
Quantitative Proteomic and Phosphoproteomic Profiling of Lung Tissues from Pulmonary Arterial Hypertension Rat Model

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
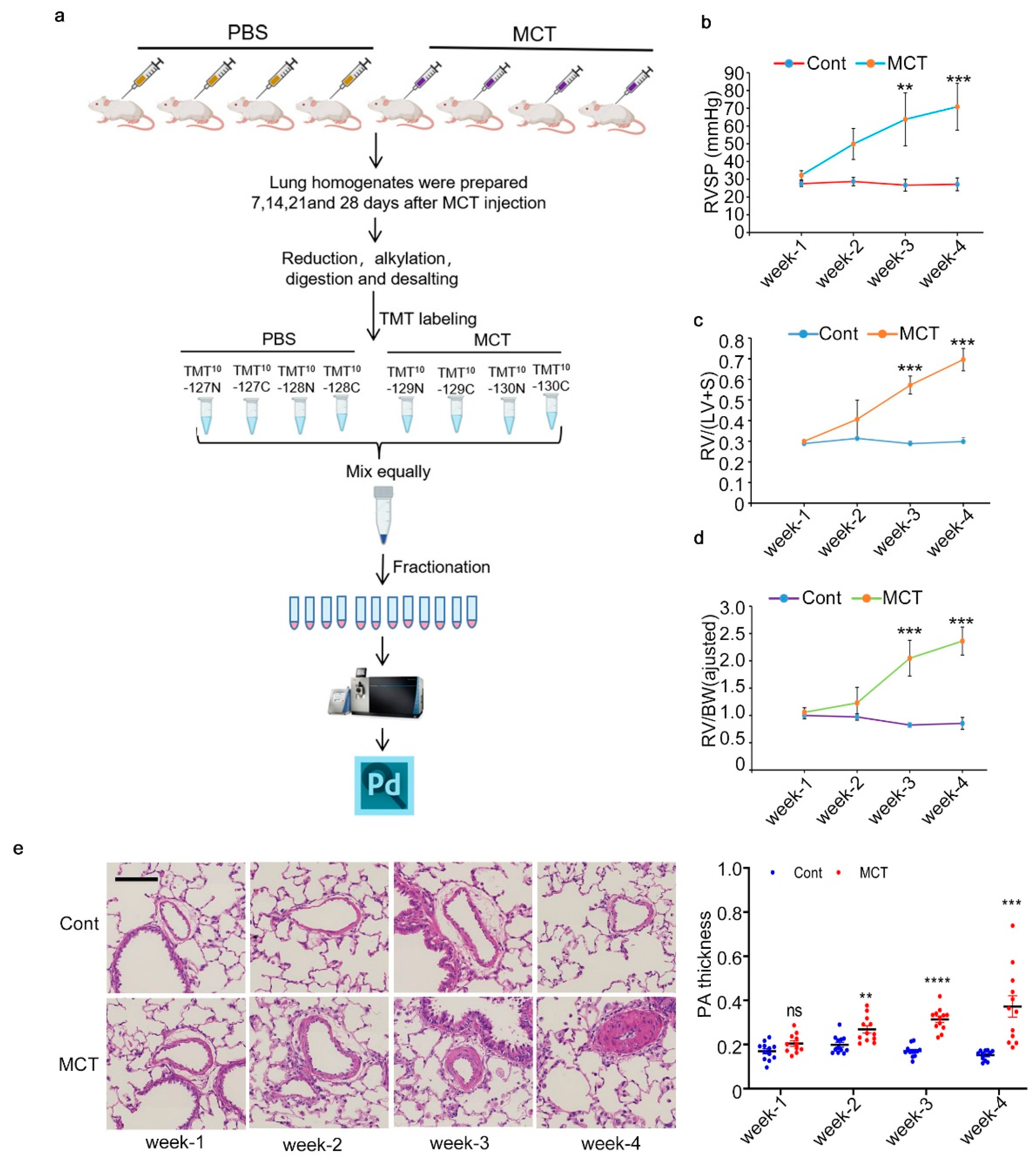
2.1. Workflow for Total Proteome Analysis and Verification of PAH Animal Model
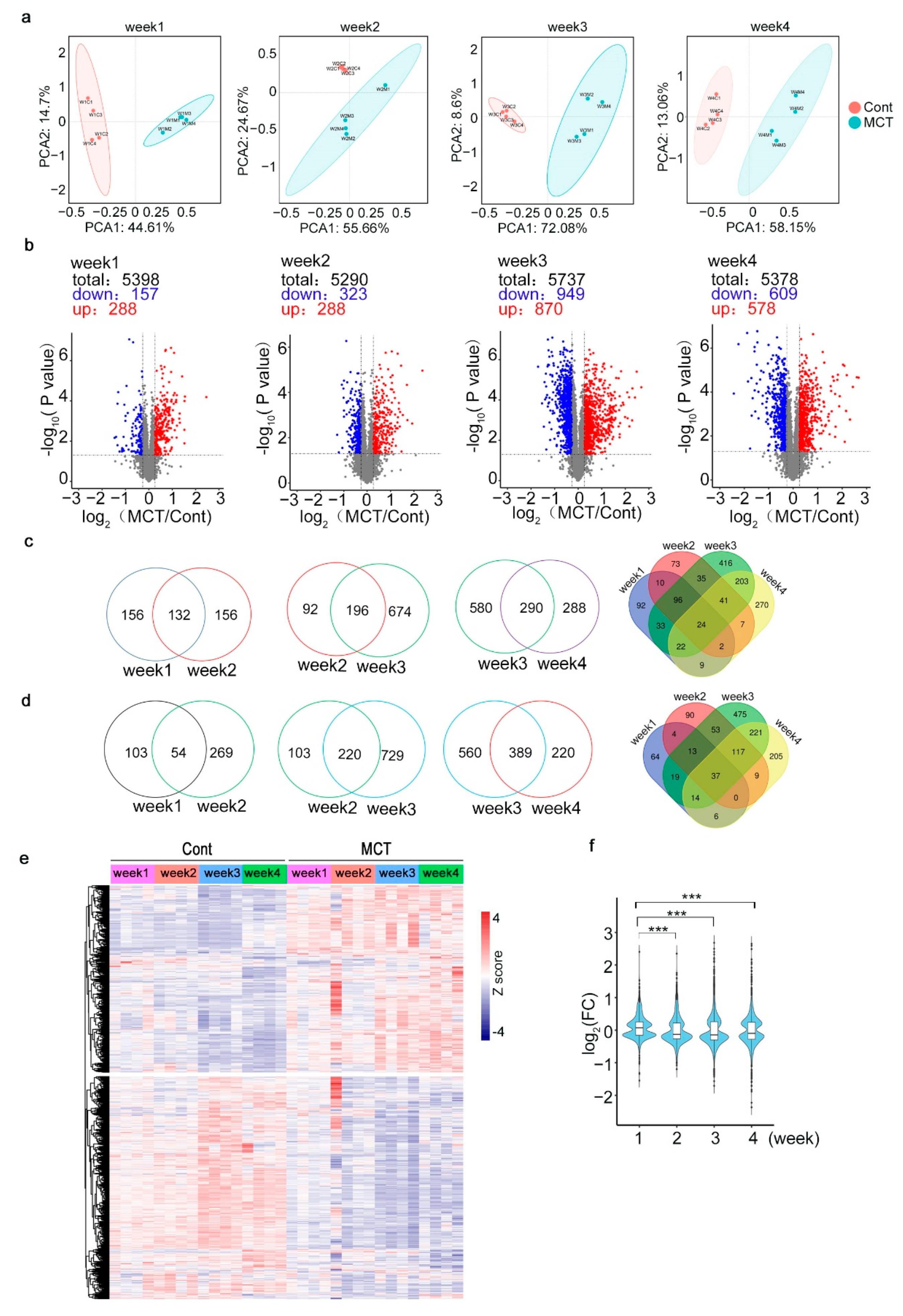
2.2. Proteomic Profiling of Lung Tissues from MCT Rat Model
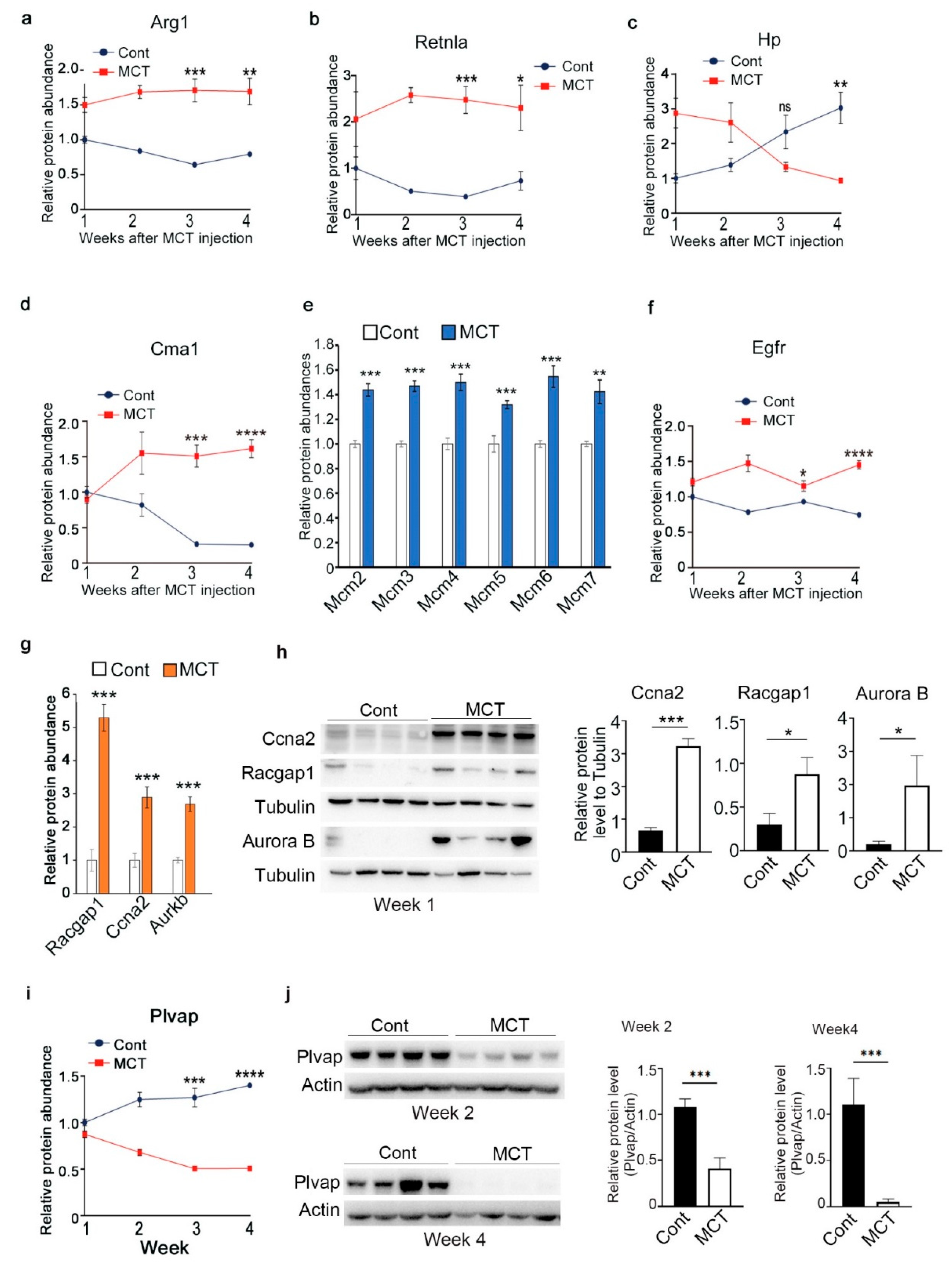
2.3. Validation of Representative Proteins Revealed by Proteomic Analysis
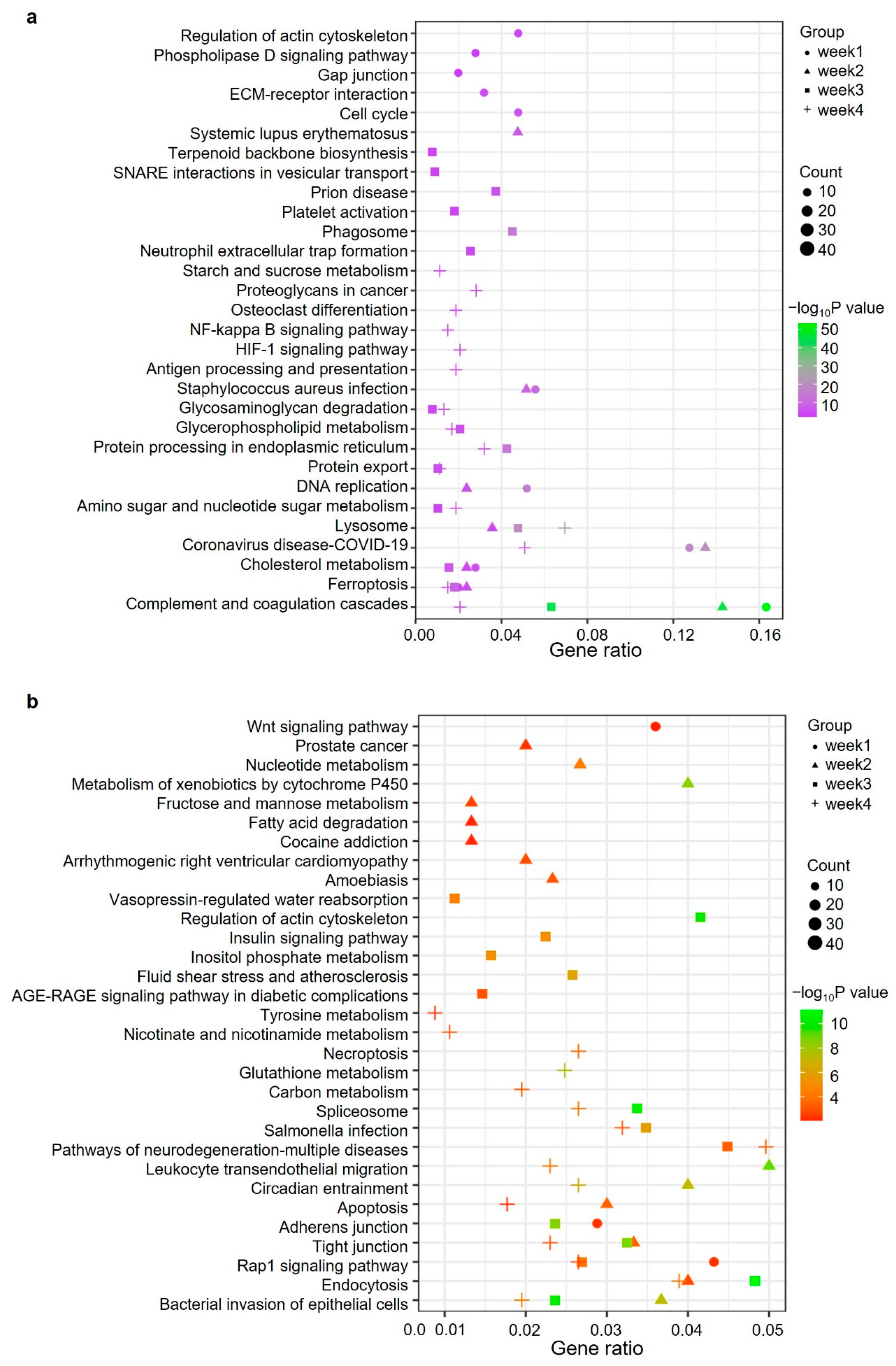
2.4. Gene Ontology and Pathway Analysis of Significantly Changed Proteins
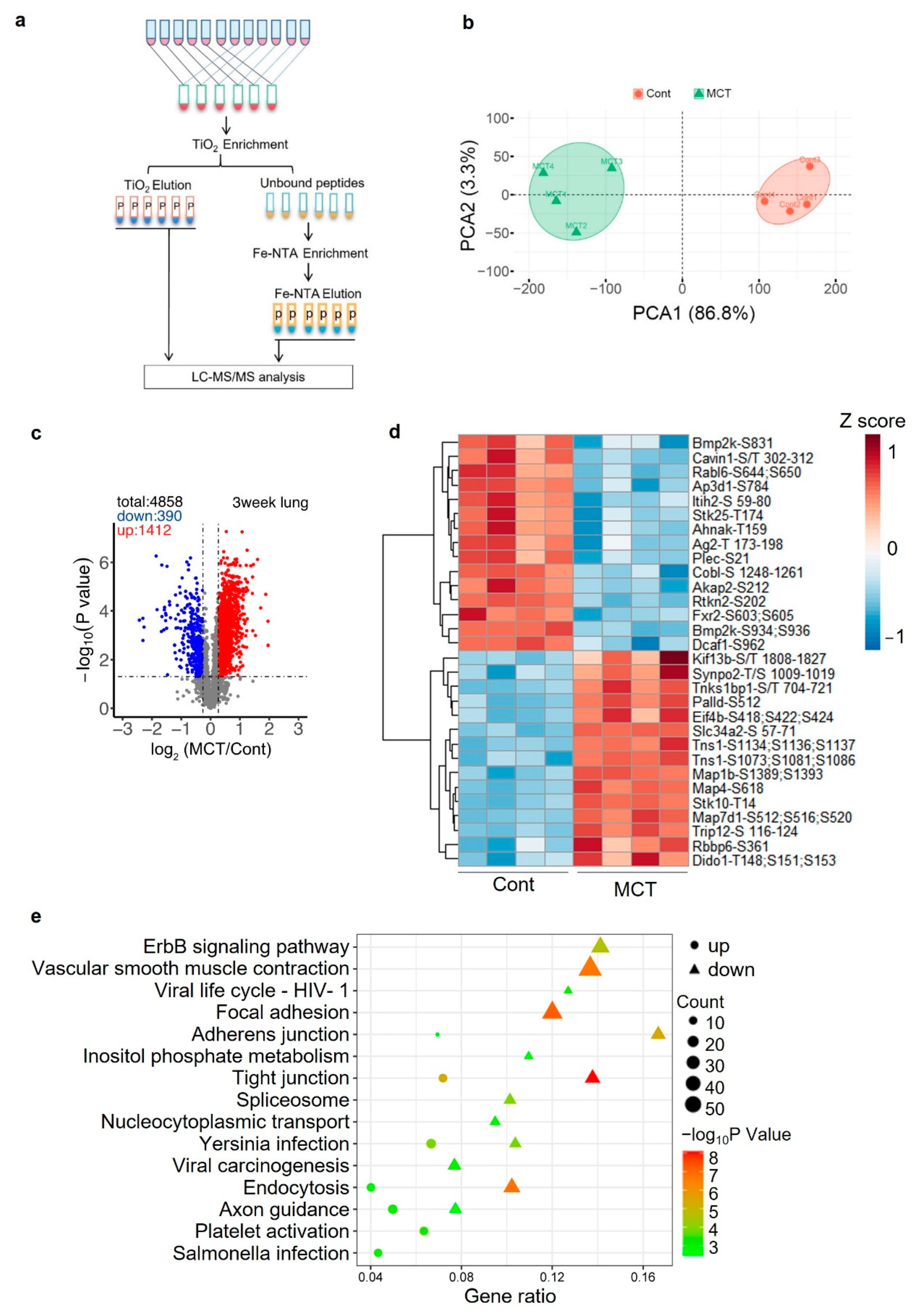
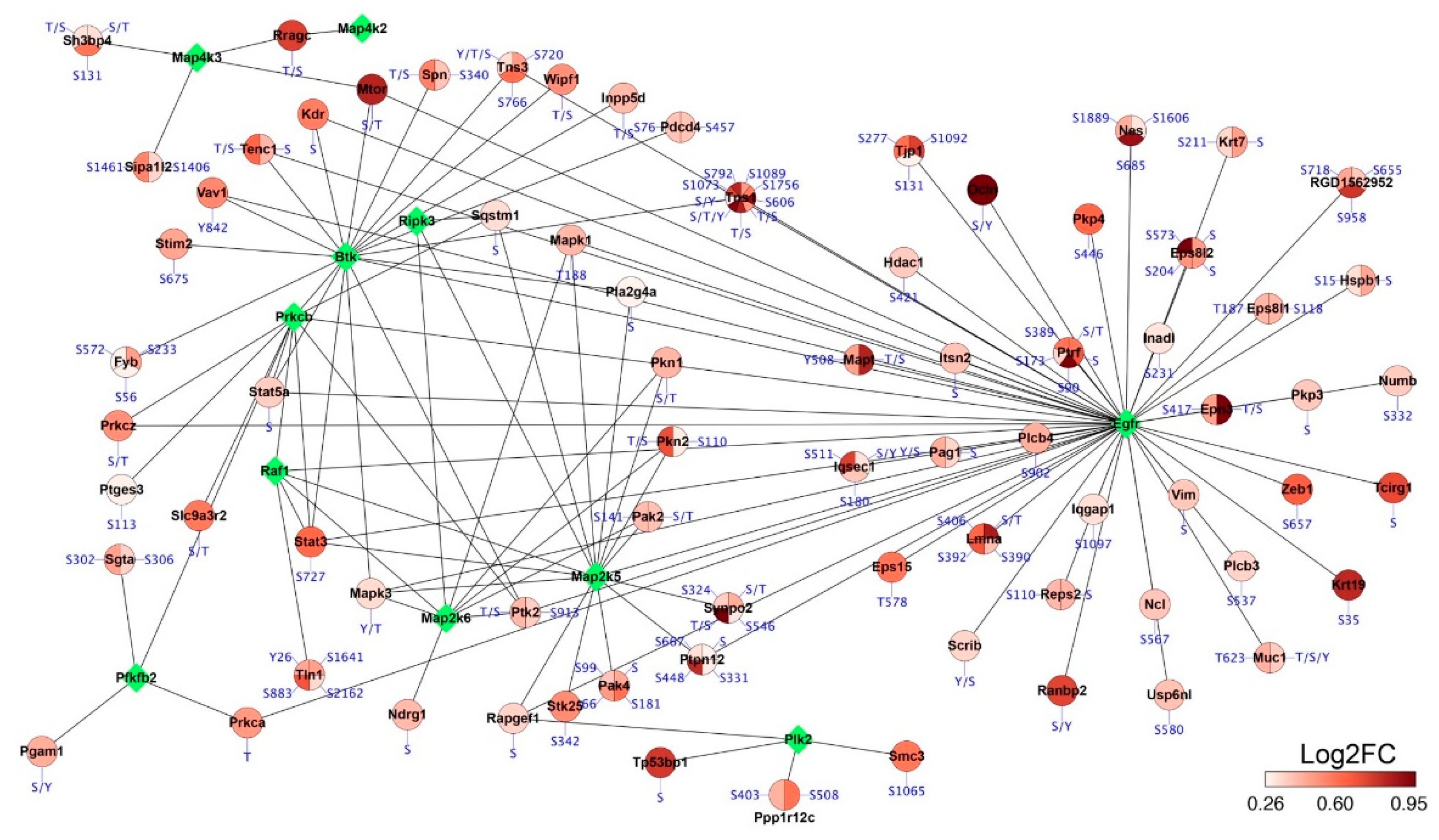
2.5. Phosphoproteomics Profiling of Lung Tissue from MCT Rat Model
3. Discussion
4. Materials and Methods
4.1. Rat Monocrotaline (MCT) Model
4.2. Sample Collection
4.3. Mass Spectrometry Sample Preparation
4.4. Peptide Fractionation
4.5. Phosphopeptide Enrichment
4.6. Mass Spectrometry (MS) Data Acquisition
4.7. MS Data Analysis
4.8. Statistical and Bioinformatics Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brusca, S.B.; Zou, Y.; Elinoff, J.M. How low should we go? Potential benefits and ramifications of the pulmonary hypertension hemodynamic definitions proposed by the 6th World Symposium. Curr. Opin. Pulm. Med. 2020, 26, 384–390. [Google Scholar] [CrossRef]
- Tonelli, A.R.; Arelli, V.; Minai, O.A.; Newman, J.; Bair, N.; Heresi, G.A.; Dweik, R.A. Causes and circumstances of death in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2013, 188, 365–369. [Google Scholar] [CrossRef]
- Nogueira-Ferreira, R.; Ferreira, R.; Henriques-Coelho, T. Cellular interplay in pulmonary arterial hypertension: Implications for new therapies. Biochim. Biophys. Acta 2014, 1843, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.M.T.; Giannoulatou, E.; Celermajer, D.S.; Humbert, M. Epidemiology and treatment of pulmonary arterial hypertension. Nat. Rev. Cardiol. 2017, 14, 603–614. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 13S–24S. [Google Scholar] [CrossRef]
- Bisserier, M.; Katz, M.G.; Bueno-Beti, C.; Brojakowska, A.; Zhang, S.; Gubara, S.; Kohlbrenner, E.; Fazal, S.; Fargnoli, A.; Dorfmuller, P.; et al. Combination Therapy with STAT3 Inhibitor Enhances SERCA2a-Induced BMPR2 Expression and Inhibits Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2021, 22, 9105. [Google Scholar] [CrossRef]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2alpha contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L256–L275. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Phan, C.; Tu, L.; Le Hiress, M.; Thuillet, R.; Jutant, E.M.; Fadel, E.; Savale, L.; Huertas, A.; Humbert, M.; et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J. Clin. Investig. 2018, 128, 1956–1970. [Google Scholar] [CrossRef]
- Leopold, J.A.; Hemnes, A.R. Integrative Omics to Characterize and Classify Pulmonary Vascular Disease. Clin. Chest Med. 2021, 42, 195–205. [Google Scholar] [CrossRef]
- Abdul-Salam, V.B.; Wharton, J.; Cupitt, J.; Berryman, M.; Edwards, R.J.; Wilkins, M.R. Proteomic analysis of lung tissues from patients with pulmonary arterial hypertension. Circulation 2010, 122, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, S.; Zeng, X.; Lin, D.; Li, Y.; Gui, L.; Lin, M.J. Revealing the pathogenic changes of PAH based on multiomics characteristics. J. Transl. Med. 2019, 17, 231. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Comhair, S.A.A.; Chen, R.; Hu, B.; Hou, Y.; Zhou, Y.; Mavrakis, L.A.; Janocha, A.J.; Li, L.; Zhang, D.; et al. Integrative proteomics and phosphoproteomics in pulmonary arterial hypertension. Sci. Rep. 2019, 9, 18623. [Google Scholar] [CrossRef]
- Zhang, J.; Dong, J.; Martin, M.; He, M.; Gongol, B.; Marin, T.L.; Chen, L.; Shi, X.; Yin, Y.; Shang, F.; et al. AMP-activated Protein Kinase Phosphorylation of Angiotensin-Converting Enzyme 2 in Endothelium Mitigates Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Chen, J.; Fraidenburg, D.R.; Song, S.; Sysol, J.R.; Drennan, A.R.; Offermanns, S.; Ye, R.D.; Bonini, M.G.; Minshall, R.D.; et al. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L208–L220. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Siamwala, J.H.; Chatterjee, S. eNOS phosphorylation in health and disease. Biochimie 2010, 92, 1186–1198. [Google Scholar] [CrossRef]
- Savai, R.; Al-Tamari, H.M.; Sedding, D.; Kojonazarov, B.; Muecke, C.; Teske, R.; Capecchi, M.R.; Weissmann, N.; Grimminger, F.; Seeger, W.; et al. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat. Med. 2014, 20, 1289–1300. [Google Scholar] [CrossRef]
- Lin, Q.; Fan, C.; Gomez-Arroyo, J.; Van Raemdonck, K.; Meuchel, L.W.; Skinner, J.T.; Everett, A.D.; Fang, X.; Macdonald, A.A.; Yamaji-Kegan, K.; et al. HIMF (Hypoxia-Induced Mitogenic Factor) Signaling Mediates the HMGB1 (High Mobility Group Box 1)-Dependent Endothelial and Smooth Muscle Cell Crosstalk in Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2505–2519. [Google Scholar] [CrossRef]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaco, R.D.; Southwood, M.; Toshner, M.; Crotty Alexander, L.E.; Morrell, N.W.; Chilvers, E.R.; et al. HIF2alpha-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef]
- Kosanovic, D.; Luitel, H.; Dahal, B.K.; Cornitescu, T.; Janssen, W.; Danser, A.H.; Garrelds, I.M.; De Mey, J.G.; Fazzi, G.; Schiffers, P.; et al. Chymase: A multifunctional player in pulmonary hypertension associated with lung fibrosis. Eur. Respir. J. 2015, 46, 1084–1094. [Google Scholar] [CrossRef]
- Nakamura, H.; Kato, M.; Nakaya, T.; Kono, M.; Tanimura, S.; Sato, T.; Fujieda, Y.; Oku, K.; Ohira, H.; Bohgaki, T.; et al. Decreased haptoglobin levels inversely correlated with pulmonary artery pressure in patients with pulmonary arterial hypertension: A cross-sectional study. Medicine 2017, 96, e8349. [Google Scholar] [CrossRef]
- Xiao, G.; Zhuang, W.; Wang, T.; Lian, G.; Luo, L.; Ye, C.; Wang, H.; Xie, L. Transcriptomic analysis identifies Toll-like and Nod-like pathways and necroptosis in pulmonary arterial hypertension. J. Cell. Mol. Med. 2020, 24, 11409–11421. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Chen, D.; Liu, J.; Chen, S.; Zhang, X.; Zhang, Y.; Li, M.; Pan, W.; Zhou, D.; Guan, L.; et al. Profiling and Molecular Mechanism Analysis of Long Non-Coding RNAs and mRNAs in Pulmonary Arterial Hypertension Rat Models. Front. Pharmacol. 2021, 12, 709816. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wu, X.; Peng, L.; Yang, M.; Zhou, H.; Xu, J.; Wang, J.; Wang, H.; Xie, W.; Kong, H. Inhibition of Bruton’s Tyrosine Kinase Alleviates Monocrotaline-Induced Pulmonary Arterial Hypertension by Modulating Macrophage Polarization. Oxid. Med. Cell. Longev. 2022, 2022, 6526036. [Google Scholar] [CrossRef] [PubMed]
- Guignabert, C.; Tu, L.; Le Hiress, M.; Ricard, N.; Sattler, C.; Seferian, A.; Huertas, A.; Humbert, M.; Montani, D. Pathogenesis of pulmonary arterial hypertension: Lessons from cancer. Eur. Respir. Rev. 2013, 22, 543–551. [Google Scholar] [CrossRef]
- Satpathy, S.; Krug, K.; Jean Beltran, P.M.; Savage, S.R.; Petralia, F.; Kumar-Sinha, C.; Dou, Y.; Reva, B.; Kane, M.H.; Avanessian, S.C.; et al. A proteogenomic portrait of lung squamous cell carcinoma. Cell 2021, 184, 4348–4371.e40. [Google Scholar] [CrossRef]
- Gillette, M.A.; Satpathy, S.; Cao, S.; Dhanasekaran, S.M.; Vasaikar, S.V.; Krug, K.; Petralia, F.; Li, Y.; Liang, W.W.; Reva, B.; et al. Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 2020, 182, 200–225.e35. [Google Scholar] [CrossRef]
- Clark, D.J.; Dhanasekaran, S.M.; Petralia, F.; Pan, J.; Song, X.; Hu, Y.; da Veiga Leprevost, F.; Reva, B.; Lih, T.M.; Chang, H.Y.; et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 2019, 179, 964–983.e31. [Google Scholar] [CrossRef]
- Wu, M.; Wu, Y.; Huang, J.; Wu, Y.; Wu, H.; Jiang, B.; Zhuang, J. Protein expression profile changes of lung tissue in patients with pulmonary hypertension. PeerJ 2020, 8, e8153. [Google Scholar] [CrossRef]
- Liang, S.; Desai, A.A.; Black, S.M.; Tang, H. Cytokines, Chemokines, and Inflammation in Pulmonary Arterial Hypertension. Adv. Exp. Med. Biol. 2021, 1303, 275–303. [Google Scholar] [CrossRef]
- Stan, R.V.; Tkachenko, E.; Niesman, I.R. PV1 is a key structural component for the formation of the stomatal and fenestral diaphragms. Mol. Biol. Cell 2004, 15, 3615–3630. [Google Scholar] [CrossRef]
- Stan, R.V.; Kubitza, M.; Palade, G.E. PV-1 is a component of the fenestral and stomatal diaphragms in fenestrated endothelia. Proc. Natl. Acad. Sci. USA 1999, 96, 13203–13207. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Engelman, J.A.; Wang, X.B.; Schubert, W.; Zhang, X.L.; Marks, C.B.; Macaluso, F.; Russell, R.G.; Li, M.; Pestell, R.G.; et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J. Biol. Chem. 2001, 276, 38121–38138. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Liu, Y.; Stan, R.V.; Fan, L.; Gu, Y.; Dalton, N.; Chu, P.H.; Peterson, K.; Ross, J., Jr.; Chien, K.R. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc. Natl. Acad. Sci. USA 2002, 99, 11375–11380. [Google Scholar] [CrossRef]
- Jones, J.H.; Friedrich, E.; Hong, Z.; Minshall, R.D.; Malik, A.B. PV1 in Caveolae Controls Lung Endothelial Permeability. Am. J. Respir. Cell Mol. Biol. 2020, 63, 531–539. [Google Scholar] [CrossRef]
- Feng, Y.H.; Li, L.F.; Zhang, Q.; Zhang, J.H.; Huang, Y.; Lv, Y.L.; Jia, J.Z.; Zhang, D.; Hu, J.Y.; Huang, Y.S. Microtubule associated protein 4 (MAP4) phosphorylation reduces cardiac microvascular density through NLRP3-related pyroptosis. Cell Death Discov. 2021, 7, 213. [Google Scholar] [CrossRef]
- Zhang, J.; Li, L.; Zhang, Q.; Yang, X.; Zhang, C.; Zhang, X.; Zhang, D.; Lv, Y.; Song, H.; Chen, B.; et al. Phosphorylation of Microtubule- Associated Protein 4 Promotes Hypoxic Endothelial Cell Migration and Proliferation. Front. Pharmacol. 2019, 10, 368. [Google Scholar] [CrossRef]
- Hedhli, N.; Kalinowski, A.; Russell, K. Cardiovascular effects of neuregulin-1/ErbB signaling: Role in vascular signaling and angiogenesis. Curr. Pharm. Des. 2014, 20, 4899–4905. [Google Scholar] [CrossRef]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., 3rd; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef]
- Dannewitz Prosseda, S.; Ali, M.K.; Spiekerkoetter, E. Novel Advances in Modifying BMPR2 Signaling in PAH. Genes 2020, 12, 8. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chen, T.; Ma, J.; Liu, Y.; Chen, Z.; Xiao, N.; Lu, Y.; Fu, Y.; Yang, C.; Li, M.; Wu, S.; et al. iProX in 2021: Connecting proteomics data sharing with big data. Nucleic Acids Res. 2022, 50, D1522–D1527. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, A.; Jia, Y.; Hao, R.; Yu, Y.; Zhou, X.; Gu, C.; Ren, M.; Tang, H. Quantitative Proteomic and Phosphoproteomic Profiling of Lung Tissues from Pulmonary Arterial Hypertension Rat Model. Int. J. Mol. Sci. 2023, 24, 9629. https://doi.org/10.3390/ijms24119629
Luo A, Jia Y, Hao R, Yu Y, Zhou X, Gu C, Ren M, Tang H. Quantitative Proteomic and Phosphoproteomic Profiling of Lung Tissues from Pulmonary Arterial Hypertension Rat Model. International Journal of Molecular Sciences. 2023; 24(11):9629. https://doi.org/10.3390/ijms24119629
Chicago/Turabian StyleLuo, Ang, Yangfan Jia, Rongrong Hao, Yafang Yu, Xia Zhou, Chenxin Gu, Meijuan Ren, and Haiyang Tang. 2023. "Quantitative Proteomic and Phosphoproteomic Profiling of Lung Tissues from Pulmonary Arterial Hypertension Rat Model" International Journal of Molecular Sciences 24, no. 11: 9629. https://doi.org/10.3390/ijms24119629
APA StyleLuo, A., Jia, Y., Hao, R., Yu, Y., Zhou, X., Gu, C., Ren, M., & Tang, H. (2023). Quantitative Proteomic and Phosphoproteomic Profiling of Lung Tissues from Pulmonary Arterial Hypertension Rat Model. International Journal of Molecular Sciences, 24(11), 9629. https://doi.org/10.3390/ijms24119629