepted | Rejected | Accepted | 434.16 | 2.953 | 1.325 | 9 | 3 | 130.4 |
| Imatinib | Accepted | Accepted | Rejected | Accepted | 493.26 | 3.805 | 3.144 | 8 | 2 | 89.51 |
| Nintedanib | Accepted | Accepted | Rejected | Rejected | 539.25 | 3.466 | 3.036 | 9 | 2 | 101.47 |
| Nilotinib | Accepted | Accepted | Rejected | Rejected | 529.18 | 4.894 | 3.881 | 8 | 2 | 100.85 |
| Radotinib | Accepted | Accepted | Rejected | Rejected | 530.18 | 4.574 | 3.643 | 9 | 2 | 113.74 |
| Flumatinib | Accepted | Accepted | Rejected | Rejected | 562.24 | 4.043 | 3.27 | 9 | 2 | 102.4 |
| Ag-13958 | Accepted | Accepted | Rejected | Accepted | 467.19 | 4.625 | 3.476 | 8 | 3 | 100.52 |
| R428_Bemcentinib | Accepted | Accepted | Rejected | Rejected | 506.29 | 4.54 | 4.017 | 8 | 3 | 101.74 |
| Bafetinib | Accepted | Accepted | Rejected | Rejected | 576.26 | 4.276 | 3.518 | 9 | 2 | 102.4 |
| Ditercalinium | Rejected | Rejected | Rejected | Rejected | 718.4 | 7.562 | 5.014 | 8 | 2 | 64.28 |
| Bms-833923 | Accepted | Accepted | Rejected | Accepted | 473.22 | 5.193 | 3.902 | 6 | 3 | 82.17 |
| Tg100-801 | Rejected | Accepted | Rejected | Rejected | 579.2 | 6.745 | 4.857 | 8 | 1 | 92.7 |
| Hispaglabridin_B | Accepted | Rejected | Rejected | Rejected | 390.18 | 6.757 | 5.324 | 4 | 1 | 47.92 |
| Crenolanib | Accepted | Accepted | Rejected | Accepted | 443.23 | 4.34 | 3.093 | 7 | 2 | 78.43 |
| Dasatinib | Accepted | Accepted | Rejected | Accepted | 487.16 | 2.807 | 2.922 | 9 | 3 | 109.74 |
| MolPort-001-740-557_Quercetin | Accepted | Accepted | Accepted | Accepted | 302.04 | 2.155 | 1.767 | 7 | 5 | 131.36 |
| MolPort-001-768-161_Binaphthalene | Accepted | Rejected | Rejected | Accepted | 254.11 | 6.02 | 4.668 | 0 | 0 | 0 |
| MolPort-001-741-358_EGCG | Rejected | Accepted | Rejected | Accepted | 458.08 | 1.893 | 0.652 | 11 | 8 | 197.37 |
| MolPort-009-018-791_Curcumin | Accepted | Accepted | Accepted | Accepted | 368.13 | 2.742 | 2.82 | 6 | 2 | 93.06 |
| MolPort-039-052-621 | Rejected | Accepted | Rejected | Rejected | 542.12 | 5.021 | 3.33 | 10 | 5 | 162.98 |
| MolPort-002-524-598 | Rejected | Accepted | Rejected | Rejected | 596.14 | −0.524 | −0.3 | 16 | 10 | 269.43 |
| MolPort-001-740-946_Luteolin-7-glucuronide | Rejected | Accepted | Rejected | Accepted | 462.08 | 0.864 | 0.745 | 12 | 7 | 207.35 |
| 29215783_iPPI | Accepted | Rejected | Rejected | Accepted | 410.08 | 5.88 | 4.167 | 4 | 0 | 48.67 |
| 24301892_iPPI | Accepted | Accepted | Rejected | Accepted | 454.13 | 5.425 | 4.104 | 7 | 1 | 77.75 |
| 47194043_iPPI | Accepted | Rejected | Rejected | Accepted | 354.14 | 4.824 | 3.525 | 4 | 2 | 61.96 |
| 24824231_div | Accepted | Accepted | Accepted | Accepted | 330.1 | 2.863 | 2.829 | 5 | 2 | 75.27 |
| Compound | QED | NP-Likeness | Caco-2 | MDCK | Carcinogenicity | SkinSen | EC | EI | Respiratory | LD50 Oral |
|---|---|---|---|---|---|---|---|---|---|---|
| Recommended Range | >0.67 | −5/5 | >−5.15 | >2.0 × 10−6 | 0/0.3 Excellent; 0.3/0.7 Medium; 0.7/1 Poor | 0 | ||||
| Benfluorex | 0.603 | −0.95 | −4.62 | 2.19 × 10−5 | 0.046 | 0.033 | 0.003 | 0.011 | 0.686 | 0 |
| Glisolamide | 0.613 | −1.57 | −5.447 | 1.56 × 10−5 | 0.026 | 0.109 | 0.003 | 0.007 | 0.007 | 0 |
| Imatinib | 0.424 | −1.477 | −5.576 | 7.27 × 10−6 | 0.044 | 0.946 | 0.003 | 0.007 | 0.992 | 0 |
| Nintedanib | 0.272 | −1.176 | −5.716 | 2 × 10−5 | 0.078 | 0.75 | 0.003 | 0.007 | 0.694 | 0 |
| Nilotinib | 0.302 | −1.657 | −5.112 | 1.12 × 10−5 | 0.027 | 0.932 | 0.003 | 0.009 | 0.928 | 0 |
| Radotinib | 0.325 | −1.62 | −5.065 | 1.06 × 10−5 | 0.021 | 0.944 | 0.003 | 0.009 | 0.954 | 0 |
| Flumatinib | 0.361 | −1.566 | −5.492 | 7.93 × 10−6 | 0.028 | 0.948 | 0.003 | 0.006 | 0.988 | 0 |
| Ag-13958 | 0.314 | −1.989 | −5.46 | 6.03 × 10−6 | 0.591 | 0.245 | 0.003 | 0.013 | 0.986 | 0 |
| R428_Bemcentinib | 0.364 | −0.866 | −5.342 | 6.99 × 10−6 | 0.95 | 0.581 | 0.003 | 0.007 | 0.74 | 0 |
| Bafetinib | 0.327 | −1.607 | −5.658 | 5.79 × 10−6 | 0.036 | 0.949 | 0.003 | 0.006 | 0.95 | 0 |
| Ditercalinium | 0.149 | 0.003 | −5.949 | 6.59 × 10−6 | 0.086 | 0.962 | 0.003 | 0.009 | 0.999 | 0 |
| Bms-833923 | 0.294 | −0.978 | −5.526 | 1.54 × 10−5 | 0.169 | 0.392 | 0.003 | 0.01 | 0.896 | 0 |
| Tg100-801 | 0.166 | −1.068 | −5.193 | 1.92 × 10−5 | 0.254 | 0.477 | 0.003 | 0.009 | 0.743 | 0 |
| Hispaglabridin_B | 0.7 | 2.088 | −5.06 | 1.87 × 10−5 | 0.906 | 0.854 | 0.003 | 0.111 | 0.884 | 0 |
| Crenolanib | 0.504 | −0.993 | −5.421 | 9.76 × 10−5 | 0.825 | 0.543 | 0.003 | 0.008 | 0.81 | 0 |
| Dasatinib | 0.493 | −1.724 | −4.871 | 1.25 × 10−5 | 0.948 | 0.139 | 0.003 | 0.008 | 0.846 | 0 |
| MolPort-001-740-557_Quercetin | 0.434 | 1.701 | −5.204 | 7.69 × 10−5 | 0.05 | 0.919 | 0.007 | 0.936 | 0.072 | 0 |
| MolPort-001-768-161_Binaphthalene | 0.411 | −0.156 | −4.691 | 1.45 × 10−5 | 0.861 | 0.945 | 0.011 | 0.983 | 0.019 | 0 |
| MolPort-001-741-358_EGCG | 0.212 | 1.65 | −6.717 | 5.05 × 10−6 | 0.034 | 0.969 | 0.003 | 0.936 | 0.021 | 0 |
| MolPort-009-018-791_Curcumin | 0.548 | 0.722 | −4.834 | 1.63 × 10−5 | 0.706 | 0.958 | 0.007 | 0.792 | 0.951 | 0 |
| MolPort-039-052-621 | 0.225 | 0.947 | −5.059 | 2.03 × 10−5 | 0.39 | 0.94 | 0.003 | 0.912 | 0.816 | 0 |
| MolPort-002-524-598 | 0.137 | 1.984 | −6.448 | 1.21 × 10−5 | 0.243 | 0.674 | 0.003 | 0.222 | 0.018 | 0 |
| MolPort-001-740-946_Luteolin-7-glucuronide | 0.253 | 1.771 | −6.471 | 3.08 × 10−5 | 0.279 | 0.079 | 0.003 | 0.013 | 0.058 | 0 |
| 29215783_iPPI | 0.388 | −0.195 | −4.916 | 3.44 × 10−5 | 0.575 | 0.033 | 0.003 | 0.513 | 0.704 | 0 |
| 24301892_iPPI | 0.471 | −1.923 | −4.942 | 1.95 × 10−5 | 0.554 | 0.042 | 0.003 | 0.054 | 0.838 | 0 |
| 47194043_iPPI | 0.499 | −0.597 | −5.034 | 7.22 × 10−6 | 0.465 | 0.882 | 0.003 | 0.623 | 0.684 | 0 |
| 24824231_div | 0.725 | −1.188 | −5.289 | 1.36 × 10−5 | 0.427 | 0.854 | 0.003 | 0.036 | 0.017 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mozzicafreddo, M.; Benfaremo, D.; Paolini, C.; Agarbati, S.; Svegliati Baroni, S.; Moroncini, G. Screening and Analysis of Possible Drugs Binding to PDGFRα: A Molecular Modeling Study. Int. J. Mol. Sci. 2023, 24, 9623. https://doi.org/10.3390/ijms24119623
Mozzicafreddo M, Benfaremo D, Paolini C, Agarbati S, Svegliati Baroni S, Moroncini G. Screening and Analysis of Possible Drugs Binding to PDGFRα: A Molecular Modeling Study. International Journal of Molecular Sciences. 2023; 24(11):9623. https://doi.org/10.3390/ijms24119623
Chicago/Turabian StyleMozzicafreddo, Matteo, Devis Benfaremo, Chiara Paolini, Silvia Agarbati, Silvia Svegliati Baroni, and Gianluca Moroncini. 2023. "Screening and Analysis of Possible Drugs Binding to PDGFRα: A Molecular Modeling Study" International Journal of Molecular Sciences 24, no. 11: 9623. https://doi.org/10.3390/ijms24119623
APA StyleMozzicafreddo, M., Benfaremo, D., Paolini, C., Agarbati, S., Svegliati Baroni, S., & Moroncini, G. (2023). Screening and Analysis of Possible Drugs Binding to PDGFRα: A Molecular Modeling Study. International Journal of Molecular Sciences, 24(11), 9623. https://doi.org/10.3390/ijms24119623