
Firing of Replication Origins Is Disturbed by a CDK4/6 Inhibitor in a pRb-Independent Manner

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Palbociclib Affects Cell Viability of pRb-Deficient Cell Lines
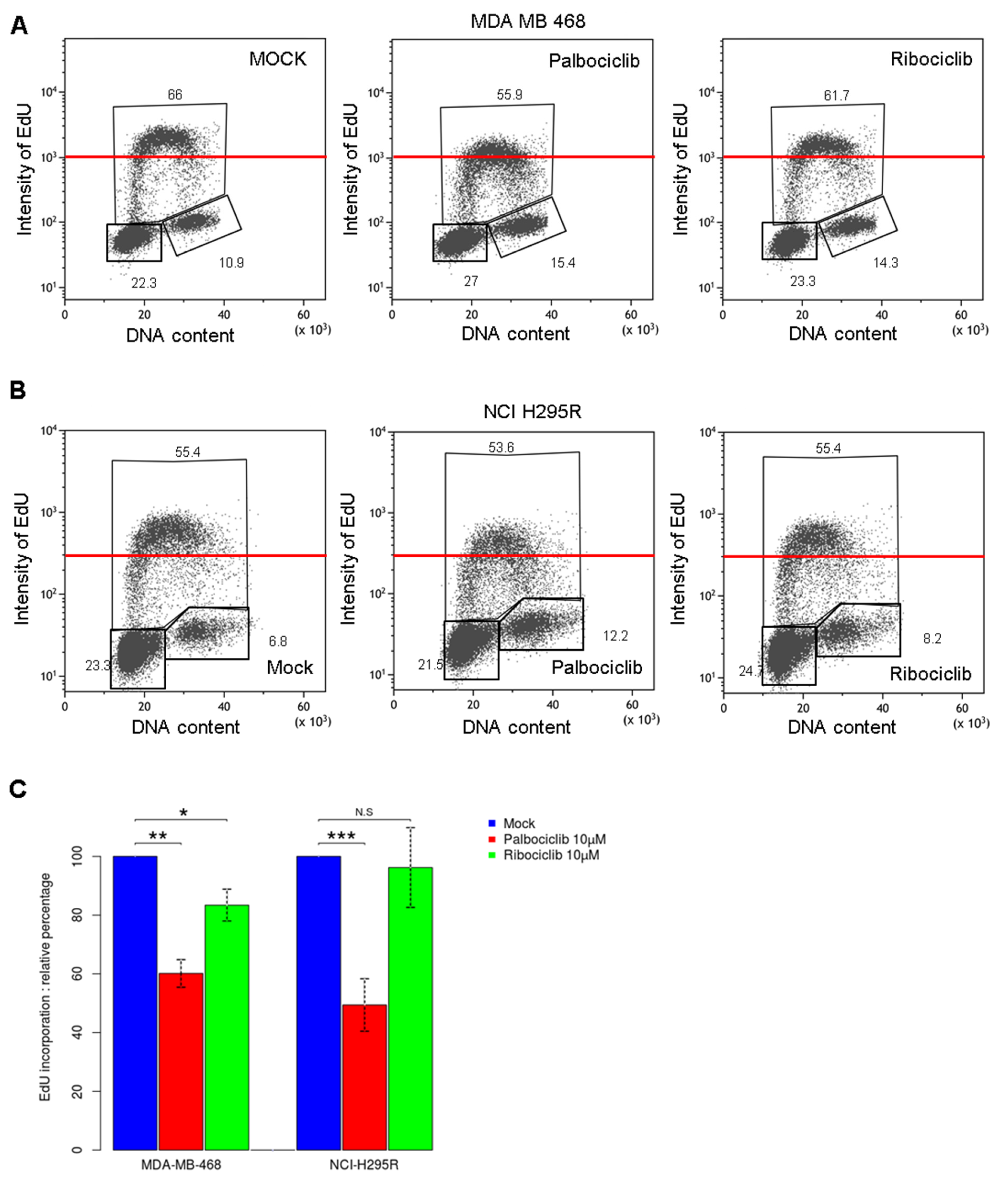
2.2. Palbociclib Treatment Impairs DNA Synthesis during the S Phase in pRb Deficient Cells
2.3. Palbociclib Alters DNA Replication Timing
2.4. Palbociclib Reduces the Number of Active Origins without Altering Replication Fork Progression
2.5. Palbociclib Acts on the Quantity of Pre-Initiation Complex Proteins
2.6. Palbociclib Affects the Expression of Genes Coding Pre-Initiation Complex Proteins
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Analysis of Cellular Viability
4.3. Apoptosis Analysis
4.4. Cell Cycle Analyses by Flow Cytometry
4.5. Total Protein Extraction
4.6. Western Blot
4.7. Analysis of the Replication Timing Program
4.8. Molecular Combing and Immunodetection
4.9. Fractionation of Chromatin-Bound Proteins
4.10. RT-qPCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, P.; Lee, N.V.; Hu, W.; Xu, M.; Ferre, R.A.; Lam, H.; Bergqvist, S.; Solowiej, J.; Diehl, W.; He, Y.A.; et al. Spectrum and degree of CDK drug interactions predicts clinical performance. Mol. Cancer Ther. 2016, 15, 2273–2281. [Google Scholar] [CrossRef]
- Spring, L.M.; Wander, S.A.; Zangardi, M.; Bardia, A. CDK 4/6 Inhibitors in Breast Cancer: Current Controversies and Future Directions. Curr. Oncol. Rep. 2019, 21, 25. [Google Scholar] [CrossRef]
- Loibl, S.; Turner, N.C.; Ro, J.; Cristofanilli, M.; Iwata, H.; Im, S.A.; Masuda, N.; Loi, S.; André, F.; Harbeck, N.; et al. Palbociclib Combined with Fulvestrant in Premenopausal Women with Advanced Breast Cancer and Prior Progression on Endocrine Therapy: PALOMA-3 Results. Oncologist 2017, 22, 1028–1038. [Google Scholar] [CrossRef]
- Chen, X.; Xu, D.; Li, X.; Zhang, J.; Xu, W.; Hou, J.; Zhang, W.; Tang, J. Latest overview of the cyclin-dependent kinases 4/6 inhibitors in breast cancer: The past, the present and the future. J. Cancer 2019, 10, 6608–6617. [Google Scholar] [CrossRef]
- Bollard, J.; Miguela, V.; Ruiz de Galarreta, M.; Venkatesh, A.; Bian, C.B.; Roberto, M.P.; Tovar, V.; Sia, D.; Molina-Sánchez, P.; Nguyen, C.B. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2016, 66, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, D.; Kim, S.J.; Denecker, T.; Ben Driss, L.; Cadoret, J.C.; Maric, C.; Baldacci, G.; Fauchereau, F. A hypothesis-driven approach identifies CDK4 and CDK6 inhibitors as candidate drugs for treatments of adrenocortical carcinomas. Aging 2017, 9, 2695–2716. [Google Scholar] [CrossRef]
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Li, Y.; Zhang, G. Cell cycle regulation and anticancer drug discovery. Cancer Biol. Med. 2017, 14, 348–362. [Google Scholar] [PubMed]
- Marzec, M.; Kasprzycka, M.; Lai, R.; Gladden, A.B.; Wlodarski, P.; Tomczak, E.; Nowell, P.; Deprimo, S.E.; Sadis, S.; Eck, S.; et al. Mantle cell lymphoma cells express predominantly cyclin D1a isoform and are highly sensitive to selective inhibition of CDK4 kinase activity. Blood 2006, 108, 1744–1750. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, T.P.; Peltier, J.; Härtlova, A.; Gierliński, M.; Jansen, V.M.; Trost, M.; Björklund, M. Thermal proteome profiling of breast cancer cells reveals proteasomal activation by CDK 4/6 inhibitor palbociclib. EMBO J. 2018, 37, e98359. [Google Scholar] [CrossRef]
- Wells, C.I.; Vasta, J.D.; Corona, C.R.; Wilkinson, J.; Zimprich, C.A.; Ingold, M.R.; Pickett, J.E.; Drewry, D.H.; Pugh, K.M.; Schwinn, M.K.; et al. Quantifying CDK inhibitor selectivity in live cells. Nat. Commun. 2020, 11, 2743. [Google Scholar] [CrossRef]
- Hafner, M.; Mills, C.E.; Subramanian, K.; Chen, C.; Chung, M.; Boswell, S.A.; Everley, R.A.; Liu, C.; Walmsley, C.S.; Juric, D.; et al. Multiomics Profiling Establishes the Polypharmacology of FDA-Approved CDK4/6 Inhibitors and the Potential for Differential Clinical Activity. Cell Chem. Biol. 2019, 26, 1067–1080. [Google Scholar] [CrossRef]
- Hsieh, F.S.; Chen, Y.L.; Hung, M.H.; Chu, P.Y.; Tsai, M.H.; Chen, L.J.; Hsiao, Y.J.; Shih, C.T.; Chang, M.J.; Chao, T.I.; et al. Palbociclib induces activation of AMPK and inhibits hepatocellular carcinoma in a CDK4/6-independent manner. Mol. Oncol. 2017, 11, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.C.; Méchali, M. DNA replication origins. Cold Spring Harb. Perspect. Biol. 2013, 5, a010116. [Google Scholar] [CrossRef] [PubMed]
- Limas, J.C.; Cook, J.G. Preparation for DNA replication: The key to a successful S phase. FEBS Lett. 2019, 593, 2853–2867. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; McDonald, D.; Hope, T.J.; Hunter, T. Mammalian Cdc7-Dbf4 protein kinase complex is essential for initiation of DNA replication. EMBO J. 1999, 18, 5703–5713. [Google Scholar] [CrossRef] [PubMed]
- Im, J.S.; Ki, S.H.; Farina, A.; Jung, D.S.; Hurwitz, J.; Lee, J.K. Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc. Nat. Acad. Sci. USA 2009, 106, 15628–15632. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Araki, H. Helicase activation and establishment of replication forks at chromosomal origins of replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010371. [Google Scholar] [CrossRef]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef]
- Bonte, D.; Lindvall, C.; Liu, H.; Dykema, K.; Furge, K.; Weinreich, M. Cdc7-Dbf4 kinase overexpression in multiple cancers and tumor cell lines is correlated with p53 inactivation. Neoplasia 2008, 10, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, D.; Denecker, T.; Guérin, E.; Kim, S.J.; Fauchereau, F.; Baldacci, G.; Maric, C.; Cadoret, J.C. Efficient, quick and easy-to-use DNA replication timing analysis with START-R suite. NAR Genom. Bioinform. 2020, 2, lqaa045. [Google Scholar] [CrossRef]
- Técher, H.; Koundrioukoff, S.; Azar, D.; Wilhelm, T.; Carignon, S.; Brison, O.; Debatisse, M.; Le Tallec, B. Replication dynamics: Biases and robustness of DNA fiber analysis. J. Mol. Biol. 2011, 425, 4845–4855. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Acebes, S.; Mourón, S.; Méndez, J. Uncoupling fork speed and origin activity to identify the primary cause of replicative stress phenotypes. J. Biol. Chem. 2018, 293, 12855–12861. [Google Scholar] [CrossRef] [PubMed]
- Diéras, V.; Rugo, H.S.; Schnell, P.; Gelmon, K.; Cristofanilli, M.; Loi, S.; Colleoni, M.; Lu, D.R.; Mori, A.; Gauthier, E.; et al. Long-term Pooled Safety Analysis of Palbociclib in Combination with Endocrine Therapy for HR+/HER2-Advanced Breast Cancer. J. Nat. Cancer Inst. 2019, 111, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.L.; Thangavel, C.; McClendon, A.K.; Reed, C.A.; Knudsen, E.S. Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene 2010, 29, 4018–4032. [Google Scholar] [CrossRef] [PubMed]
- Briu, L.M.; Maric, C.; Cadoret, J.C. Replication stress, genomic instability, and replication timing: A complex relationship. Int. J. Mol. Sci. 2021, 22, 4764. [Google Scholar] [CrossRef]
- Podust, V.N.; Podust, L.M.; Goubin, F.; Ducommun, B.; Hübscher, U. Mechanism of inhibition of proliferating cell nuclear antigen-dependent DNA synthesis by the cyclin-dependent kinase inhibitor p21. Biochemistry 1995, 34, 8869–8875. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Hutcheson, J.; Vail, P.; Witkiewicz, A.K. Biological specificity of CDK4/6 inhibitors: Dose response relationship, in vivo signaling, and composite response signature. Oncotarget 2017, 8, 43678–43691. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Witkiewicz, A.K. The transcriptome of CDK4/6 inhibition. Aging 2017, 9, 1859–1860. [Google Scholar] [CrossRef]
- Raspé, E.; Coulonval, K.; Pita, J.M.; Paternot, S.; Rothé, F.; Twyffels, L.; Brohée, S.; Craciun, L.; Larsimont, D.; Kruys, V.; et al. CDK 4 phosphorylation status and a linked gene expression profile predict sensitivity to palbociclib. EMBO Mol. Med. 2017, 9, 1052–1066. [Google Scholar] [CrossRef]
- Pack, L.R.; Daigh, L.H.; Chung, M.; Meyer, T. Clinical CDK4/6 inhibitors induce selective and immediate dissociation of p21 from cyclin D-CDK4 to inhibit CDK2. Nat. Commun. 2021, 12, 3356. [Google Scholar] [CrossRef]
- Dick, F.A.; Rubin, S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 2013, 14, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Dean, D.C. Chromatin remodeling and Rb activity. Curr. Opin. Cell Biol. 2000, 12, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. Rb Loss Causes Cancer by Driving Mitosis Mad. Cancer Cell 2007, 11, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Van Harn, T.; Foijer, F.; van Vugt, M.; Banerjee, R.; Yang, F.; Oostra, A.; Joenje, H.; te Riele, H. Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes Dev. 2010, 24, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Xing, E.P.; Yang, G.Y.; Wang, L.D.; Shi, S.T.; Yang, C.S. Loss of heterozygosity of the Rb gene correlates with pRb protein expression and associates with p53 alteration in human esophageal cancer. Clin. Cancer Res. 1999, 5, 1231–1240. [Google Scholar] [PubMed]
- Petropoulos, M.; Tsaniras, S.C.; Taraviras, S.; Lygerou, Z. Replication Licensing Aberrations, Replication Stress, and Genomic Instability. Trends Biochem. Sci. 2019, 44, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Fei, L.; Xu, H. Role of MCM2-7 protein phosphorylation in human cancer cells. Cell Biosci. 2018, 8, 43. [Google Scholar] [CrossRef]
- Rodriguez-Acebes, S.; Proctor, I.; Loddo, M.; Wollenschlaeger, A.; Rashid, M.; Falzon, M.; Prevost, A.T.; Sainsbury, R.; Stoeber, K.; Williams, G. Targeting DNA replication before it starts: Cdc7 as a therapeutic target in p53-mutant breast cancers. Am. J. Pathol. 2010, 177, 2034–2045. [Google Scholar] [CrossRef]
- Montagnoli, A.; Tenca, P.; Sola, F.; Carpani, D.; Brotherton, D.; Albanese, C.; Santocanale, C. Cdc7 inhibition reveals a p53-dependent replication checkpoint that is defective in cancer cells. Cancer Res. 2004, 64, 7110–7116. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Chung, S.; Brough, R.; Vail, P.; Franco, J.; Lord, C.J.; Knudsen, E.S. Targeting the Vulnerability of RB Tumor Suppressor Loss in Triple-Negative Breast Cancer. Cell Rep. 2018, 22, 1185–1199. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Concentration | Reference | Manufacturer |
|---|---|---|---|
| Mouse-anti-Chk1 | 1/1000 | #2360 | Cell signaling |
| Rabbit-anti-Chk1 pS345 | 1/500 | #2348 | Cell signaling |
| Rabbit-anti-γH2AX | 1/1000 | #9718 | Cell signaling |
| Mouse-anti-αTubulin | 1/2000 | T9026 | Sigma |
| Rabbit-anti-Orc1 | 1/500 | Ab88530 | Abcam |
| Rabbit-anti-Cdt1 | 1/250 | D10F11 | Cell signaling |
| Rabbit-anti-Mcm2 | 1/1000 | 4461 | Abcam |
| Rabbit-anti-Mcm2 phosphoS53 | 1/2000 | Ab109133 | Abcam |
| Mouse-anti-Cdc7 | 1/1000 | Sc 56275 | Santa Cruz |
| Rabbit-anti-Dbf4 | 1/20,000 | Ab 124707 | Abcam |
| Mouse-anti-Cdc45 | 1/1000 | Sc 55569 | Santa Cruz |
| Rabbit-anti-Histone H4 | 1/2000 | 07-108 | Millipore |
| Genes | Sequence (3′->5′) |
|---|---|
| Tbp | Forward:cacgaaccacggcactgatt Reverse:ttttcttgctgccagtctggac |
| Cdc7 | Forward:caaagtgccccaatcaaact Reverse:tgggccaaagcagttaaatc |
| Dbf4 | Forward:aaaccacttcacctcatccc Reverse:tttactccccatgacaaggc |
| Cdc45 | Forward:gcaaacacctgctcaagtcc Reverse:tcccaaaaaagttcttcctgtc |
| Orc1 | Forward:gccaaagaagagtctcaagcc Reverse:acagcagaaacatgcagcc |
| Mcm2 | Forward:cgtatccgaatccaggagag Reverse:gtgttgagggagccatcatag |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-J.; Maric, C.; Briu, L.-M.; Fauchereau, F.; Baldacci, G.; Debatisse, M.; Koundrioukoff, S.; Cadoret, J.-C. Firing of Replication Origins Is Disturbed by a CDK4/6 Inhibitor in a pRb-Independent Manner. Int. J. Mol. Sci. 2023, 24, 10629. https://doi.org/10.3390/ijms241310629
Kim S-J, Maric C, Briu L-M, Fauchereau F, Baldacci G, Debatisse M, Koundrioukoff S, Cadoret J-C. Firing of Replication Origins Is Disturbed by a CDK4/6 Inhibitor in a pRb-Independent Manner. International Journal of Molecular Sciences. 2023; 24(13):10629. https://doi.org/10.3390/ijms241310629
Chicago/Turabian StyleKim, Su-Jung, Chrystelle Maric, Lina-Marie Briu, Fabien Fauchereau, Giuseppe Baldacci, Michelle Debatisse, Stéphane Koundrioukoff, and Jean-Charles Cadoret. 2023. "Firing of Replication Origins Is Disturbed by a CDK4/6 Inhibitor in a pRb-Independent Manner" International Journal of Molecular Sciences 24, no. 13: 10629. https://doi.org/10.3390/ijms241310629
APA StyleKim, S. -J., Maric, C., Briu, L. -M., Fauchereau, F., Baldacci, G., Debatisse, M., Koundrioukoff, S., & Cadoret, J. -C. (2023). Firing of Replication Origins Is Disturbed by a CDK4/6 Inhibitor in a pRb-Independent Manner. International Journal of Molecular Sciences, 24(13), 10629. https://doi.org/10.3390/ijms241310629