

Decipher the Immunopathological Mechanisms and Set Up Potential Therapeutic Strategies for Patients with Lupus Nephritis
 , , ,
, , ,
Abstract
:1. Introduction
2. Recent Advances in the Pathophysiologic Mechanisms for LN
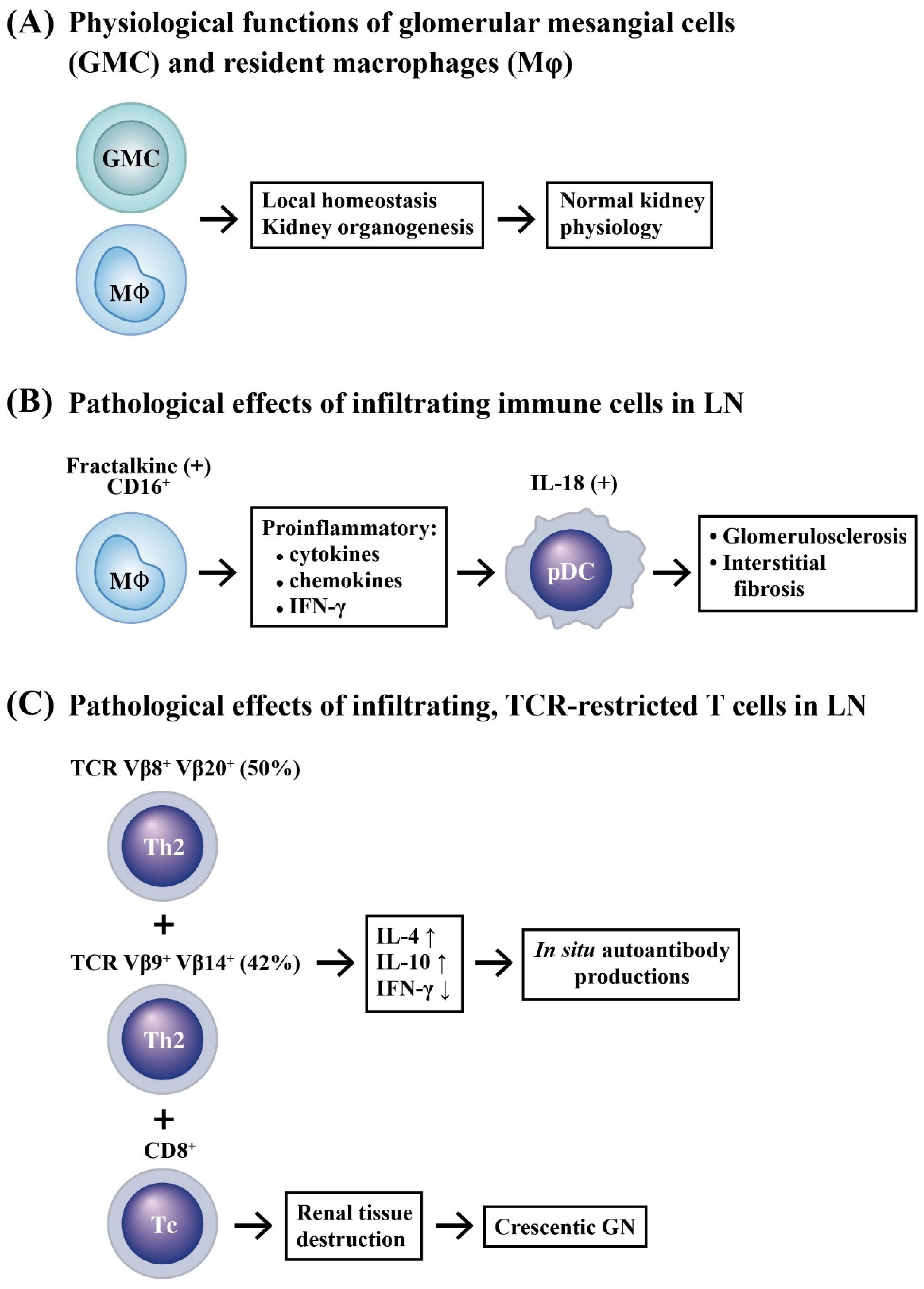
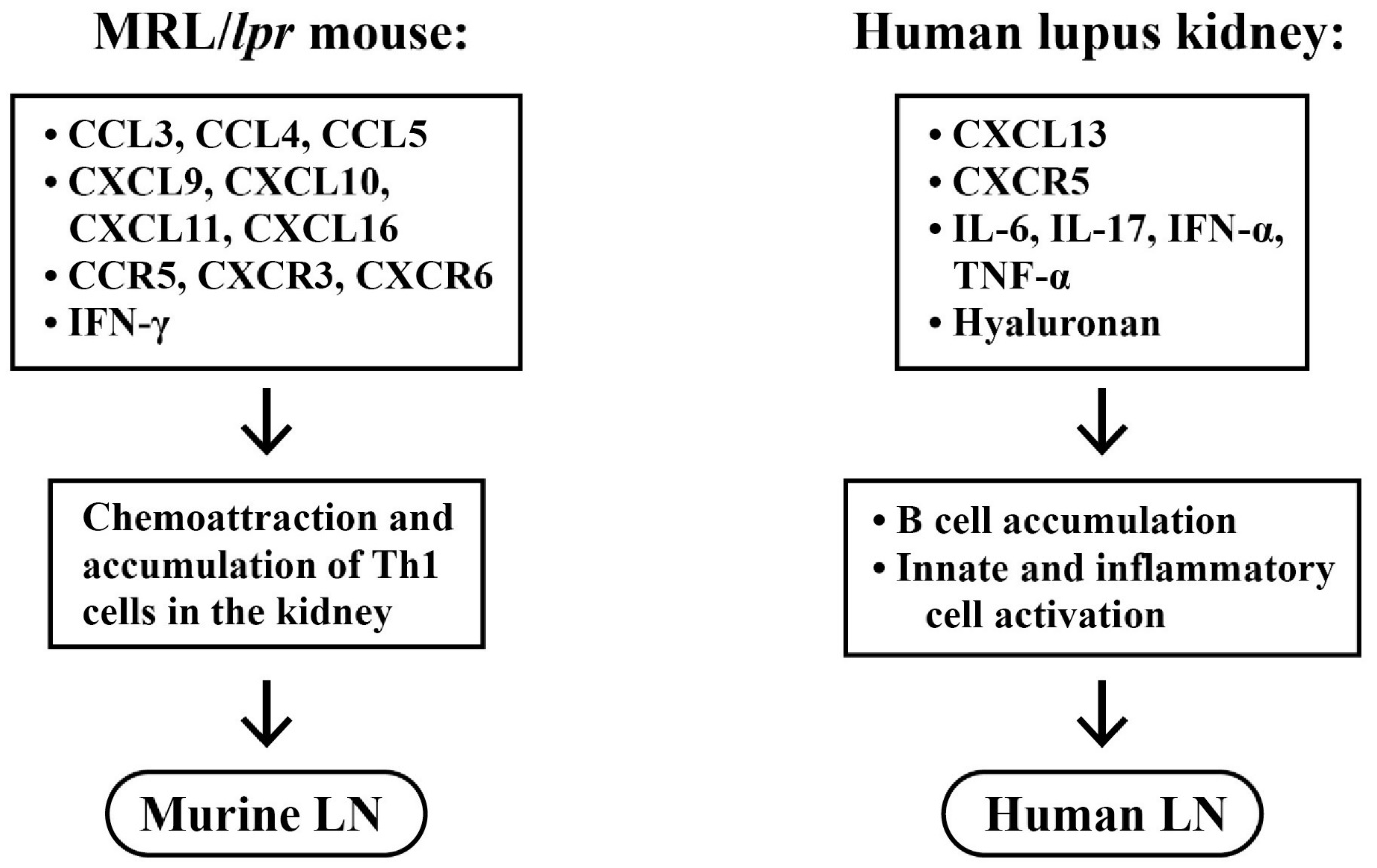
2.1. Selective Accumulation of Adaptive Immune Cells in LN
2.2. Innate Immune Cell Infiltration in LN
2.3. Renal Resident Cell Activation in Causing Podocytopathy and Mesangial Proliferation
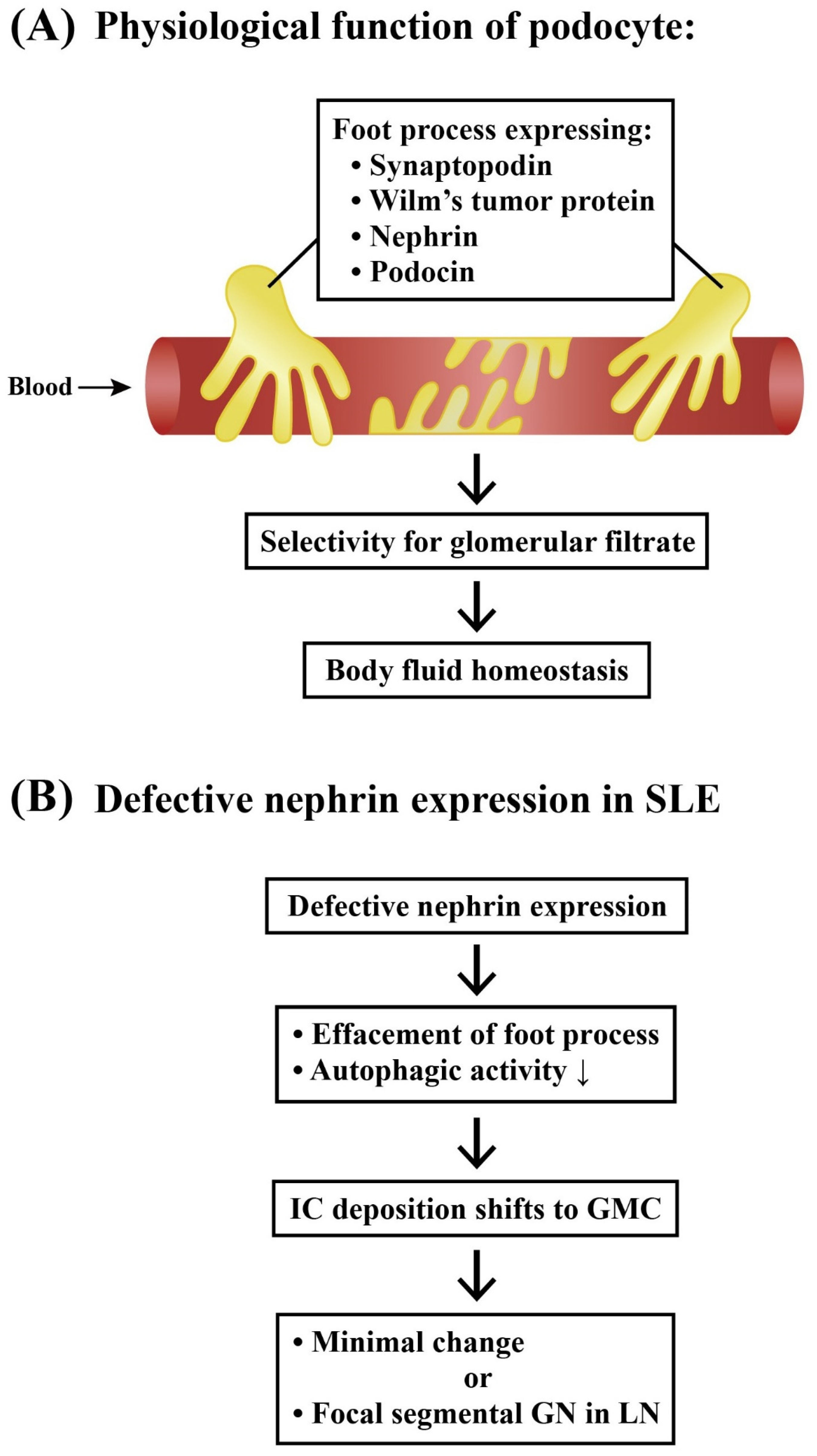
2.3.1. Podocytes and Lupus Podocytopathy
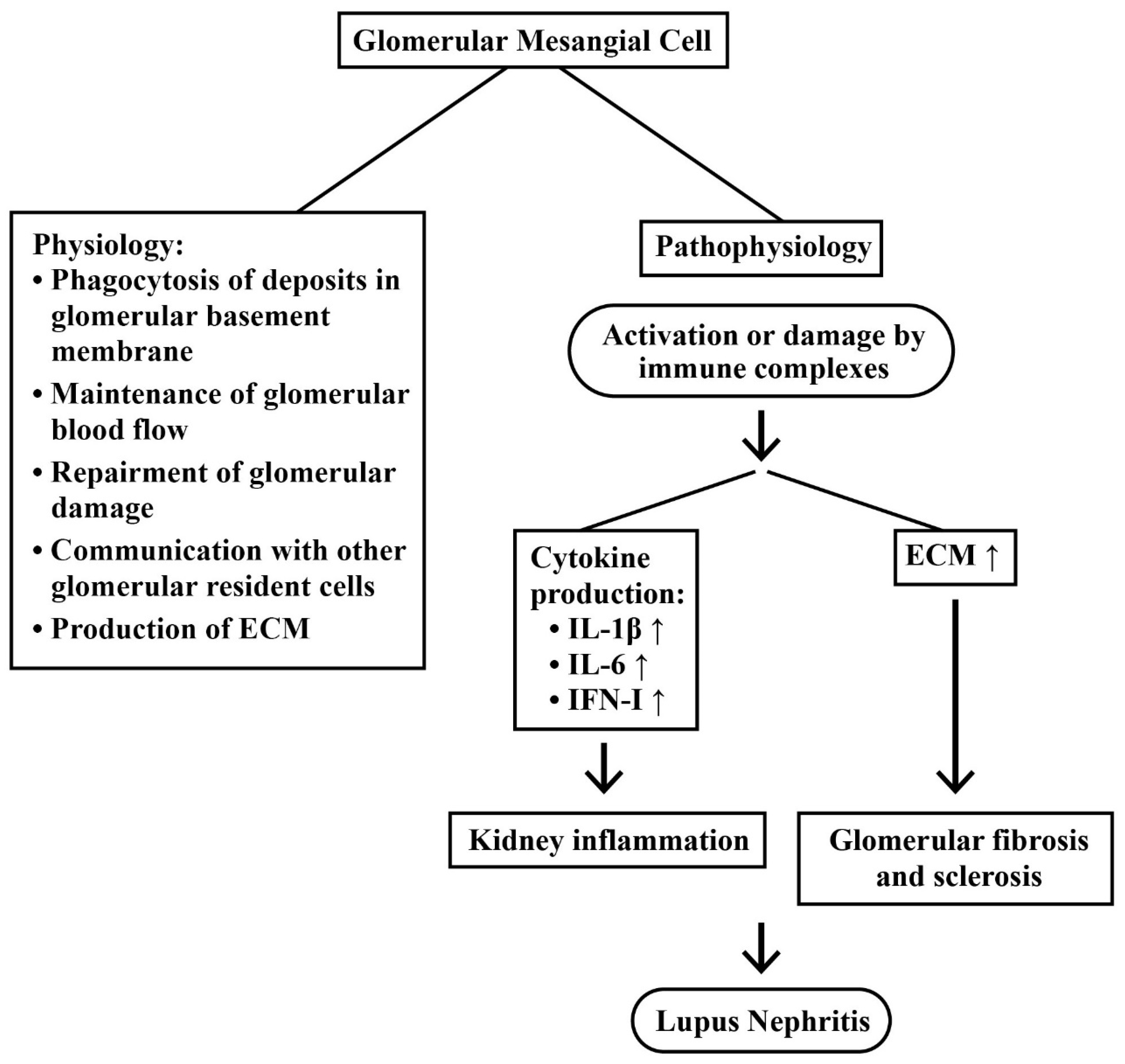
2.3.2. Mesangial Cells (MC) in LN
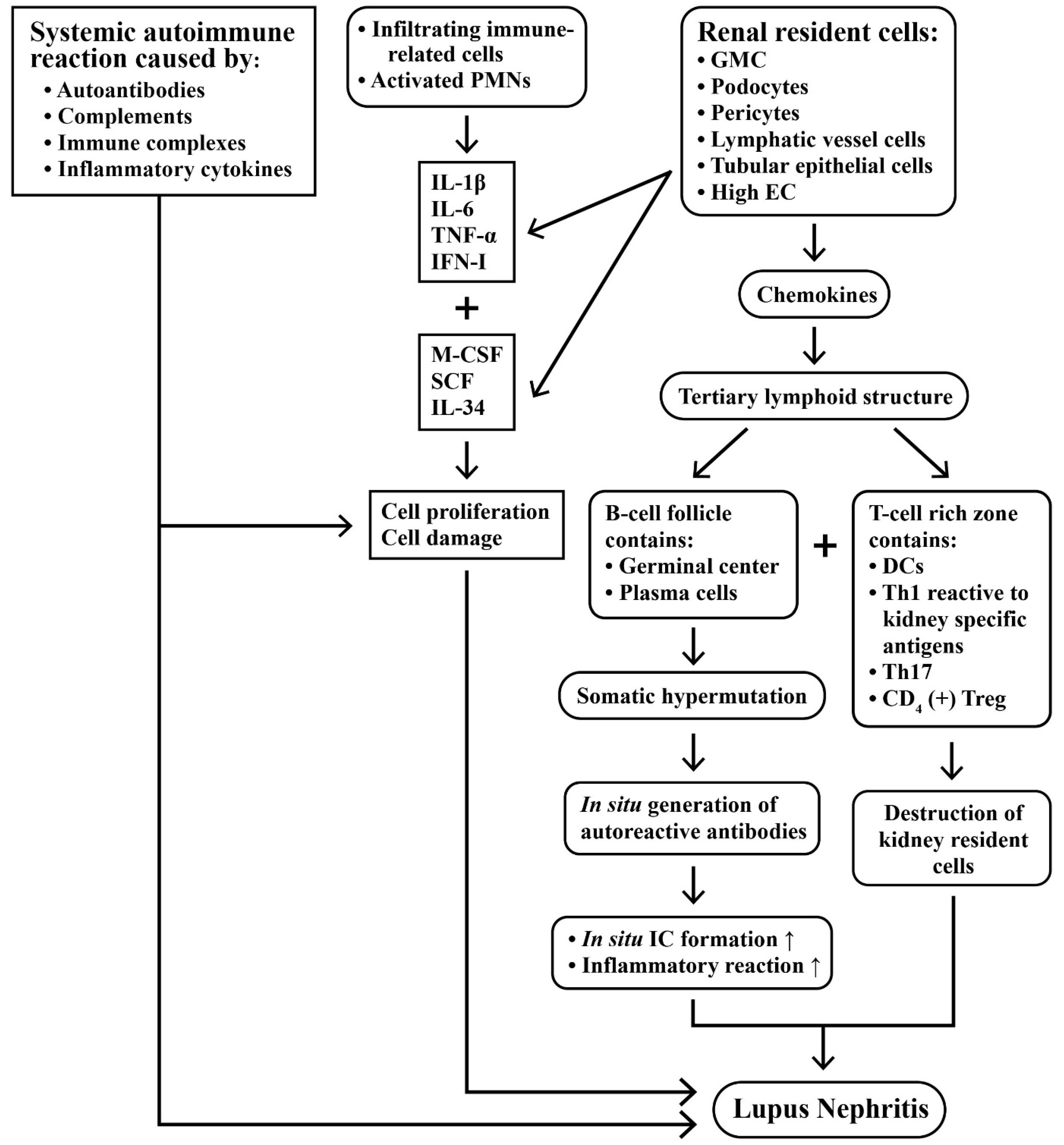
2.4. Intricate Interactions between Infiltrating Immune Cells and Renal Resident Cells in Causing Kidney Inflammation
2.5. Roles of Chemokines, Cytokinnes and Inflammation-Related Molecules in the Pathogenesis of LN
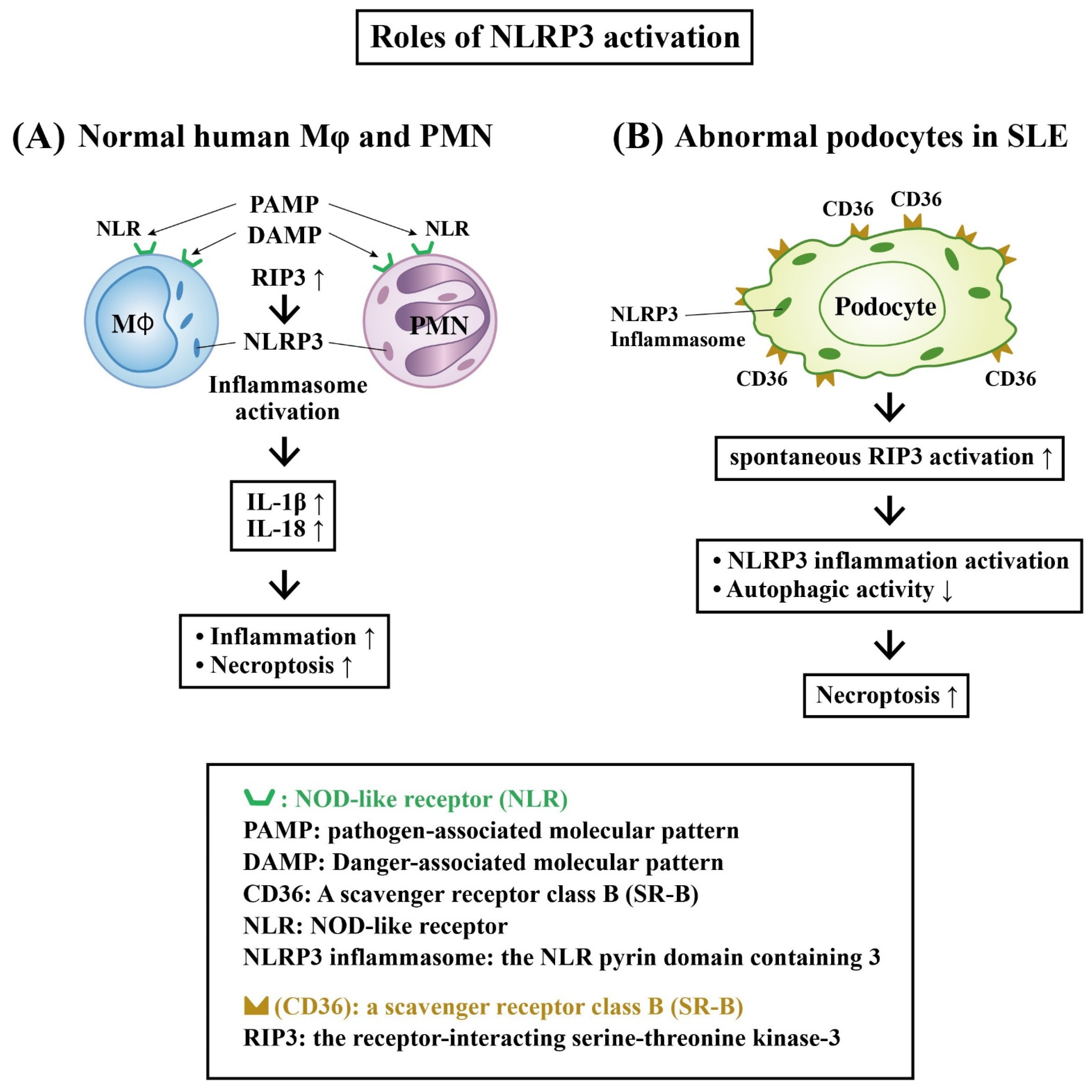
Role of Receptor-Interacting Serine–Threonine Kinase 3 (RIP-3) and NLRP3 Inflammasome Activation in LN
2.6. Involvement of Autoantigens and Autoantibodies in LN
2.6.1. Pathogenic Anti-dsDNA Antibodies
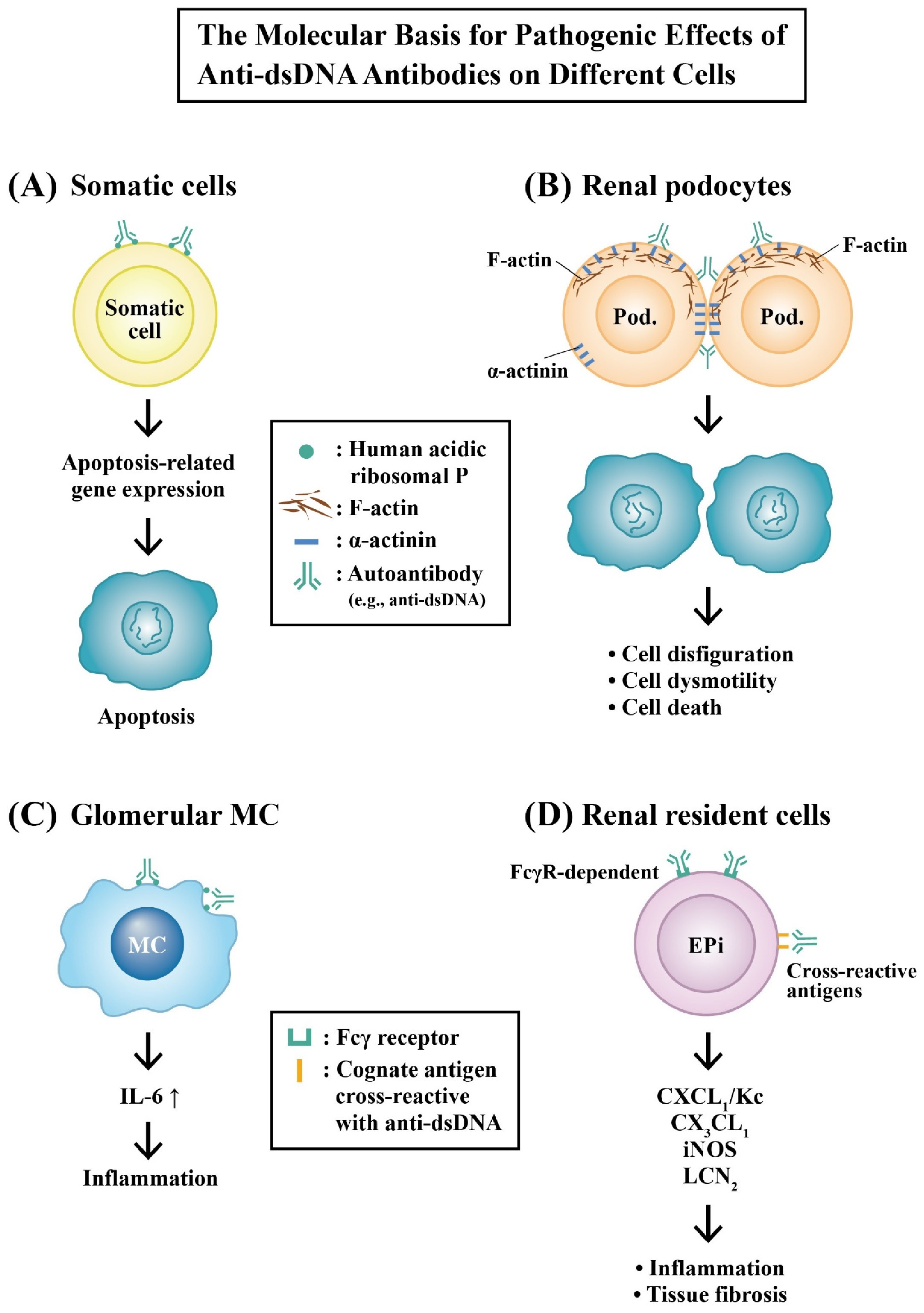
2.6.2. Molecular Mechanisms of Pathogenic Anti-dsDNA Autoantibodies in the Development of LN
Molecular Basis for the Pathologic Effects of Anti-dsDNA Autoantibodies
Involvement of Glycosylation of Anti-dsDNA Autoantibodies in LN and Its Possible Mechanisms
2.6.3. The Autoantibodies Co-Existing with Anti-dsDNA That Are Involved in LN
3. Current Useful Biomarkers in the Renal Tissues of Patients with LN
3.1. Immune Gene Expression in Renal Tissue as Disease Activity Biomarkers for LN
3.1.1. Expression of BAFF, APRIL and Their Corresponding Receptors as Disease Activity Biomarkers for LN
3.1.2. Expression of IFN-α and TNF-α Genes in Renal Tissues as the Biomarkers for Disease Activity in LN
3.1.3. Immune Gene Expression in the Infiltrating Immune Cells in LN
3.2. Potentially Useful Urinary Biomarkers in LN
4. Potential Novel Therapeutic Strategy for LN in Future
4.1. Novel Potential Therapeutic Targets or Checkpoints in the Treatment of LN
4.1.1. Antagonists Targeting the CX3CL1 Family
4.1.2. NLRP3 Inflammasome and NF-κB Pathway as Targets of Phytochemicals
4.1.3. JAK/STAT Signaling Pathways as Targets
4.1.4. Complement Components as Targets
4.1.5. Increased Autophagy by mTOR Inhibitors, Rapamycin or Sirolimus, for Cytoprotection of Podocyte Injury
4.1.6. Type I IFN Signaling Pathway Inhibitors
4.2. The Pathogenic and Therapeutic Role of Non-Coding RNAs in LN
4.2.1. Molecular Mechanisms of Different miRs in Suppressing LN
4.2.2. Molecular Mechanism of Different lncRNAs and circRNAs in the Treatment of LN
4.2.3. Molecular Mechanisms for Other Unique ncRNA Inhibitors in the Treatment of LN
5. Conclusions and Prospects
- (1)
- The ncRNA mimics or antagonists can be taken by the infiltrated and/or resident cells in the lupus kidneys.
- (2)
- Major adverse effects can be avoided.
- (3)
- Precision medicine for LN can be established depending on highly specific serum or urinary biomarkers.
- (4)
- Personalized regimens for LN should be identified for modulating the individual pathological factor(s).
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aCL | anti-cardiolipin antibody |
| Akt | protein kinase B derived from AKR mouse thymoma |
| ANA | anti-nuclear antibody |
| APRIL | a proliferation-inducing ligand |
| BAFF | B cell activating factor of TNF Family |
| BAFF-R | receptor for BAFF |
| BCMA | B cell maturation antigen |
| BR3 | BLyS receptor 3 |
| C1q | Q fragment of complement component 1 |
| CCL | C-C chemokine Receptor |
| CCR | receptor of cysteine-cysteine motif containing chemokine |
| CD | cluster of differentiation |
| circRNA | circular ribonucleic acid |
| CKD | chronic kidney disease |
| CSF | colony stimulating factor |
| CXC | cysteine-X amino acid-cysteine motif containing chemokine |
| CXCL | ligand for CXC chemokine |
| DAMP | damage associated molecular pattern |
| DC | dendritic cell |
| DEPs | differential expressed proteins |
| dsDNA | double-stranded deoxyribonucleic acid |
| EC | endothelial cell |
| ECM | extracellular matrix |
| Erb-B | receptor tyrosine kinase derived from epidermal growth factor |
| ESRD | end-stage renal disease |
| EZH-2 | enhancer of zeste 2 polycomb repressive complex 2 subunit |
| Fab | a and b fragment of immunoglobulin |
| Fc | c (or constant) fragment of immunoglobulin |
| Fh2 | type II follicular helper T cell |
| GMC | glomerular mesangial cell |
| GN | glomerulonephritis |
| HIF | hypoxia inducible factor |
| HLA-DR | R chain of human leukocyte antigen-class D |
| HRMC | human renal mesangial cell |
| IC | immune complex |
| IFN | interferon |
| IFN-I | type I interferon |
| Ig | immunoglobulin |
| IL | interleukin |
| IL-22BP | interleukin 22 binding protein |
| iNOS | inducible nitric oxide synthase |
| IP-10 | Interferon gamma-induced protein 10 (=CXCL10) |
| ISG | interferon stimulated gene |
| ISN | International Society of Nephrology |
| IVIG | intravenous immunoglobulin G infusion |
| JAK | Janus kinase |
| JSLE | juvenile systemic lupus erythematosus |
| KC | keratinocyte-derived cytokine (=CXCL1) |
| LAIR1 | leukocyte-associated immunoglobulin-like receptor 1 |
| LCN-2 | lipocalin 2 |
| LN | lupus nephritis |
| LNA | locked nucleic acid |
| lncRNA | long non-coding RNA |
| LPS | lipopolysaccharide |
| LRR | leucine rich repeat |
| MC | mesangial cell |
| MCP-1 | monocyte chemoattractant protein 1 |
| M-CSF | myeloid colony stimulating factor |
| mDC | myeloid dendritic cell |
| MHC-II | major histocompatibility complex Class II |
| miR | microRNA |
| MPAA | mycophenolic acid analogue |
| MPGN | membrano-proliferative glomerulonephritis |
| mRNA | messenger ribonucleic acid |
| mTOR | mechanistic target of rapamycin |
| ncRNAs | non-coding RNAs |
| NETs | neutrophil extracellular traps |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cell |
| NGAL | neutrophil gelatinase-associated lipocalin |
| NLR | NOD-like receptor |
| NLRP3 | nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain-containing receptor 3 |
| NMDAR | N-methyl-D-aspartate receptor |
| NOD | nucleotide-binding oligomerization domain-containing protein |
| PAMP | Pathogen-Associated Molecular Pattern |
| PBL | peripheral blood lymphocyte |
| PBMC | peripheral blood mononuclear cell |
| pcDNA | mammalian expressing vector |
| pDC | plasmacytoid dendritic cell |
| PI3K | phosphoinositide 3 kinase |
| PMN | polymorphonuclear leukocyte |
| PRR | pattern recognition receptor |
| RIG-1 | retinoic acid-inducible gene 1 |
| RIP3 | receptor-interacting serine–threonine kinase 3 |
| RLR | retinoic acid-inducible gene 1 like receptor |
| RMC | rodent glomerular mesangial cell |
| RRP8 | ribosomal RNA-processing protein 8 |
| RT-PCR | reverse transcriptase assisted polymerase chain reaction |
| SCF | stem cell factor |
| ScRNA-Seq | single-cell analysis of RNA sequence |
| SIA | sialic acid |
| SLE | systemic lupus erythematosus |
| SLEDAI | systemic lupus erythematosus disease activity index |
| sncRNA | short non-coding ribonucleic acid |
| SOCS | suppressor of cytokine signaling |
| STAT | signal transducer and activators of transcription |
| TACI | transmembrane activator and calcium modulator and cytophilin ligand interactor |
| TCR | T cell receptor |
| TEC | tubular epithelial cell |
| TGF-β | transforming growth factor beta |
| Th | T helper cells |
| THP | Tamm-Horsfall glycoprotein |
| TLR | toll-like receptor |
| TLS | tertiary lymphoid structure |
| TNF-a | tumor necrosis factor |
| TNP-1 | Spermatid nuclear transition protein 1 |
| TPL | triptolide |
| tRNA | transfer ribonucleic acid |
| tsRNA | tRNA derived small RNA |
| Tyk | tyrosine kinase |
| UTR | untranslated region |
| UV | ultraviolet |
| VCAM-1 | vascular cell adhesion molecule 1 |
References
- Hoover, P.J.; Costenbader, K.H. Insights into the epidemiology and management of lupus nephritis from the US rheumatologist’s perspective. Kidney Int. 2016, 90, 487–492. [Google Scholar] [CrossRef] [Green Version]
- Hanly, J.G.; O’Keeffe, A.G.; Su, L.; Urowitz, M.B.; Romero-Diaz, J.; Gordon, C.; Bae, S.-C.; Bernatsky, S.; Clarke, A.E.; Wallace, D.J.; et al. The frequency and outcome of lupus nephritis: Results from an international inception cohort study. Rheumatology 2016, 55, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Weening, J.J.; D’Agati, V.D.; Schwartz, M.M.; Seshan, S.V.; Alpers, C.E.; Appel, G.B.; Balow, J.E.; Bruijn, J.A.; Cook, T.; Ferrario, F.; et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004, 65, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moroni, G.; Depetri, F.; Ponticelli, C. Lupus nephritis: When and how often to biopsy and what does it mean? J. Autoimmun. 2016, 74, 27–40. [Google Scholar] [CrossRef]
- Bastian, H.M.; Roseman, J.M.; McGwin, G., Jr.; Alarcón, G.S.; Friedman, A.W.; Fessler, B.J.; Baethge, B.A.; Reveille, J.D.; LUMINA Study Group. Systemic lupus erythematosus in three ethnic groups. XII. Risk factors for lupus nephritis after diagnosis. Lupus 2002, 11, 152–160. [Google Scholar] [CrossRef]
- Doria, A.; Iaccarino, L.; Ghirardello, A.; Zampieri, S.; Arienti, S.; Sarzi-Puttini, P.; Atzeni, F.; Piccoli, A.; Todesco, S. Long-term prognosis and causes of death in systemic lupus erythematosus. Am. J. Med. 2006, 119, 700–706. [Google Scholar] [CrossRef]
- Anders, H.-J.; Saxena, R.; Zhao, M.-H.; Parodis, I.; Salmon, J.E.; Mohan, C. Lupus nephritis. Nat. Rev. Dis. Primers 2020, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Sciascia, S.; Cozzi, M.; Barinotti, A.; Radin, M.; Cecchi, I.; Fenoglio, R.; Mancardi, D.; Jones, G.W.; Rossi, D.; Roccatello, D. Renal fibrosis in lupus nephritis. Int. J. Mol. Sci. 2022, 23, 14317. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, M.; Gatto, M.; Binda, V.; Doria, A.; Moroni, G. Lupus nephritis: Clinical presentations and outcomes in the 21st century. Rheumatology 2020, 59 (Suppl. S5), v39–v51. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Li, P.; Dang, X.; Zhang, X.; Mao, Y.; Chen, X. Lupus nephritis: New progress in diagnosis and treatment. J. Autommun. 2022, 132, 102871. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Fueyo, A.; Bradley, S.J.; Klatzmann, D.; Tsokos, G.C. T cells and autoimmune kidney disease. Nat. Rev. Nephrol. 2017, 13, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Bhat, P.; Radhakrishnan, J. B lymphocytes and lupus nephritis: New insights into pathogenesis and targeted therapies. Kidney Int. 2008, 73, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.; Henderson, S.G.; Brandt, D.; Liu, N.; Guttikonda, R.; Hsieh, C.; Kaverina, N.; Utset, T.O.; Meehan, S.M.; Quigg, R.J.; et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J. Immunol. 2011, 186, 1849–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, M.; Yagita, H.; Inoue, H.; Takanashi, T.; Matsuda, H.; Munechika, E.; Kanamaru, Y.; Shirato, I.; Tomino, Y.; Matsushima, K.; et al. Selective accumulation of CCR4+ T lymphocytes into renal tissue of patients with lupus nephritis. Arthritis Rheum. 2002, 46, 735–740. [Google Scholar] [CrossRef]
- Murata, H.; Matsumura, R.; Koyama, A.; Sugiyama, T.; Sueishi, M.; Shibuya, K.; Tsutsumi, A.; Sumida, T. T cell receptor repertoire of T cells in the kidneys of patients with lupus nephritis. Arthritis Rheum. 2002, 46, 2141–2147. [Google Scholar] [CrossRef]
- Crispin, J.C.; Oukka, M.; Bayliss, G.; Cohen, R.A.; Van Beek, C.A.; Stillman, I.E.; Kyttaris, V.C.; Juang, Y.-T.; Tsokos, G.C. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J. Immunol. 2008, 181, 8761–8766. [Google Scholar] [CrossRef] [Green Version]
- Enghard, P.; Humrich, J.Y.; Rudolph, B.; Rosenberger, S.; Biesen, R.; Kuhn, A.; Manz, R.; Hiepe, F.; Radbruch, A.; Burmester, G.-R.; et al. CXCR3+CD4+ T cells are enriched in inflamed kidneys and urine and provide a new biomarker for acute nephritis flares in systemic lupus erythematosus patients. Arthritis Rheum. 2009, 60, 199–206. [Google Scholar] [CrossRef]
- Allison, S.J. Breaches in the Bowman’s capsule and CD8+ T cell infiltration in crescentic GN. Nat. Rev. Nephrol. 2018, 14, 597. [Google Scholar] [CrossRef]
- Munro, D.A.D.; Hughes, J. The origins and functions of tissue-resident macrophages in kidney development. Front. Physiol. 2017, 8, 837. [Google Scholar] [CrossRef] [Green Version]
- Der, E.; Suryawanshi, H.; Morozov, P.; Kustagi, M.; Goilav, B.; Ranabothu, S.; Izmirly, P.; Clancy, R.; Belmont, H.M.; Koenigsberg, M.; et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat. Immunol. 2019, 20, 915–927. [Google Scholar] [CrossRef]
- Arazi, A.; Rao, D.A.; Berthier, C.C.; Davidson, A.; Liu, Y.; Hoover, P.J.; Chicoine, A.; Eisenhaure, T.M.; Jonsson, A.H.; Li, S.; et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol. 2019, 20, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A. Renal mononuclear phagocytes in lupus nephritis. ACR Open Rheumatol. 2021, 3, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, E.; Seron, D.; Hartley, R.B.; Cameron, J.S. Lupus nephritis: Correlation of interstitial cells with glomerular function. Kidney Int. 1990, 37, 100–169. [Google Scholar] [CrossRef] [Green Version]
- D’Agati, V.D.; Appel, G.B.; Estes, D.; Knowles, D.M., II; Pirani, C.L. Monoclonal antibody identification of infiltrating mononuclear leukocytes in lupus nephritis. Kidney Int. 1986, 30, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, S.; Nakatani, K.; Iwano, M.; Asai, O.; Samejima, K.-i.; Sakan, H.; Terada, M.; Harada, K.; Akai, Y.; Shiiki, H.; et al. Elevated levels of fractalkine expression and accumulation of CD16+ monocytes in glomeruli of active lupus nephritis. Am. J. Kidney Dis. 2007, 50, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, K.; Yoshimoto, S.; Iwano, M.; Asai, O.; Samejima, K.-i.; Sakan, H.; Terada, M.; Hasegawa, H.; Nose, M.; Saito, Y. Fractalkine expression and CD16+ monocyte accumulation in glomerular lesions: Association with their severity and diversity in lupus models. Am. J. Physiol. Renal Physiol. 2010, 299, F207–F216. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.; Chang, A.; Brandt, D.; Guttikonda, R.; Utset, T.O.; Clark, M.R. Predicting outcomes of lupus nephritis with tubulointerstitial inflammation and scarring. Arthritis Care Res. 2011, 63, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Blanco, P.; Palucka, A.K.; Gill, M.; Pascual, V.; Banchereau, J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science 2001, 294, 1540–1543. [Google Scholar] [CrossRef]
- Scheinecker, C.; Zwölfer, B.; Köller, M.; Männer, G.; Smolen, J.S. Alterations of dendritic cells in systemic lupus erythematosus: Phenotypic and functional deficiencies. Arthritis Rheum. 2001, 44, 856–865. [Google Scholar] [CrossRef]
- Tucci, M.; Quatraro, C.; Lombardi, L.; Pellegrino, C.; Dammacco, F.; Silvestris, F. Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: Role of interleukin-18. Arthritis Rheum. 2008, 58, 251–262. [Google Scholar] [CrossRef]
- Maria, N.I.; Davidson, A. Renal macrophages and dendritic cells in SLE nephritis. Curr. Rheumatol. Rep. 2017, 19, 81. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Tang, W.; Tang, W. Immune cell infiltration characteristics and related core genes in lupus nephritis: Results from bioinformatic analysis. BMC Immunol. 2019, 20, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perico, L.; Conti, S.; Benigni, A.; Remuzzi, G. Podocyte actin dynamics in health and disease. Nat. Rev. Nephrol. 2016, 12, 692–710. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.-J.; Klionsky, D.J.; Zhang, H. Podocytes and autophagy: A potential therapeutic target in lupus nephritis. Autophagy 2019, 15, 908–912. [Google Scholar] [CrossRef]
- Jin, J.; Ye, M.; Zhao, L.; Zou, W.; Shen, W.; Zhang, H.; Gong, J.; He, Q. The novel involvement of podocyte autophagic activity in the pathogenesis of lupus nephritis. Histol. Histopathol. 2018, 33, 803–814. [Google Scholar]
- Oliva-Damaso, N.; Payan, J.; Oliva-Damaso, E.; Pereda, T.; Bomback, A.S. Lupus podocytopathy: An overview. Adv. Chronic Kidney Dis. 2019, 26, 369–375. [Google Scholar] [CrossRef]
- Avraham, S.; Korin, B.; Chung, J.-J.; Oxburgh, L.; Shaw, A.S. The mesangial cell-the glomerular stromal cell. Nat. Rev. Nephrol. 2021, 17, 855–864. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, L.; Wang, Y.; Hu, W.; Wang, C.; Wen, Z. Mesangial cell: A hub in lupus nephritis. Front. Immunol. 2022, 13, 1063497. [Google Scholar] [CrossRef]
- Yu, C.-L.; Sun, K.-H.; Tsai, C.-Y.; Hsieh, S.-C.; Yu, H.-S. Anti-dsDNA antibody up-regulates interleukin 6, but not cyclo-oxygenase, gene expression in glomerular mesangial cells: A marker of immune-mediated renal damage? Inflamm. Res. 2001, 50, 12–18. [Google Scholar] [CrossRef]
- Nowling, T.K. Mesangial cells in lupus nephritis. Curr. Rheumatol. Rep. 2022, 23, 83. [Google Scholar] [CrossRef] [PubMed]
- Kwok, S.-K.; Tsokos, G.C. New insights into the role of renal resident cells in the pathogenesis of lupus nephritis. Korean J. Intern. Med. 2018, 33, 284–289. [Google Scholar] [CrossRef] [Green Version]
- Kolios, A.G.; Tsokos, G.C. Skin-kidney crosstalk in SLE. Nat. Rev. Rheumatol. 2021, 17, 253–254. [Google Scholar] [CrossRef]
- Sung, S.-S.J.; Fu, S.M. Interactions among glomerulus infiltrating macrophages and intrinsic cells via cytokines in chronic lupus glomerulonephritis. J. Autoimmun. 2020, 106, 102331. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, R.; Li, H.; Tsokos, G.C. Pathogenesis of lupus nephritis: The contribution of immune and kidney resident cells. Curr. Opin. Rheumatol. 2023, 35, 107–116. [Google Scholar] [CrossRef]
- Jamaly, S.; Rakaee, M.; Abdi, R.; Tsokos, G.C.; Fenton, K.A. Interplay of immune and kidney resident cells in the formation of tertiary lymphoid structures in lupus nephritis. Autoimmun. Rev. 2021, 20, 102980. [Google Scholar] [CrossRef]
- Dorraji, S.E.; Kanapathippillai, P.; Hovd, A.-M.K.; Stenersrød, M.R.; Horvei, K.D.; Ursvik, A.; Figenschau, S.L.; Thiyagarajan, D.; Fenton, C.G.; Pedersen, H.L.; et al. Kidney tertiary lymphoid structures in lupus nephritis develop into large interconnected networks and resemble lymph nodes in gene signature. Am. J. Pathol. 2020, 190, 2203–2225. [Google Scholar] [CrossRef] [PubMed]
- Fakhfakh, R.; Zian, Z.; Elloumi, N.; Abida, O.; Bouallegui, E.; Houssaini, H.; Volpe, E.; Capone, A.; Hachicha, H.; Marzouk, S.; et al. Th17 and Th1 cells in systemic lupus erythematosus with focus on lupus nephritis. Immunol. Res. 2022, 70, 644–653. [Google Scholar] [CrossRef]
- Pisitkun, P.; Ha, H.-L.; Wang, H.; Claudio, E.; Tivy, C.C.; Zhou, H.; Mayadas, T.N.; Illei, G.G.; Siebenlist, U. Interleukin-17 cytokines are critical in development of fatal lupus glomerulonephritis. Immunity 2012, 37, 1104–1115. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Kyttaris, V.C.; Tsokos, G.C. The role of IL-23/IL-17 axis in lupus nephritis. J. Immunol. 2009, 183, 3160–3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Adamopoulos, I.E.; Moulton, V.R.; Stillman, I.E.; Herbert, Z.; Moon, J.J.; Sharabi, A.; Krishfield, S.; Tsokos, M.G.; Tsokos, G.C. Systemic lupus erythematosus favors the generation of IL-17 producing double negative T cells. Nat. Commun. 2020, 11, 2859. [Google Scholar] [CrossRef]
- Chen, A.; Lee, K.; D’Agati, V.D.; Wei, C.; Fu, J.; Guan, T.-J.; He, J.C.; Schlondorff, D.; Agudo, J. Bowman’s capsule provides a protective niche for podocytes from cytotoxic CD8+ T cells. J. Clin. Investig. 2018, 128, 3413–3424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Tsokos, M.G.; Bhargava, R.; Adamopoulos, I.E.; Menn-Josephy, H.; Stillman, I.E.; Rosenstiel, P.; Jordan, J.; Tsokos, G.C. IL-23 reshapes kidney resident cell metabolism and promotes local kidney inflammation. J. Clin. Investig. 2021, 131, e142428. [Google Scholar] [CrossRef]
- Teramoto, K.; Negoro, N.; Kitamoto, K.; Iwai, T.; Iwao, H.; Okamura, M.; Miura, K. Microarray analysis of glomerular gene expression in murine lupus nephritis. J. Pharmacol. Sci. 2008, 106, 56–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinmetz, O.M.; Turner, J.-E.; Paust, H.-J.; Lindner, M.; Peters, A.; Heiss, K.; Velden, J.; Hopfer, H.; Fehr, S.; Krieger, T.; et al. CXCR3 mediates renal Th1 and Th17 immune response in murine lupus nephritis. J. Immunol. 2009, 183, 4693–4704. [Google Scholar] [CrossRef] [Green Version]
- Tshilela, K.A.; Ikeuchi, H.; Matsumoto, T.; Kuroiwa, T.; Sakurai, N.; Sakairi, T.; Kaneko, Y.; Maeshima, A.; Hiromura, K.; Nojima, Y. Glomerular cytokine expression in murine lupus nephritis. Clin. Exp. Nephrol. 2016, 20, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-Y.; Wu, T.-H.; Huang, S.-F.; Sun, K.-H.; Hsieh, S.-C.; Han, S.-H.; Yu, H.-S.; Yu, C.-L. Abnormal thymic and splenic IL-4 and TNF-α expression in MRL-lpr/lpr mice. Scand. J. Immunol. 1995, 41, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-T.; Shiao, Y.-M.; Wu, T.-H.; Chen, W.-S.; Hsu, Y.-H.; Tsai, S.-F.; Tsai, C.-Y. Serum BLC/CXCL13 concentration and renal expression of CXCL13/CXCR5 in patients with systemic lupus erythematosus and lupus nephritis. J. Rheumatol. 2010, 37, 45–52. [Google Scholar] [CrossRef]
- Wang, Y.; Ito, S.; Chino, Y.; Goto, D.; Matsumoto, I.; Murata, H.; Tsutsumi, A.; Hayashi, T.; Uchida, K.; Usui, J.; et al. Laser microdissection-based analysis of cytokine balance in the kidneys of patients with lupus nephritis. Clin. Exp. Immunol. 2010, 159, 1–10. [Google Scholar] [CrossRef]
- Yung, S.; Cheung, K.F.; Zhang, Q.; Chan, T.M. Mediators of inflammation and their effect on resident renal cells: Implications in lupus nephritis. Clin. Dev. Immunol. 2013, 2013, 317682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paquissi, F.C.; Absensur, H. The Th17/IL-17 axis and kidney disease, with focus on lupus nephritis. Front. Med. 2021, 8, 654912. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflamasome: An overview of mechanism of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeoka, A.A.; Mueller, J.L.; Kambo, A.; Mathison, J.C.; King, A.J.; Hall, W.F.; da Silva Correia, J.; Ulevitch, R.J.; Hoffman, H.M.; Mckay, D.B. An inflammasome-independent role for epithelial-expressed NLRP3 in renal ischemia-reperfusion injury. J. Immunol. 2010, 185, 6277–6285. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, G.; Darisipudi, M.N.; Anders, H.-J. Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis. Nephrol. Dial. Transplant. 2014, 29, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, C.B.; Lima, C.A.D.; Vajgel, G.; Sandrin-Garcia, P. The role of NLRP3 inflammasome in lupus nephritis. Int. J. Mol. Sci. 2021, 22, 12476. [Google Scholar] [CrossRef]
- Anton-Pampols, P.; Diaz-Requena, C.; Martinez-Valenzuela, L.; Gomez-Preciado, F.; Fulladosa, X.; Vidal-Alabro, A.; Torras, J.; Lloberas, N.; Draibe, J. The role of inflammasomes in glomerulonephritis. Int. J. Mol. Sci. 2022, 23, 4208. [Google Scholar] [CrossRef]
- Guo, C.; Fu, R.; Zhou, M.; Wang, S.; Huang, Y.; Hu, H.; Zhao, J.; Gaskin, F.; Yang, N.; Fu, S.M. Pathogenesis of lupus nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome activation. J. Autoimmun. 2019, 103, 112286. [Google Scholar] [CrossRef]
- Fu, R.; Guo, C.; Wang, S.; Huang, Y.; Jin, O.; Hu, H.; Chen, J.; Xu, B.; Zhou, M.; Zhao, J.; et al. Podocyte activation of NLRP3 inflammasomes contributes to the development of proteinuria in lupus nephritis. Arthritis Rheumatol. 2017, 69, 1636–1646. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.-W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.-J.; Lin, S.-C.; Dong, M.-Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.; He, Y.; Xu, H.; Li, Y.; Han, L.; Yan, L.; Lang, H.; Zhao, Y.; Zhao, Z.; Qi, Y. CD36 aggravates podocyte injury by activating NLRP3 inflammasome and inhibiting autophagy in lupus nephritis. Cell Death Dis. 2022, 13, 729. [Google Scholar] [CrossRef] [PubMed]
- Tsao, Y.-P.; Tseng, F.-Y.; Chao, C.-W.; Chen, M.-H.; Yeh, Y.-C.; Abdulkareem, B.O.; Chen, S.-Y.; Chuang, W.-T.; Chang, P.-C.; Chen, I.-C.; et al. NLRP12 is an innate immune checkpoint for repressing IFN signatures and attenuating lupus nephritis progression. J. Clin. Investig. 2023, 133, e157272. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Harada, T.; Mizushima, Y.; Sakane, T. Possible pathogenic role of cationic anti-DNA autoantibodies in the development of nephritis in patients with systemic lupus erythematosus. J. Immunol. 1993, 151, 1128–1136. [Google Scholar] [CrossRef]
- Mostoslavsky, G.; Fischel, R.; Yachimovich, N.; Yarkoni, Y.; Rosenmann, E.; Monestier, M.; Baniyash, M.; Eilat, D. Lupus anti-DNA autoantibodies cross-react with a glomerular structural protein: A case for tissue injury by molecular mimicry. Eur. J. Immunol. 2001, 31, 1221–1227. [Google Scholar] [CrossRef]
- Deocharan, B.; Qing, X.; Lichauco, J.; Putterman, C. Alpha-actinin is cross reactive renal target for pathogenic anti-DNA antibodies. J. Immunol. 2002, 168, 3072–3078. [Google Scholar] [CrossRef] [Green Version]
- Yung, S.; Cheung, K.F.; Zhang, Q.; Chan, T.M. Anti-dsDNA antibodies bind to mesangial annexin II in lupus nephritis. J. Am. Soc. Nephrol. 2010, 21, 1912–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Cheng, H.; Zhang, Y.; Fang, C.; Xia, Y. The antigen-binding fragment of anti-double-stranded DNA IgG enhances F-actin formation in mesangial cells by binding to alpha-actinin-4. Exp. Biol. Med. 2012, 237, 1023–1031. [Google Scholar] [CrossRef]
- Franchin, G.; Son, M.; Kim, S.J.; Ben-Zvi, I.; Zhang, J.; Diamond, B. Anti-DNA antibodies cross-react with C1q. J. Autoimmun. 2013, 44, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.L.; Huang, M.H.; Tsai, C.Y.; Tsai, Y.Y.; Tsai, S.T.; Sun, K.H.; Han, S.H.; Yu, H.S. The effect of human polyclonal anti-dsDNA autoantibodies on apoptotic gene expression in cultured rat glomerular mesangial cells. Scand. J. Rheumatol. 1998, 27, 54–60. [Google Scholar]
- Yung, S.; Tsang, R.C.W.; Leung, J.K.H.; Chan, T.M. Increased mesangial cell hyaluronan expression in lupus nephritis is mediated by anti-DNA antibody-induced IL-1beta. Kidney Int. 2006, 69, 272–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, X.; Zavadil, J.; Crosby, M.B.; Hogarth, M.P.; Hahn, B.H.; Mohan, C.; Gilkeson, G.S.; Bottinger, E.P.; Putterman, C. Nephritogenic anti-DNA antibodies regulate gene expression in MRL/lpr mouse glomerular mesangial cells. Arthritis Rheum. 2006, 54, 2198–2210. [Google Scholar] [CrossRef] [PubMed]
- Qing, X.; Pitashny, M.; Thomas, D.B.; Barrat, F.J.; Hogarth, M.P.; Putterman, C. Pathogenic anti-DNA antibodies modulate gene expression in mesangial cells: Involvement of HMGB1 in anti-DNA antibody-induced renal injury. Immunol. Lett. 2008, 121, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, S.; Zhang, Q.; Zhang, C.Z.; Chan, K.W.; Lui, S.L.; Chan, T.M. Anti-DNA antibody induction of protein kinase C phosphorylation and fibronectin synthesis in human and murine lupus and the effect of mycophenolic acid. Arthritis Rheum. 2009, 60, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, C.; Wang, S.; Huang, Y.; Wang, H.; Zhao, J.; Yang, N. Anti-dsDNA antibodies induce inflammation via endoplasmic reticulum stress in human mesangial cells. J. Transl. Med. 2015, 13, 178. [Google Scholar] [CrossRef] [Green Version]
- Yung, S.; Ng, C.Y.C.; Ho, S.K.; Cheung, K.F.; Chan, K.W.; Zhang, Q.; Chau, M.K.M.; Chan, T.M. Anti-dsDNA antibody induces soluble fibronectin secretion by proximal renal tubular epithelial cells and downstream increase of TGF-β1 and collagen synthesis. J. Autoimmun. 2015, 58, 111–122. [Google Scholar] [CrossRef]
- Yung, S.; Chan, T.M. Anti-dsDNA antibodies and resident renal cells—Their putative roles in pathogenesis of renal lesions in lupus nephritis. Clin. Immunol. 2017, 185, 40–50. [Google Scholar] [CrossRef]
- Hu, Z.; Leppla, S.H.; Li, B.; Elkins, C.A. Antibodies specific for nucleic acids and applications in genomic detection and clinical diagnostics. Expert Rev. Mol. Diagn. 2014, 14, 895–916. [Google Scholar] [CrossRef]
- Akberova, N.I.; Zhmurov, A.A.; Nevzorova, T.A.; Litvinov, R.I. An anti-DNA antibody prefers damaged dsDNA over native. J. Biomol. Struct. Dyn. 2017, 35, 219–232. [Google Scholar] [CrossRef]
- Wang, X.; Xia, Y. Anti-double stranded DNA antibodies: Origin, pathogenicity, and targeted therapies. Front. Immunol. 2019, 10, 1667. [Google Scholar] [CrossRef] [Green Version]
- Yung, S.; Chan, T.M. Mechanisms of kidney injury in lupus nephritis—The role of anti-dsDNA antibodies. Front. Immunol. 2015, 6, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.-H.; Lui, W.-T.; Tsai, C.-Y.; Tang, S.-J.; Han, S.H.; Yu, C.-L. Anti-dsDNA antibodies cross-react with ribosomal P proteins expressed on the surface of glomerular mesangial cells to exert a cytostatic effect. Immunology 1995, 85, 262–269. [Google Scholar]
- Sun, K.-H.; Liu, W.-T.; Tang, S.-J.; Tsai, C.-Y.; Hsieh, S.-C.; Wu, T.-H.; Han, S.-H.; Yu, C.-L. The expression of acidic ribosomal phosphoproteins on the surface membrane of different tissues in autoimmune and normal mice which are the target molecules for anti-double stranded DNA antibodies. Immunology 1996, 87, 362–371. [Google Scholar]
- Otey, C.A.; Carpen, O. Alpha-actinin revisited: A fresh look at an old player. Cell Motil. Cytoskeleton 2004, 58, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Weinstein, E.; Tuzova, M.; Davidson, A.; Mundel, P.; Marambio, P.; Putterman, C. Cross-reactivity of human lupus anti-DNA antibodies with α-actinin and nephritogenic potential. Arthritis Rheum. 2005, 52, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Riboldi, P.; Gerosa, M.; Moroni, G.; Radice, A.; Allegri, F.; Sinico, A.; Tincani, A.; Meroni, P.L. Anti-DNA antibodies: A diagnostic and prognostic tool for systemic lupus erythematosus? Autoimmunity 2005, 38, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Devey, M.E.; Lee, S.R.; Le Page, S.; Feldman, R.; Isenberg, D.A. Serial studies of the IgG subclass and functional affinity of DNA antibodies in systemic lupus erythematosus. J. Autoimmun. 1998, 1, 483–494. [Google Scholar] [CrossRef]
- Kaneko, Y.; Nimmerjahn, F.; Ravetch, J.V. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 2006, 313, 670–673. [Google Scholar] [CrossRef] [Green Version]
- Käsermann, F.; Boerema, D.J.; Rüegsegger, M.; Hofmann, A.; Wymann, S.; Zuercher, A.W.; Miescher, S. Analysis and functional consequences of increased Fab-sialylation of intravenous immunoglobulin (IVIG) after lectin fractionation. PLoS ONE 2012, 7, e37243. [Google Scholar] [CrossRef]
- Otani, M.; Kuroki, A.; Kikuchi, S.; Kihara, M.; Nakata, J.; Ito, K.; Furukawa, J.-i. Sialylation determines the nephritogenicity of IgG3 cryoglobulins. J. Am. Soc. Nephrol. 2012, 23, 1869–1878. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Zhou, Z.; Zhang, R.; You, Y.; Guo, Z.; Huang, J.; Wang, F.; Sun, Y.; Liu, H.; Cheng, X.; et al. Fucosylation of anti-dsDNA IgG1 correlates with disease activity of treatment-naïve systemic lupus erythematosus patients. eBioMedicine 2022, 77, 103883. [Google Scholar] [CrossRef] [PubMed]
- Liou, L.-B.; Chen, C.-C.; Chiang, W.-Y.; Chen, M.-H. De-sialylated and sialylated IgG anti-dsDNA antibodies respectively worsen and mitigate experimental mouse lupus proteinuria and possible mechanisms. Int. Immunopharmacol. 2022, 109, 108837. [Google Scholar] [CrossRef] [PubMed]
- Liou, L.-B.; Wang, T.-Y.; Lui, I.-J.; Wu, H.-C.; Ke, P.-Y.; Fang, Y.-F.; Chen, Y.-F. α-2,6-sialic acid/IgG anti-dsDNA ratios correlate with human lupus disease activity and possible mechanisms: A pilot study. Lupus 2022, 31, 927–938. [Google Scholar] [CrossRef]
- Tsai, C.-Y.; Yu, C.-L.; Wu, T.-H.; Lu, J.-Y.; Lair, T.-S.; Tsai, Y.-Y.; Chou, C.-T. Polyclonal anticardiolipin antibodies purified from sera of patients with active systemic lupus erythematosus induce apoptosis of the cultured glomerular mesangial cells. Scand. J. Rheumatol. 2000, 29, 370–379. [Google Scholar] [PubMed]
- Oelzner, P.; Deliyska, B.; Fünfstück, R.; Hein, G.; Herrmann, D.; Stein, G. Anti-C1q antibodies and antiendothelial cell antibodies in systemic lupus erythematosus—Relationship with disease activity and renal involvement. Clin. Rheumatol. 2003, 22, 271–278. [Google Scholar] [CrossRef]
- Kianmehr, N.; Khoshmirsafa, M.; Shekarabi, M.; Falak, R.; Haghighi, A.; Masoodian, M.; Seif, F.; Omidi, F.; Shirani, F.; Dadfar, N. High frequency of concurrent anti-C1q and anti-dsDNA but not anti-C3b antibodies in patients with lupus nephritis. J. Immunoassay Immunothem. 2021, 42, 406–423. [Google Scholar] [CrossRef]
- Renaudineau, Y.; Deocharan, B.; Jousse, S.; Renaudineau, E.; Putterman, C.; Youinou, P. Anti-alpha-actinin antibodies: A new marker of lupus nephritis. Autoimmun. Rev. 2007, 6, 464–468. [Google Scholar] [CrossRef]
- Onishi, S.; Adnan, E.; Ishizaki, J.; Miyazaki, T.; Tanaka, Y.; Matsumoto, T.; Suemori, K.; Shudou, M.; Okura, T.; Takeda, H.; et al. Novel autoantigens associated with lupus nephritis. PLoS ONE 2015, 10, e0126564. [Google Scholar] [CrossRef]
- Sarfaraz, S.; Anis, S.; Ahmed, E.; Muzaffar, R. Clinical significance of anti-ribosomal P protein antibodies in patients with lupus nephritis. Rev. Recent Clin. Trials 2018, 13, 281–286. [Google Scholar] [CrossRef]
- Wakamatsu, A.; Sato, H.; Kaneko, Y.; Cho, T.; Ito, Y.; Kurosawa, Y.; Hasegawa, E.; Kobayashi, D.; Nakatsue, T.; Kuroda, T.; et al. Association of coexisting anti-ribosomal P and anti-dsDNA antibodies with histology and renal prognosis in lupus nephritis patients. Lupus 2021, 30, 448–458. [Google Scholar] [CrossRef]
- Shang, X.; Ren, L.; Sun, G.; Yu, T.; Yao, Y.; Wang, L.; Liu, F.; Zhang, L.; He, X.; Liu, M. Anti-dsDNA, anti-nucleosome, anti-C1q, and anti-histone antibodies as markers of active lupus nephritis and systemic lupus erythematosus disease activity. Immun. Inflamm. Dis. 2021, 9, 407–418. [Google Scholar] [CrossRef]
- Soliman, S.; Mohan, C. Lupus nephritis biomarkers. Clin. Immunol. 2017, 185, 10–20. [Google Scholar] [CrossRef]
- Wu, T.; Ding, H.; Han, J.; Arriens, C.; Wei, C.; Han, W.; Pedroza, C.; Jiang, S.; Anolik, J.; Petri, M.; et al. Antibody-array-based proteomic screening of serum markers in systemic lupus erythematosus: A discovery study. J. Proteome. Res. 2016, 15, 2102–2114. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Duran, V.; Vanarsa, K.; Mohan, C. Targeted urine proteomics in lupus nephritis—A meta-analysis. Expert Rev. Proteom. 2020, 17, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, L.; Lindblom, J.; Mohan, C.; Parodis, I. Current insights on biomarkers in lupus nephritis: A systematic review of the literature. J. Clin. Med. 2022, 11, 5759. [Google Scholar] [CrossRef] [PubMed]
- Vincent, F.B.; Saulep-Easton, D.; Figgett, W.A.; Fairfax, K.A.; Mackay, F. The BAFF/APRIL system: Emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev. 2013, 24, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Vincent, F.B.; Morand, E.F.; Mackay, F. BAFF and innate immunity: New therapeutic targets for systemic lupus erythematosus. Immunol. Cell Biol. 2012, 90, 293–303. [Google Scholar] [CrossRef]
- Schwarting, A.; Relle, M.; Meineck, M.; Föhr, B.; Triantafyllias, K.; Weinmann, A.; Roth, W.; Weimann-Menke, J. Renal tubular epithelial cell-derived BAFF expression mediates kidney damage and correlates with activity of proliferative lupus nephritis in mouse and men. Lupus 2018, 27, 243–256. [Google Scholar] [CrossRef]
- Suso, J.P.; Posso-Osorio, I.; Jiménez, C.A.; Naranjo-Escobar, J.; Ospina, F.E.; Sánchez, A.; Caňas, C.A.; Tobón, G.J. Profile of BAFF and its receptors’ expression in lupus nephritis is associated with pathological classes. Lupus 2018, 27, 708–715. [Google Scholar] [CrossRef]
- Aguirre-Valencia, D.; Ríos-Serna, L.J.; Posso-Osorio, I.; Naranjo-Escobar, J.; López, D.; Bedoya-Joaqui, V.; Nieto-Aristizábal, I.; Castro, A.M.; Diáz-Ordoňez, L.; Navarro, E.P.; et al. Expression of BAFF, APRIL, and cognate receptor genes in lupus nephritis and potential use as urinary biomarkers. J. Transl. Autoimmun. 2019, 3, 100027. [Google Scholar] [CrossRef] [PubMed]
- Marín-Rosales, M.; Palafox-Sánchez, C.A.; Franco-Topete, R.A.; Carrillo-Ballesteros, F.J.; Cruz, A.; Salazar-Camarena, D.C.; Muňoz-Valle, J.F.; Ramos-Solano, F. Renal tissue expression of BAFF and BAFF receptors is associated with proliferative lupus nephritis. J. Clin. Med. 2022, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Mejia-Vilet, J.M.; Parikh, S.V.; Song, H.; Fadda, P.; Shapiro, J.P.; Ayoub, I.; Yu, L.; Zhang, J.; Uribe-Uribe, N.; Rovin, B.H. Immune gene expression in kidney biopsies of lupus nephritis patients at diagnosis and at a renal flare. Nephrol. Dial. Transplant. 2019, 34, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Fairhurst, A.-M.; Xie, C.; Fu, Y.; Wang, A.; Boudreaux, C.; Zhou, X.J.; Cibotti, R.; Coyle, A.; Connolly, J.E.; Wakeland, E.K.; et al. Type I interferons produced by resident renal cells may promote end-organ disease in autoantibody-mediated glomerulonephritis. J. Immuol. 2009, 183, 6831–6838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Ren, Y.; He, X. IFN-1 mediates lupus nephritis from the beginning to renal fibrosis. Front. Immunol. 2021, 12, 676082. [Google Scholar] [CrossRef]
- Mavragani, C.P.; Kirou, K.A.; Seshan, S.V.; Crow, M.K. Type 1 interferon and neutrophil transcripts in lupus nephritis renal biopsies: Clinical and histopathological association. Rheumatology 2022, 1–5. [Google Scholar] [CrossRef]
- Wang, L.; Yang, Z.; Yu, H.; Lin, W.; Wu, R.; Yang, H.; Yang, K. Predicting diagnostic gene expression profiles associated with immune infiltration in patients with lupus nephritis. Front. Immunol. 2022, 13, 839197. [Google Scholar] [CrossRef]
- Gilmore, A.C.; Wilson, H.R.; Cairns, T.D.; Botto, M.; Lightstone, L.; Bruce, I.N.; Cook, H.T.; Pickering, M.C. Immune gene expression and functional networks in distinct lupus nephritis. Lupus Sci. Med. 2022, 9, e000615. [Google Scholar] [CrossRef]
- Parikh, S.; Malvar, A.; Song, H.; Shapiro, J.; Mejia-Vilet, J.M.; Ayoub, I.; Almaani, S.; Madhavan, S.; Alberton, V.; Besso, C.; et al. Molecular profiling of kidney compartments from serial biopsies differentiate treatment responders from non-responders in lupus nephritis. Kidney Int. 2022, 102, 845–865. [Google Scholar] [CrossRef]
- Hammad, A.M.; Youssef, H.M.; El-Arman, M.M. Transforming growth factor beta 1 in children with systemic lupus erythematosus: A possible relation with clinical presentation of lupus nephritis. Lupus 2006, 15, 608–612. [Google Scholar] [CrossRef]
- Chan, R.W.-Y.; Tam, L.-S.; Li, E.K.-M.; Lai, F.M.-M.; Chow, K.-M.; Lai, K.-B.; Li, P.K.-T.; Szeto, C.-C. Inflammatory cytokine gene expression in the urinary sediment of patients with lupus nephritis. Arthritis Rheum. 2003, 48, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Song, H.; Birmingham, D.J.; Hebert, L.A.; Yu, C.Y.; Nagaraja, H.N. Urine chemokines as biomarkers of human systemic lupus erythematosus activity. J. Am. Soc. Nephrol. 2005, 16, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avihingsanon, Y.; Phumesin, P.; Benjachat, T.; Akkasilpa, S.; Killikowit, V.; Praditpomsilpa, K.; Wongpiyabavorn, J.; Eiam-Ong, S.; Hemachudha, T.; Tungsanga, K.; et al. Measurement of urinary chemokine and growth factor messenger RNAs: A noninvasive monitoring in lupus nephritis. Kidney Int. 2006, 69, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, N.; Rubinstein, T.; Burkly, L.C.; Collins, C.E.; Blanco, I.; Su, L.; Hojaili, B.; Mackay, M.; Aranow, C.; Stohl, W.; et al. Urinary TWEAK as a biomarker of lupus nephritis: A multicenter cohort study. Arthritis Res. Ther. 2009, 11, R143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Xie, C.; Wang, H.W.; Zhou, X.J.; Schwartz, N.; Calixto, S.; Mackay, M.; Aranow, C.; Putterman, C.; Mohan, C. Elevated urinary VCAM-1, P-selectin, soluble TNF receptor, and CXC chemokine ligand 16 in multiple murine lupus strains and human lupus nephritis. J. Immunol. 2007, 179, 7166–7175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolignano, D.; Donato, V.; Coppolino, G.; Campo, S.; Buemi, A.; Lacquaniti, A.; Buemi, M. Neutrophil gelatinase-associated lipocalin (NGAL) as a marker of kidney damage. Am. J. Kidney Dis. 2008, 52, 595–605. [Google Scholar] [CrossRef]
- Rubinstein, T.; Pitashny, M.; Putterman, C. The novel role of neutrophil gelatinase-B associated lipocalin (NGAL)/lipocalin -2 as a biomarker of lupus nephritis. Autoimmun. Rev. 2008, 7, 229–234. [Google Scholar] [CrossRef]
- Yang, C.-C.; Hsieh, S.-C.; Li, K.-J.; Wu, C.-H.; Lu, M.-C.; Tsai, C.-Y.; Yu, C.-L. Urinary neutrophil gelatinase-associated lipocalin is a potential biomarker for renal damage in patients with systemic lupus erythematosus. J. Biomed. Biotechnol. 2012, 2012, 759313. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, S.-C.; Tsai, C.-Y.; Yu, C.-L. Potential serum and urine biomarkers in patients with lupus nephritis and the unsolved problems. Open Access Rheumatol. 2016, 8, 81–91. [Google Scholar]
- Wu, T.-H.; Li, K.-J.; Yu, C.-L.; Tsai, C.-Y. Tamm-Horsfall protein is a potent immunomodulatory molecule and a disease biomarker in the urinary system. Molecules 2018, 23, 200. [Google Scholar] [CrossRef] [Green Version]
- Badr, A.M.M.; Farag, Y.; Abdelshafy, M.; Riad, N.M. Urinary interleukin 22 binding protein as a marker of lupus nephritis in Egyptian children with juvenile systemic lupus erythematosus. Clin. Rheumatol. 2018, 37, 451–458. [Google Scholar] [CrossRef]
- Bertolo, M.; Baumgart, S.; Durek, P.; Peddinghaus, A.; Mei, H.; Rose, T.; Enghard, P.; Crützkau, A. Deep phenotyping of urinary leukocytes by mass cytometry reveals a leukocyte signature for early and non-invasive prediction of response to treatment in active lupus nephritis. Front. Immunol. 2020, 11, 256. [Google Scholar] [CrossRef] [PubMed]
- Fava, A.; Rao, D.A.; Mohan, C.; Zhang, T.; Rosenberg, A.; Fenaroli, P.; Belmont, H.M.; Izmirly, P.; Clancy, R.; Trujillo, J.M.; et al. Urine proteomics and renal single-cell transcriptomics implicate interkeukine-16 in lupus nephritis. Arthritis Rheumatol. 2022, 74, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Burbano, C.; Gómez-Puerta, J.A.; Muñoz-Vahos, C.; Vanegas-García, A.; Rajas, M.; Vásquez, G.; Castaño, D. HMGB1+ microparticles present in urine are hallmarks of nephritis in patients with systemic lupus erythematosus. Eur. J. Immunol. 2019, 49, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Hu, Z.B.; Chen, P.P.; Lu, C.C.; Zhang, J.X.; Li, X.Q.; Yuan, B.Y.; Huang, S.J.; Ma, K.L. Urinary podocyte microparticles are associated with disease activity and renal injury in systemic lupus erythematosus. BMC Nephrol. 2019, 20, 303. [Google Scholar] [CrossRef] [Green Version]
- Gudehithlu, K.P.; Hart, P.; Joshi, A.; Garcia-Gomez, I.; Cimbaluk, D.J.; Dunea, G.; Arruda, J.A.L.; Singh, A.K. Urine exosomal ceruloplasmin: A potential early biomarker of underlying kidney disease. Clin. Exp. Nephrol. 2019, 23, 1013–1021. [Google Scholar] [CrossRef]
- Solé, C.; Cortés-Hernández, J.; Felip, M.L.; Vidal, M.; Ordi-Ros, J. miR-29c in urinary exosomes as predictor of early renal fibrosis in lupus nephritis. Nephrol. Dial. Transplant. 2015, 30, 1488–1496. [Google Scholar] [CrossRef] [Green Version]
- Solé, C.; Moliné, T.; Vidal, M.; Ordi-Ros, J.; Cortés-Hernández, J. An exosomal urinary miRNA signature for early diagnosis of renal fibrosis in lupus nephritis. Cells 2019, 8, 773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tangtanatakul, P.; Klinchanhom, S.; Sodsai, P.; Sutichet, T.; Promjeen, C.; Avihingsanon, Y.; Hirankarn, N. Down-regulation of let-7a and miR-21 in urine exosomes from lupus nephritis patients during disease flare. Asian Pac. J. Allergy Immunol. 2019, 37, 189–197. [Google Scholar]
- Garcia-Vives, E.; Solé, C.; Moliné, T.; Vidal, M.; Agraz, I.; Ordi-Ros, J.; Cortés-Hernández, J. The urinary exosomal miRNA expression profile is predictive of clinical response in lupus nephritis. Int. J. Mol. Sci. 2020, 21, 1372. [Google Scholar] [CrossRef] [Green Version]
- Perez-Hernandez, J.; Martinez-Arroyo, O.; Ortega, A.; Galera, M.; Solis-Salguero, M.A.; Chaves, F.J.; Redon, J.; Forner, M.J.; Cortes, R. Urinary exosomal miR-146a as a marker of albuminuria, activity changes and disease flares in lupus nephritis. J. Nephrol. 2021, 34, 1157–1167. [Google Scholar] [CrossRef]
- Cheng, C.; Guo, F.; Yang, H.; Ma, J.; Li, H.; Yin, L.; Li, M.; Liu, S. Identification and analysis of the predictive urinary exosomal miR-195-5p in lupus nephritis based on renal miRNA-mRNA co-expression network. Lupus 2022, 31, 1786–1799. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, X.; Meng, K.; Sun, Y.; Shu, R.; Han, Y.; Feng, Q.; Li, Z.; Yang, P.; Liang, J. Urinary exosome tsRNAs as novel markers for diagnosis and prediction of lupus nephritis. Front. Immunol. 2023, 14, 1077645. [Google Scholar] [CrossRef]
- Aragón, C.C.; Tafúr, R.-A.; Suárez-Avellaneda, A.; Martínez, M.T.; de las Salas, A.; Tobón, G.J. Urinary biomarkers in lupus nephritis. J. Transl. Autoimmun. 2020, 3, 100042. [Google Scholar] [CrossRef]
- Morell, M.; Pérez-Cózar, F.; Maraňón, C. Immune-related urine biomarkers for the diagnosis of lupus nephritis. Int. J. Mol. Sci. 2021, 22, 7143. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.Y.H.; Mok, C.C. Novel and emerging treatment strategies for lupus nephritis. Expert Rev. Clin. Pharmacol. 2022, 15, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Parodis, I.; Gomez, A.; Lindblom, J.; Chow, J.W.; Sjöwall, C.; Sciascia, S.; Gatto, M. B cell kinetics upon therapy commencement for active extrarenal systemic lupus erythematosus in relation to development of renal flares: Results from three phase III clinical trials of belimumab. Int. J. Mol. Sci. 2022, 23, 13941. [Google Scholar] [CrossRef] [PubMed]
- Mejia-Vilet, J.M.; Malvar, A.; Arazi, A.; Rovin, B.H. The lupus nephritis management renaissance. Kidney Int. 2022, 101, 242–255. [Google Scholar] [CrossRef]
- Furuichi, K.; Wada, T.; Iwata, Y.; Sakai, N.; Yoshimoto, K.; Shimizu, M.; Kobayashi, K.; Takasawa, K.; Kida, H.; Takeda, S.; et al. Upregulation of fractalkine in human crescentic glomerulonephritis. Nephron 2001, 87, 314–320. [Google Scholar] [CrossRef]
- Segerer, S.; Hughes, E.; Hudkins, K.L.; Mack, M.; Goodpaster, T.; Alpers, C.E. Expression of the fractalkine receptor (CX3CR1) in human kidney diseases. Kidney Int. 2002, 62, 488–495. [Google Scholar] [CrossRef] [Green Version]
- Bazan, J.F.; Bacon, K.B.; Hardiman, G.; Wang, W.; Soo, K.; Rossi, D.; Greaves, D.R.; Zlotnik, A.; Schall, T.J. A new class of membrane-bound chemokine with a CX3C motif. Nature 1997, 385, 640–644. [Google Scholar] [CrossRef]
- Inoue, A.; Hasegawa, H.; Kohno, M.; Ito, M.R.; Terada, M.; Imai, T.; Yoshie, O.; Nose, M.; Fujita, S. Antagonist of fractalkine (CX3CL1) delays the initiation and ameliorates the progression of lupus nephritis in MRL/lpr mice. Arthritis Rheum. 2005, 52, 1522–1533. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef]
- Su, B.; Ye, H.; You, X.; Ni, H.; Chen, X.; Li, L. Icariin alleviates murine lupus nephritis via inhibiting NF-κB activation pathway and NLRP3 inflammasome. Life Sci. 2018, 208, 26–32. [Google Scholar] [CrossRef]
- Wu, D.; Ai, L.; Sun, Y.; Yang, B.; Chen, S.; Wang, Q.; Kuang, H. Role of NLPR3 inflammasome in lupus nephritis and therapeutic targeting by phytochemicals. Front. Pharmacol. 2021, 12, 621300. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, S.; Wu, P.; Ren, Q.; Wei, P.; Hong, M.; Feng, Y.; Wong, C.K.; Tang, H.; Zeng, H. A novel potential target of IL-35-regulated JAK/STAT signaling pathway in lupus nephritis. Clin. Transl. Med. 2021, 11, e309. [Google Scholar] [CrossRef]
- Shi, D.; Li, Y.; Shi, X.; Yao, M.; Wu, D.; Zheng, Y.; Lin, Q.; Yang, Y. Transcriptional expression of CXCL10 and STAT1 in lupus nephritis and the intervention effect of triptolide. Clin. Rheumatol. 2023, 42, 539–548. [Google Scholar] [CrossRef]
- Liang, D.; Mai, H.; Ruan, F.; Fu, H. The efficacy of triptolide in preventing diabetic kidney diseases: A systematic review and meta-analysis. Front. Pharmacol. 2021, 12, 728758. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Guo, S.; Niu, F.; Liu, D.; Zhuang, Y. Complement 1q protects MRL/lpr mice against lupus nephritis via inhibiting the nuclear factor-kB pathway. Mol. Med. Rep. 2020, 22, 5436–5443. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Deng, Y.; Shang, S.; Tang, L.; Li, Q.; Bai, X.; Chen, X. Complement factor B inhibitor LNP023 improves lupus nephritis in MRL/lpr mice. Biomed. Pharmacother. 2022, 153, 113433. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Gao, C.; Chen, Y.; Feng, Y.; Liu, W.J.; Liu, H.-F. Update on the role of autophagy in systemic lupus erythematosus: A novel therapeutic target. Biomed. Pharmacother. 2015, 71, 190–193. [Google Scholar] [CrossRef]
- Hartleben, B.; Gödel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Köbler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Gros, F.; Muller, S. Pharmacological regulators of autophagy and their link with modulators of lupus disease. Br. J. Pharmacol. 2014, 171, 4337–4359. [Google Scholar] [CrossRef] [Green Version]
- Stylianou, K.; Petrakis, I.; Mavroeidi, V.; Stratakis, S.; Vardaki, E.; Perakis, K.; Stratigis, S.; Passam, A.; Papadogiorgaki, E.; Giannakakis, K.; et al. The PI3K/Akt/mTOR pathway is activated in murine lupus nephritis and downregulated by rapamycin. Nephrol. Dial. Transplant. 2011, 26, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.-Y.; Zhou, X.-J.; Cheng, F.-J.; Hou, P.; Ren, Y.-L.; Wang, S.-X.; Zhao, M.-H.; Yang, L.; Martínez, J.; Zhang, H. Increased autophagy is cytoprotective against podocyte injury induced by antibody and interferon-α in lupus nephritis. Ann. Rheum. Dis. 2018, 77, 1799–1809. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Coit, P.; Dozmorov, M.G.; Merrill, J.T.; McCune, W.J.; Maksimowicz-McKinnon, K.; Wren, J.D.; Sawalha, A.H. Epigenetic reprogramming in naive CD4+ T cells favoring cell activation and non-Th1 effector T cell immune responses as an early event in lupus flares. Arthritis Rheumatol. 2016, 68, 2200–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Kim, M.; Woo, D.-H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Iwata, S.; Hajime, M.; Ohkubo, N.; Todoroki, Y.; Miyata, H.; Ueno, M.; Hao, H.; Zhang, T.; Fan, J.; et al. Methionine commits cells to differentiate into plasmablasts through epigenetic regulation of BTB and CNC homolog 2 by the methyltransferase EZH2. Arthritis Rheumatol. 2020, 72, 1143–1153. [Google Scholar] [CrossRef]
- Wu, L.; Jiang, X.; Qi, C.; Zhang, C.; Qu, B.; Shen, N. EZH2 inhibition interferes with the activation of type I interferon signaling pathway and ameliorates lupus nephritis in NZB/NZW F1 mice. Front. Immunol. 2021, 12, 653989. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-Y.; Shen, C.-Y.; Liu, C.-W.; Hsieh, S.-C.; Liao, H.-T.; Li, K.-J.; Lu, C.-S.; Lee, H.-T.; Lin, C.-S.; Wu, C.-H.; et al. Aberrant non-coding RNA expression in patients with systemic lupus erythematosus: Consequences for immune dysfunctions and tissue damage. Biomolecules 2020, 10, 1641. [Google Scholar] [CrossRef]
- So, B.Y.F.; Yap, D.Y.H.; Chan, T.M. MicroRNAs in lupus nephritis- role in disease pathogenesis and clinical applications. Int. J. Mol. Sci. 2021, 22, 10737. [Google Scholar] [CrossRef]
- Mihaylova, G.; Vasilev, V.; Kosturkova, M.B.; Stoyanov, G.S.; Radanova, M. Long non-coding RNAs as new biomarkers in lupus nephritis: A connection between present and future. Cureus 2020, 12, e9003. [Google Scholar] [CrossRef]
- Liang, H.; Liu, Q. The role of non-coding RNA in lupus nephritis. Hum. Cell 2023, 36, 923–936. [Google Scholar] [CrossRef]
- Ahmed, R.F.; Shaker, O.G.; Abdelghany, H.M.; Abdallah, N.H.; Elsayed, S.H.; Kamel, B.A. Role of micro-RNA 132 and its long non-coding SOX2 in diagnosis of lupus nephritis. Lupus 2022, 31, 89–96. [Google Scholar] [CrossRef]
- Eitner, F.; Westerhuis, R.; Burg, M.; Weinhold, B.; Gröne, H.J.; Ostendorf, T.; Rüther, U.; Koch, K.M.; Rees, A.J.; Floege, J. Role of interleukin-6 in mediating mesangial cell proliferation and matrix production in vivo. Kidney Int. 1997, 51, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, H.-Y.; Chung, J.-W.; Kim, H.-A.; Yun, J.-M.; Jeon, J.-Y.; Ye, Y.-M.; Kim, S.-H.; Park, H.-S.; Suh, C.-H. Cytokine IL-6 and IL-10 as biomarkers in systemic lupus erythematosus. J. Clin. Immunol. 2007, 27, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, N.; Zhang, J.; Zhao, H.; Wang, X. miR-410 suppresses the expression of interleukin-6 as well as renal fibrosis in the pathogenesis of lupus nephritis. Clin. Exp. Pharmacol. Physiol. 2016, 43, 616–625. [Google Scholar] [CrossRef]
- Jacob, N.; Yang, H.; Pricop, L.; Liu, Y.; Gao, X.; Zheng, S.G.; Wang, J.; Gao, H.-X.; Putterman, C.; Koss, M.N.; et al. Accelerated pathological and clinical nephritis in system lupus erythematosus-prone New Zealand mixed 2328 mice doubly deficient in TNF receptor 1 and TNF receptor 2 via a Th17-associated pathway. J. Immunol. 2009, 182, 2532–2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Peng, W.; Ouyang, X.; Li, W.; Dai, Y. Circulating microRNAs as candidate biomarkers in patients with systemic lupus erythematosus. Transl. Res. 2012, 160, 198–206. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Deng, Y. miR-125a-3p decreases levels of interleukin-17 and suppresses renal fibrosis via down-regulating TGF-β1 in systemic lupus erythematosus mediated lupus nephritic mice. Am. J. Transl. Res. 2019, 11, 1843–1853. [Google Scholar]
- Rönnblom, L.; Leonard, D. Interferon pathway in SLE: One key to unlocking the mystery of the disease. Lupus Sci. Med. 2019, 6, e000270. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Han, X.; Jiang, X.; Ding, H.; Qi, C.; Yin, Z.; Xiao, J.; Xiong, L.; Guo, Q.; Ye, Z.; et al. Downregulation of renal Hsa-miR-127-3p contributes to the overactivation of type I interferon signaling pathway in the kidney of lupus nephritis. Front. Immunol. 2021, 12, 747616. [Google Scholar] [CrossRef]
- Menke, J.; Amann, K.; Cavagna, L.; Blettner, M.; Weinmann, A.; Schwarting, A.; Kelley, V.R. Colony-stimulating factor-1: A potential biomarker for lupus nephritis. J. Am. Soc. Nephrol. 2015, 26, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, R.D.; Florquin, S.; Singer, G.G.; Brennan, D.C.; Kelley, V.R. Colony stimulating factor-1 in the induction of lupus nephritis. Kidney Int. 1993, 43, 1000–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; He, X.-J.; Zhang, W.; Chen, Y.-L.; Yang, J.; Xiang, W.; Ding, Y. MiR-145 participates in the development of lupus nephritis by targeting CSF1 to regulate the JAK/STAT signaling pathway. Cytokine 2022, 154, 155877. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, F.; Ma, J.; Zhang, X.; Wu, L.; Qu, B.; Xia, S.; Chen, S.; Tang, Y.; Shen, N. Association of large intergenic non-coding RNA expression with disease activity and organ damage in systemic lupus erythematosus. Arthritis Res. Ther. 2015, 17, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Z.; Ye, Z.; Xue, Z.; Wu, L.; Ouyang, Y.; Yao, C.; Cui, C.; Xu, N.; Ma, J.; Hu, G.; et al. Identification of renal long non-coding RNA RP11-2B6.2 as a positive regulator of type I interferon signaling pathway in lupus nephritis. Front. Immunol. 2019, 10, 975. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Tang, X.; Wang, S. Roles of circRNAs in autoimmune diseases. Front. Immunol. 2019, 10, 639. [Google Scholar] [CrossRef]
- Xu, Z.-Q.; Ding, Y.; Huang, X.-Y.; Xiang, W.; He, X.-J. CircELK4 contributes to lupus nephritis by acting as a miR-27b-3p sponge to regulate STING/IRF3/IFN-I signaling. Inflammation 2021, 44, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Hasni, S.A.; Perez, P.; Tandon, M.; Jang, S.-I.; Zheng, C.; Kopp, J.B.; Austin, H., 3rd; Balow, J.E.; Alevizos, I.; et al. miR-150 promotes renal fibrosis in lupus nephritis by downregulating SOCS1. J. Am. Soc. Nephrol. 2013, 24, 1073–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Wang, C.; Yuan, Y.; Zhu, Y.; Yin, Z.; Xia, Z.; Zhang, C. Differentially expressed microRNAs in kidney biopsies from various subtypes of nephrotic children. Exp. Mol. Pathol. 2015, 99, 590–595. [Google Scholar] [CrossRef]
- Van Craenenbroeck, A.H.; Van Craenenbroeck, E.M.; Van Ackeren, K.; Hoymans, V.Y.; Verpooten, G.A.; Vrints, C.J. Impaired vascular function contributes to exercise intolerance in chronic kidney disease. Nephrol. Dial. Transplant. 2016, 31, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Fu, B.; Chen, D.; Hong, Q.; Cui, J.; Li, J.; Bai, X.; Chen, X. miR-184 and miR-150 promote renal glomerular mesangial cell aging by targeting Rab1a and Rab31. Exp. Cell Res. 2015, 336, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Fu, J.; Chen, C.; Jiao, C.; Kong, W.; Zhang, Y.; Chang, Q.; Wang, Y.; Li, D.; Illei, G.G.; et al. LNA-anti-miR-150 ameliorated kidney injury of lupus nephritis by inhibiting renal fibrosis and macrophage infiltration. Arthritis Res. Ther. 2019, 21, 276. [Google Scholar] [CrossRef] [Green Version]
- Hao, X.; Luan, J.; Jiao, C.; Ma, C.; Feng, Z.; Zhu, L.; Zhang, Y.; Fu, J.; Lai, E.; Zhang, B.; et al. LNA-anti-miR-150 alleviates renal interstitial fibrosis by reducing pro-inflammatory M1/M2 macrophage polarization. Front. Immunol. 2022, 13, 913007. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cross-Reactive with Different Molecules: |
|---|
| • Chromatin substances [88,89,90]: |
| • dsDNA, ssDNA, Z-form DNA, bent or elongated dsDNA |
| • DNA-RNA hybrids, peptide-nucleic acid hybrids, locked nucleic acid |
| • Extracellular matrix components [74,75,76,77,78]: |
| • α-actinin |
| • Annexin II |
| • Laminin |
| • Heparan sulfate proteoglycan |
| • Collagen III and IV |
| • C1q |
| • Ribosomal P |
| • N-methyl-D-aspartate receptor |
| • Others |
| • Cross-reactive with resident renal cells [79,80,81,82,83,84,85,86,87]: |
| • Glomerular mesangial cells |
| • Podocytes |
| • Vascular endothelial cells |
| • Proximal tubular epithelial cells |
|
|
|
|
|
|
| Co-Existence with Anti-dsDNA Antibodies | Clinical Significance |
|---|---|
|
|
| |
| |
|
|
| |
| |
| |
|
|
| Potential Biomarkers from
Renal Resident Cells | Effect on Infiltrated Immune Cells
in the Kidney |
|---|---|
|
|
|
|
|
|
|
|
|
|
|
| I | Targeting JAK/STAT Signaling Pathways |
| • IL-35-related JAK/STAT signaling [166] | |
| • IFN-γ-induced JAK/STAT signaling and CXCL10 expression [167] | |
| II | Modulation of complement levels [169] or complement factor B level [170] |
| III | Targeting proinflammatory cytokines or growth factors by microRNAs |
| • Suppression of IL-6 expression by miR-410 [189] | |
| • Suppression of IL-17 and TGF-β expression by miR-125a-3p [192] | |
| • Suppression of IFN-I and JAK1 signaling by hsa-miR-127-3p [194] | |
| • Suppression of CSF-1 expression by miR-145 [195,196,197] | |
| • Decreased M1/M2 polarization (ratio) by locked nucleic acid—anti-miR-150, via SOCS1/JAK1/STAT1 signaling [206,207] | |
| IV | Application of lncRNA and circRNA in the treatment of LN |
| • Suppression of IFN-induced signaling by knockdown of lncRNA RP11-2B6.2 [199] | |
| • Suppression of miR-27b-3p/STING/IRF3/IFN-I signaling by circELK4 [200] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, C.-Y.; Li, K.-J.; Shen, C.-Y.; Lu, C.-H.; Lee, H.-T.; Wu, T.-H.; Ng, Y.-Y.; Tsao, Y.-P.; Hsieh, S.-C.; Yu, C.-L. Decipher the Immunopathological Mechanisms and Set Up Potential Therapeutic Strategies for Patients with Lupus Nephritis. Int. J. Mol. Sci. 2023, 24, 10066. https://doi.org/10.3390/ijms241210066
Tsai C-Y, Li K-J, Shen C-Y, Lu C-H, Lee H-T, Wu T-H, Ng Y-Y, Tsao Y-P, Hsieh S-C, Yu C-L. Decipher the Immunopathological Mechanisms and Set Up Potential Therapeutic Strategies for Patients with Lupus Nephritis. International Journal of Molecular Sciences. 2023; 24(12):10066. https://doi.org/10.3390/ijms241210066
Chicago/Turabian StyleTsai, Chang-Youh, Ko-Jen Li, Chieh-Yu Shen, Cheng-Hsun Lu, Hui-Ting Lee, Tsai-Hung Wu, Yee-Yung Ng, Yen-Po Tsao, Song-Chou Hsieh, and Chia-Li Yu. 2023. "Decipher the Immunopathological Mechanisms and Set Up Potential Therapeutic Strategies for Patients with Lupus Nephritis" International Journal of Molecular Sciences 24, no. 12: 10066. https://doi.org/10.3390/ijms241210066
APA StyleTsai, C. -Y., Li, K. -J., Shen, C. -Y., Lu, C. -H., Lee, H. -T., Wu, T. -H., Ng, Y. -Y., Tsao, Y. -P., Hsieh, S. -C., & Yu, C. -L. (2023). Decipher the Immunopathological Mechanisms and Set Up Potential Therapeutic Strategies for Patients with Lupus Nephritis. International Journal of Molecular Sciences, 24(12), 10066. https://doi.org/10.3390/ijms241210066