
Biochemical Recurrence in Prostate Cancer Is Associated with the Composition of Lactobacillus: Microbiome Analysis of Prostatic Tissue

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Patient Profiles
2.2. Sample Analysis and Integrity of the Sequenced Data
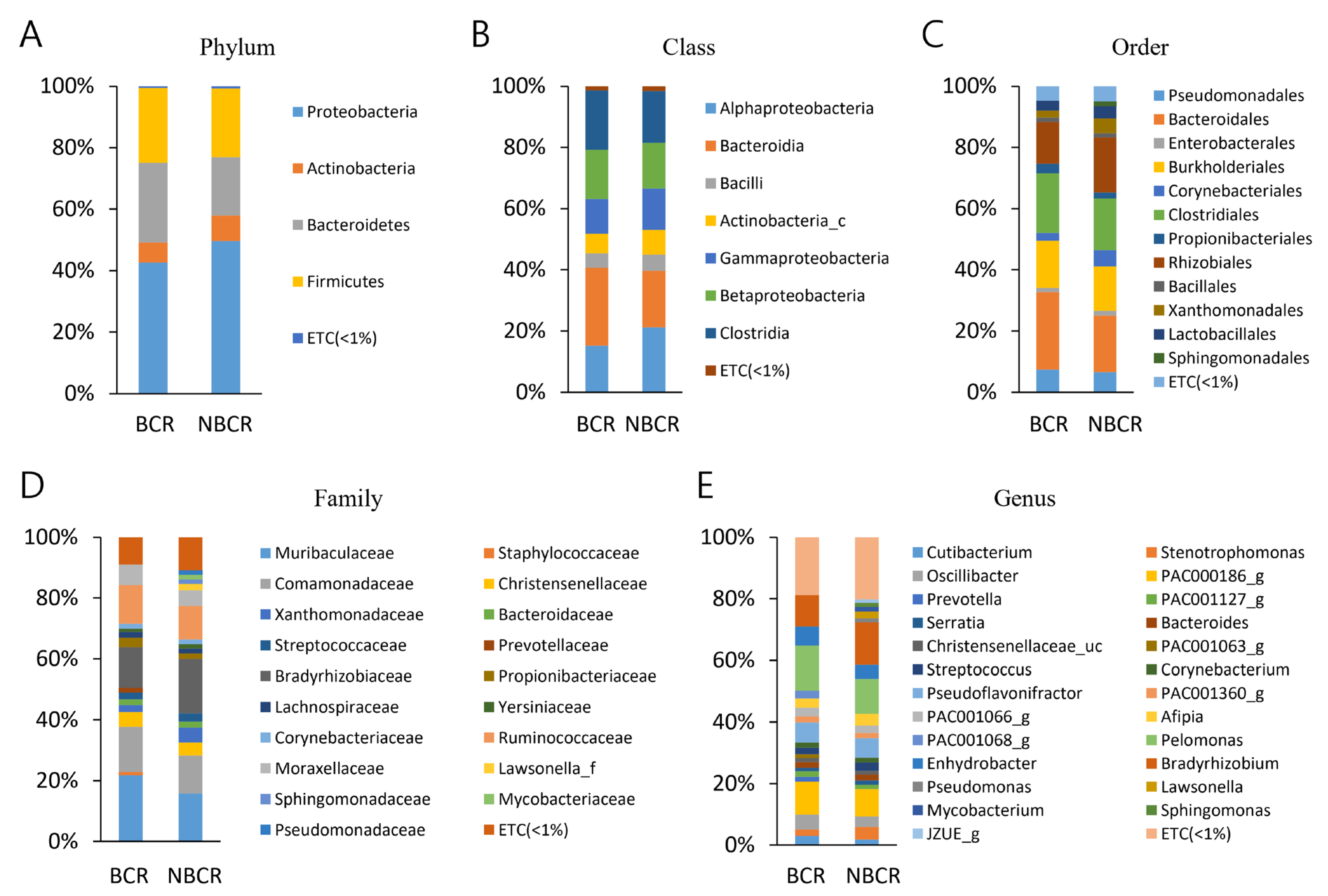
2.3. Bacterial Taxa (BCR vs. NBCR)
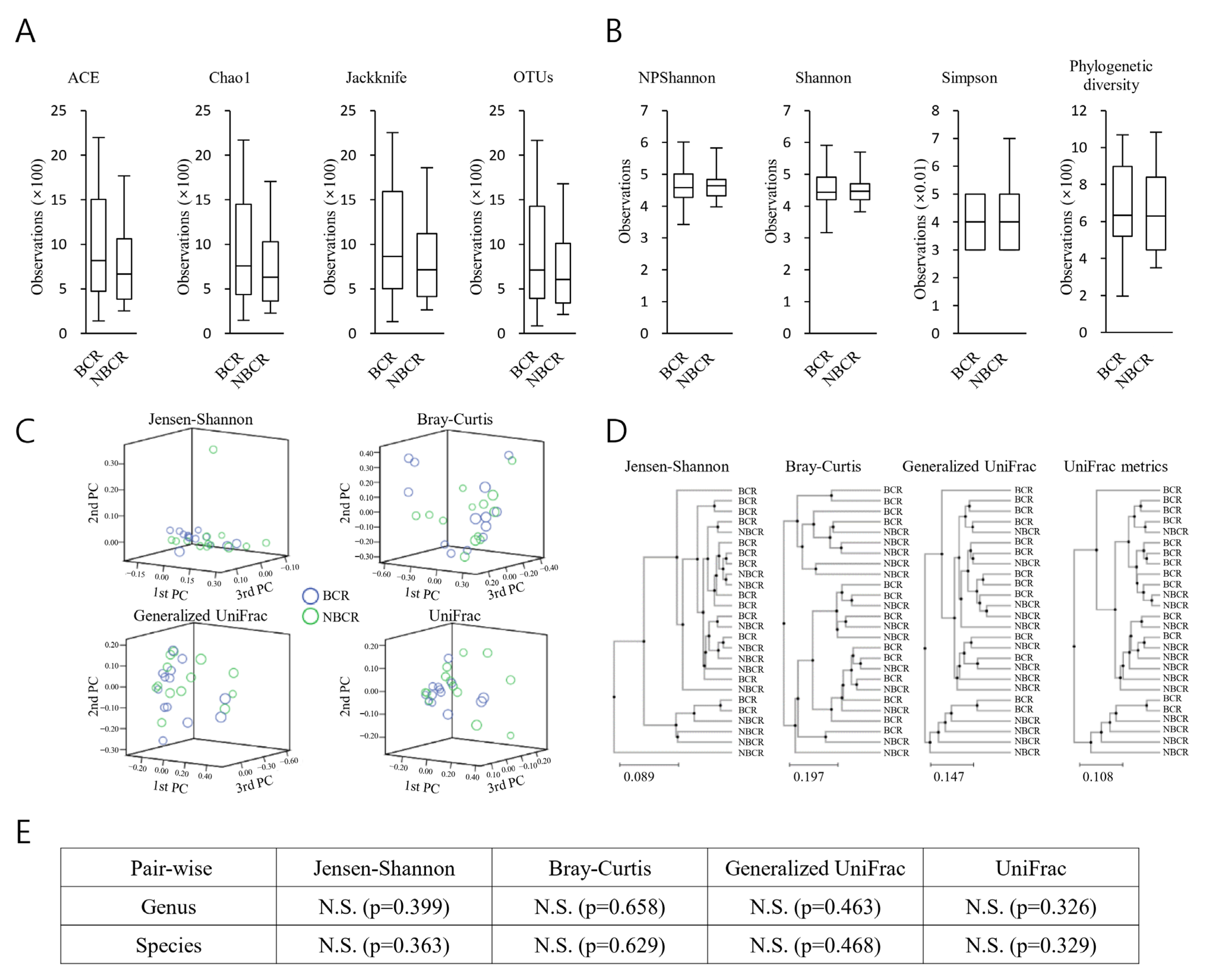
2.4. Richness and Diversity (BCR vs. NBCR)
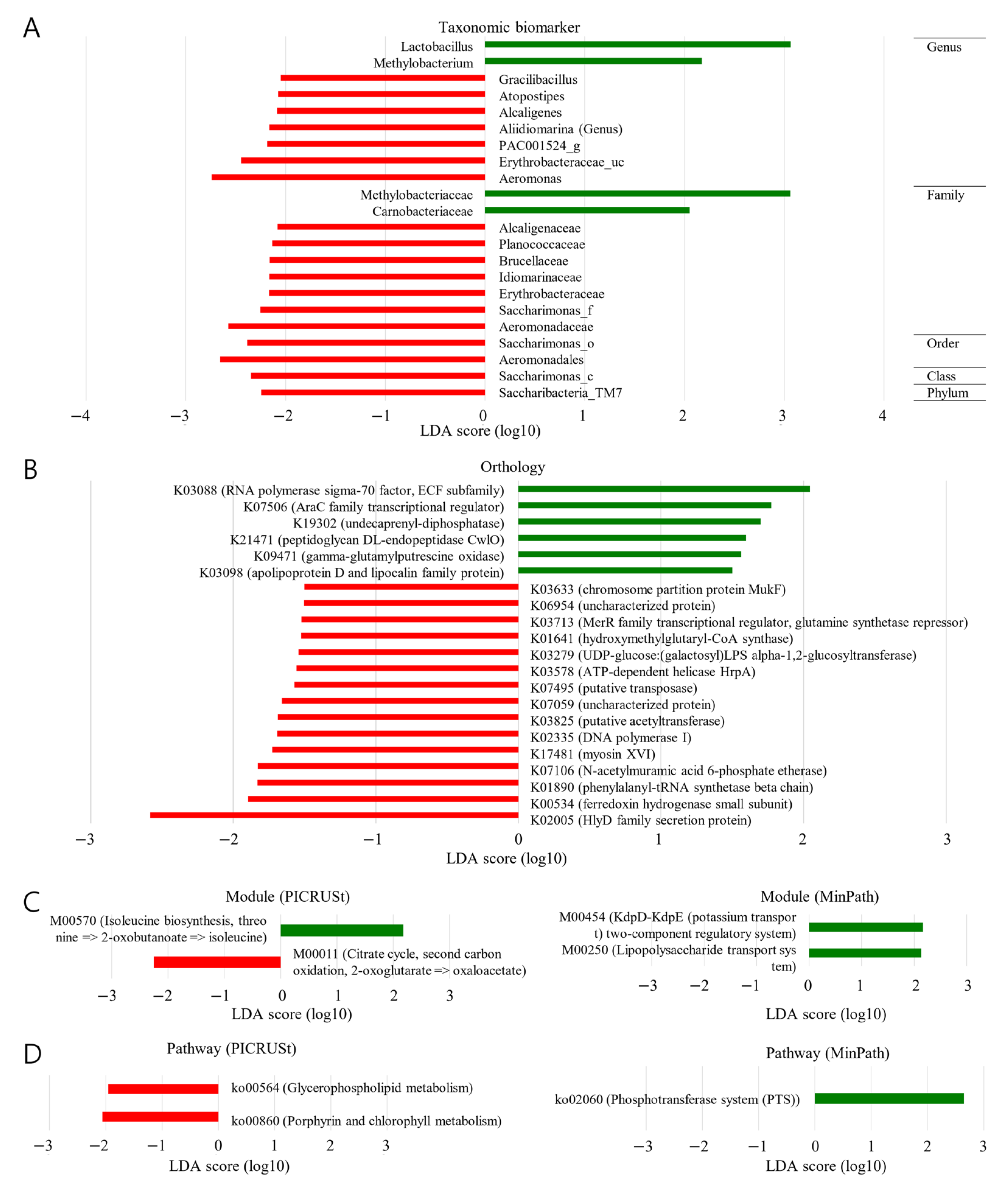
2.5. Taxonomical Biomarker (BCR vs. NBCR)
2.6. Functional Biomarker (BCR vs. NBCR)
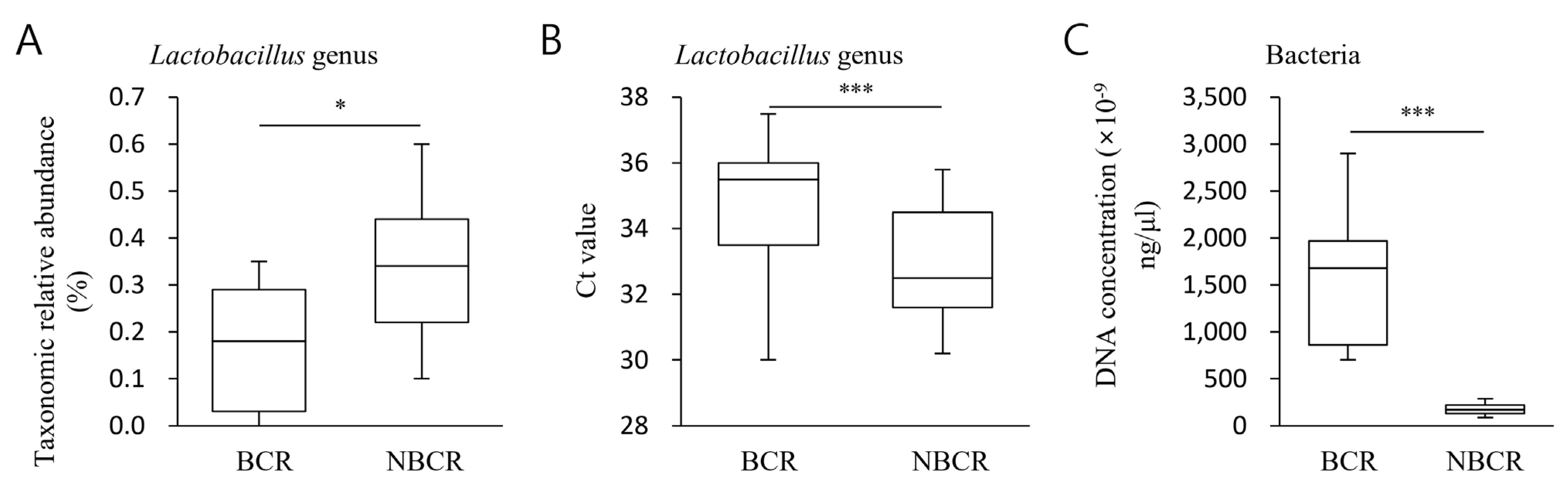
2.7. Quantitative Evaluation of Lactobacillus Abundance (BCR vs. NBCR)
3. Discussion
4. Materials and Methods
4.1. Subject Recruitment and Sample Collection
4.2. DNA Extraction
4.3. Illumina Sequencing and Bioinformatics Analysis of 16S rRNA Gene Amplicons
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kudo, Y.; Tada, H.; Fujiwara, N.; Tada, Y.; Tsunematsu, T.; Miyake, Y.; Ishimaru, N.J.G. Environment. Oral Environ. Cancer 2016, 38, 1–6. [Google Scholar]
- Perdigon, G.; Alvarez, S.; Rachid, M.; Agüero, G.; Gobbato, N. Immune System Stimulation by Probiotics. J. Dairy Sci. 1995, 78, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Buermans, H.P.J.; den Dunnen, J.T. Next generation sequencing technology: Advances and applications. BBA-Mol. Basis Dis. 2014, 1842, 1932–1941. [Google Scholar] [CrossRef] [Green Version]
- Ha, Y.S.; Kim, S.Y.; Chung, J.I.; Choi, H.; Kim, J.H.; Yu, H.S.; Cho, I.C.; Kim, H.J.; Chung, H.C.; Koh, J.S.; et al. Trends in End-of-Life Resource Utilization and Costs among Prostate Cancer Patients from 2006 to 2015: A Nationwide Population-Based Study. World J. Mens Health 2021, 39, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Braga-Basaria, M.; Dobs, A.S.; Muller, D.C.; Carducci, M.A.; John, M.; Egan, J.; Basaria, S. Metabolic syndrome in men with prostate cancer undergoing long-term androgen-deprivation therapy. J. Clin. Oncol. 2006, 24, 3979–3983. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Kim, J.H. Role of microbiome and its metabolite, short chain fatty acid in prostate cancer. Investig. Clin. Urol. 2023, 64, 3–12. [Google Scholar] [CrossRef]
- Miyake, M.; Tatsumi, Y.; Ohnishi, K.; Fujii, T.; Nakai, Y.; Tanaka, N.; Fujimoto, K. Prostate diseases and microbiome in the prostate, gut, and urine. Prostate Int. 2022, 10, 96–107. [Google Scholar] [CrossRef]
- Peisch, S.F.; Van Blarigan, E.L.; Chan, J.M.; Stampfer, M.J.; Kenfield, S.A. Prostate cancer progression and mortality: A review of diet and lifestyle factors. World J. Urol. 2017, 35, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Hullar, M.A.J.; Burnett-Hartman, A.N.; Lampe, J.W. Gut Microbes, Diet, and Cancer. In Advances in Nutrition and Cancer; Springer: Berlin/Heidelberg, Germany, 2014; pp. 377–399. [Google Scholar]
- Holmes, E.; Li, J.V.; Athanasiou, T.; Ashrafian, H.; Nicholson, J.K. Understanding the role of gut microbiome–host metabolic signal disruption in health and disease. Trends Microbiol. 2011, 19, 349–359. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Giorgetti, G.; Brandimarte, G.; Fabiocchi, F.; Ricci, S.; Flamini, P.; Sandri, G.; Trotta, M.C.; Elisei, W.; Penna, A.; Lecca, P.G.; et al. Interactions between Innate Immunity, Microbiota, and Probiotics. J. Immunol. Res. 2015, 2015, 501361. [Google Scholar] [CrossRef]
- Shinohara, D.B.; Vaghasia, A.M.; Yu, S.H.; Mak, T.N.; Bruggemann, H.; Nelson, W.G.; De Marzo, A.M.; Yegnasubramanian, S.; Sfanos, K.S. A mouse model of chronic prostatic inflammation using a human prostate cancer-derived isolate of Propionibacterium acnes. Prostate 2013, 73, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- Cohen, R.J.; Shannon, B.A.; McNeal, J.E.; Shannon, T.O.M.; Garrett, K.L. Propionibacterium Acnes Associated with Inflammation in Radical Prostatectomy Specimens: A Possible Link to Cancer Evolution? J. Urol. 2005, 173, 1969–1974. [Google Scholar] [CrossRef] [Green Version]
- Alfano, M.; Canducci, F.; Nebuloni, M.; Clementi, M.; Montorsi, F.; Salonia, A. The interplay of extracellular matrix and microbiome in urothelial bladder cancer. Nat. Rev. Urol. 2016, 13, 77–90. [Google Scholar] [CrossRef]
- Caini, S.; Gandini, S.; Dudas, M.; Bremer, V.; Severi, E.; Gherasim, A. Sexually transmitted infections and prostate cancer risk: A systematic review and meta-analysis. Cancer Epidemiol. 2014, 38, 329–338. [Google Scholar] [CrossRef]
- Yoon, B.I.; Kim, S.; Han, D.-S.; Ha, U.S.; Lee, S.-J.; Kim, H.W.; Han, C.-H.; Cho, Y.-H. Acute bacterial prostatitis: How to prevent and manage chronic infection? J. Infect. Chemother. 2012, 18, 444–450. [Google Scholar] [CrossRef]
- Cox, A.J.; West, N.P.; Cripps, A.W. Obesity, inflammation, and the gut microbiota. Lancet Diabetes Endocrinol. 2015, 3, 207–215. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Viaud, S.; Vétizou, M.; Daillère, R.; Merad, M.; Kroemer, G. Cancer and the gut microbiota: An unexpected link. Sci. Transl. Med. 2015, 7, 271ps271. [Google Scholar] [CrossRef] [Green Version]
- Mandar, R. Microbiota of male genital tract: Impact on the health of man and his partner. Pharmacol. Res. 2013, 69, 32–41. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Sauvageot, J.; Fedor, H.L.; Dick, J.D.; De Marzo, A.M.; Isaacs, W.B. A molecular analysis of prokaryotic and viral DNA sequences in prostate tissue from patients with prostate cancer indicates the presence of multiple and diverse microorganisms. Prostate 2008, 68, 306–320. [Google Scholar] [CrossRef]
- Khatoon, J.; Rai, R.P.; Prasad, K.N. Role of Helicobacter pylori in gastric cancer: Updates. World J. Gastrointest. Oncol. 2016, 8, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, E609–E616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brierley, J.D.; Gospodarowicz, M.K.; Wittekind, C. TNM Classification of Malignant Tumours; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Cavarretta, I.; Ferrarese, R.; Cazzaniga, W.; Saita, D.; Luciano, R.; Ceresola, E.R.; Locatelli, I.; Visconti, L.; Lavorgna, G.; Briganti, A.; et al. The Microbiome of the Prostate Tumor Microenvironment. Eur. Urol. 2017, 72, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Dallal, M.M.S.; Mojarrad, M.; Baghbani, F.; Raoofian, R.; Mardaneh, J.; Salehipour, Z. Effects of Probiotic Lactobacillus acidophilus and Lactobacillus casei on Colorectal Tumor Cells Activity (CaCo-2). Arch. Iran. Med. 2015, 18, 167–172. [Google Scholar]
- Zhuo, Q.; Yu, B.H.; Zhou, J.; Zhang, J.Y.; Zhang, R.L.; Xie, J.Y.; Wang, Q.L.; Zhao, S.L. Lysates of Lactobacillus acidophilus combined with CTLA-4-blocking antibodies enhance antitumor immunity in a mouse colon cancer model. Sci. Rep. 2019, 9, 20128. [Google Scholar] [CrossRef] [Green Version]
- Salva, S.; Marranzino, G.; Villena, J.; Aguero, G.; Alvarez, S. Probiotic Lactobacillus strains protect against myelosuppression and immunosuppression in cyclophosphamide-treated mice. Int. Immunopharmacol. 2014, 22, 209–221. [Google Scholar] [CrossRef]
- Shin, R.; Itoh, Y.; Kataoka, M.; Iino-Miura, S.; Miura, R.; Mizutani, T.; Fujisawa, T. Anti-tumor activity of heat-killed Lactobacillus plantarum BF-LP284 on Meth-A tumor cells in BALB/c mice. Int. J. Food Sci. Nutr. 2016, 67, 641–649. [Google Scholar] [CrossRef]
- Dai, Z.W.; Coker, O.O.; Nakatsu, G.; Wu, W.K.K.; Zhao, L.Y.; Chen, Z.G.; Chan, F.K.L.; Kristiansen, K.; Sung, J.J.Y.; Wong, S.H.; et al. Multi-cohort analysis of colorectal cancer metagenome identified altered bacteria across populations and universal bacterial markers. Microbiome 2018, 6, 70. [Google Scholar] [CrossRef] [Green Version]
- Sugimura, N.; Li, Q.; Chu, E.S.H.; Lau, H.C.H.; Fong, W.; Liu, W.X.; Liang, C.; Nakatsu, G.; Su, A.C.Y.; Coker, O.O.; et al. Lactobacillus gallinarum modulates the gut microbiota and produces anti-cancer metabolites to protect against colorectal tumourigenesis. Gut 2022, 71, 2011–2021. [Google Scholar] [CrossRef]
- Komiyama, S.; Yamada, T.; Takemura, N.; Kokudo, N.; Hase, K.; Kawamura, Y.I. Profiling of tumour-associated microbiota in human hepatocellular carcinoma. Sci. Rep. 2021, 11, 10589. [Google Scholar] [CrossRef]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]
- Giralt, J.; Regadera, J.P.; Verges, R.; Romero, J.; de la Fuente, I.; Biete, A.; Villoria, J.; Cobo, J.M.; Guarner, F. Effects of probiotic Lactobacillus casei DN-114 001 in prevention of radiation-induced diarrhea: Results from multicenter, randomized, placebo-controlled nutritional trial. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 1213–1219. [Google Scholar] [CrossRef]
- Singh, N.K.; Beckett, J.M.; Kalpurath, K.; Ishaq, M.; Ahmad, T.; Eri, R.D. Synbiotics as Supplemental Therapy for the Alleviation of Chemotherapy-Associated Symptoms in Patients with Solid Tumours. Nutrients 2023, 15, 1759. [Google Scholar] [CrossRef]
- Chandel, D.; Sharma, M.; Chawla, V.; Sachdeva, N.; Shukla, G. Isolation, characterization and identification of antigenotoxic and anticancerous indigenous probiotics and their prophylactic potential in experimental colon carcinogenesis. Sci. Rep. 2019, 9, 14769. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.N.; Altemus, J.; Niazi, F.; Green, H.; Calhoun, B.C.; Sturgis, C.; Grobmyer, S.R.; Eng, C. Breast tissue, oral and urinary microbiomes in breast cancer. Oncotarget 2017, 8, 88122–88138. [Google Scholar] [CrossRef] [Green Version]
- Samkari, A.A.; Alsulami, M.; Bataweel, L.; Altaifi, R.; Altaifi, A.; Saleem, A.M.; Farsi, A.H.; Iskanderani, O.; Akeel, N.Y.; Malibary, N.H.; et al. Body Microbiota and Its Relationship With Benign and Malignant Breast Tumors: A Systematic Review. Cureus J. Med. Sci. 2022, 14, e25473. [Google Scholar] [CrossRef]
- Xuan, C.; Shamonki, J.M.; Chung, A.; DiNome, M.L.; Chung, M.; Sieling, P.A.; Lee, D.J. Microbial Dysbiosis Is Associated with Human Breast Cancer. PLoS ONE 2014, 9, e83744. [Google Scholar] [CrossRef] [Green Version]
- Yazdi, H.R.; Movafagh, A.; Fallah, F.; Alizadeh Shargh, S.; Mansouri, N.; Heidary Pour, A.; Hashemi, M. Evaluation of Methylobacterium radiotolerance and Sphyngomonas yanoikoaie in Sentinel Lymph Nodes of Breast Cancer Cases. Asian Pac. J. Cancer Prev. 2016, 17, 279–285. [Google Scholar]
- Kim, J.H.; Seo, H.; Kim, S.; Ul-Haq, A.; Song, H.Y.; Song, Y.S. Malignant Prostate Tissue Is Associated with Different Microbiome Gene Functions. Diagnostics 2023, 13, 278. [Google Scholar] [CrossRef]
- Rickham, P.P. Human experimentation. Code of ethics of the world medical association. Declaration of Helsinki. Br. Med. J. 1964, 2, 177. [Google Scholar]
- Araújo, L.S.S.; Silva, S.Q.; Teixeira, M.C. Developing a biosurfactant to attenuate arsenic contamination in mining tailings. Heliyon 2021, 7, e06093. [Google Scholar] [CrossRef] [PubMed]
- Ul-Haq, A.; Lee, K.A.; Seo, H.; Kim, S.; Jo, S.; Ko, K.M.; Moon, S.J.; Kim, Y.S.; Choi, J.R.; Song, H.Y.; et al. Characteristic alterations of gut microbiota in uncontrolled gout. J. Microbiol. 2022, 60, 1178–1190. [Google Scholar] [CrossRef]
- Ul-Haq, A.; Seo, H.; Jo, S.; Park, H.; Kim, S.; Lee, Y.; Lee, S.; Jeong, J.H.; Song, H.Y. Characterization of Fecal Microbiomes of Osteoporotic Patients in Korea. Pol. J. Microbiol. 2022, 71, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 1091. [Google Scholar] [CrossRef]
- Bacci, G.; Bani, A.; Bazzicalupo, M.; Ceccherini, M.T.; Galardini, M.; Nannipieri, P.; Pietramellara, G.; Mengoni, A. Evaluation of the Performances of Ribosomal Database Project (RDP) Classifier for Taxonomic Assignment of 16S rRNA Metabarcoding Sequences Generated from Illumina-Solexa NGS. J. Genom. 2015, 3, 36–39. [Google Scholar] [CrossRef] [Green Version]
- Chao, A.; Lee, S.M. Estimating the Number of Classes Via Sample Coverage. J. Am. Stat. Assoc. 1992, 87, 210–217. [Google Scholar] [CrossRef]
- Chao, A.; Shen, T.J. Nonparametric estimation of Shannon's index of diversity when there are unseen species in sample. Environ. Ecol. Stat. 2003, 10, 429–443. [Google Scholar] [CrossRef]
- Wang, L.L.; Zhang, F.Y.; Dong, W.W.; Wang, C.L.; Liang, X.Y.; Suo, L.L.; Cheng, J.; Zhang, M.; Guo, X.S.; Jiang, P.H.; et al. A novel approach for the forensic diagnosis of drowning by microbiological analysis with next-generation sequencing and unweighted UniFrac-based PCoA. Int. J. Leg. Med. 2020, 134, 2149–2159. [Google Scholar] [CrossRef]
- Anderson, M.J.; Walsh, D.C.I. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecol. Monogr. 2013, 83, 557–574. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Jiang, Y.H.; Yang, Y.F.; He, Z.L.; Luo, F.; Zhou, J.Z. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Beiko, R.G.; Langille, M.G.I. Predicting the Functional Potential of the Microbiome from Marker Genes Using PICRUSt. Methods Mol. Biol. 2018, 1849, 169–177. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.H.; Seo, H.; Kim, S.; Ul-Haq, A.; Rahim, M.A.; Jo, S.; Song, H.-Y.; Song, Y.S. Biochemical Recurrence in Prostate Cancer Is Associated with the Composition of Lactobacillus: Microbiome Analysis of Prostatic Tissue. Int. J. Mol. Sci. 2023, 24, 10423. https://doi.org/10.3390/ijms241310423
Kim JH, Seo H, Kim S, Ul-Haq A, Rahim MA, Jo S, Song H-Y, Song YS. Biochemical Recurrence in Prostate Cancer Is Associated with the Composition of Lactobacillus: Microbiome Analysis of Prostatic Tissue. International Journal of Molecular Sciences. 2023; 24(13):10423. https://doi.org/10.3390/ijms241310423
Chicago/Turabian StyleKim, Jae Heon, Hoonhee Seo, Sukyung Kim, Asad Ul-Haq, Md Abdur Rahim, Sujin Jo, Ho-Yeon Song, and Yun Seob Song. 2023. "Biochemical Recurrence in Prostate Cancer Is Associated with the Composition of Lactobacillus: Microbiome Analysis of Prostatic Tissue" International Journal of Molecular Sciences 24, no. 13: 10423. https://doi.org/10.3390/ijms241310423
APA StyleKim, J. H., Seo, H., Kim, S., Ul-Haq, A., Rahim, M. A., Jo, S., Song, H. -Y., & Song, Y. S. (2023). Biochemical Recurrence in Prostate Cancer Is Associated with the Composition of Lactobacillus: Microbiome Analysis of Prostatic Tissue. International Journal of Molecular Sciences, 24(13), 10423. https://doi.org/10.3390/ijms241310423