
The Implications of Cannabinoid-Induced Metabolic Dysregulation for Cellular Differentiation and Growth



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Endocannabinoid Signaling
2. Cellular Differentiation
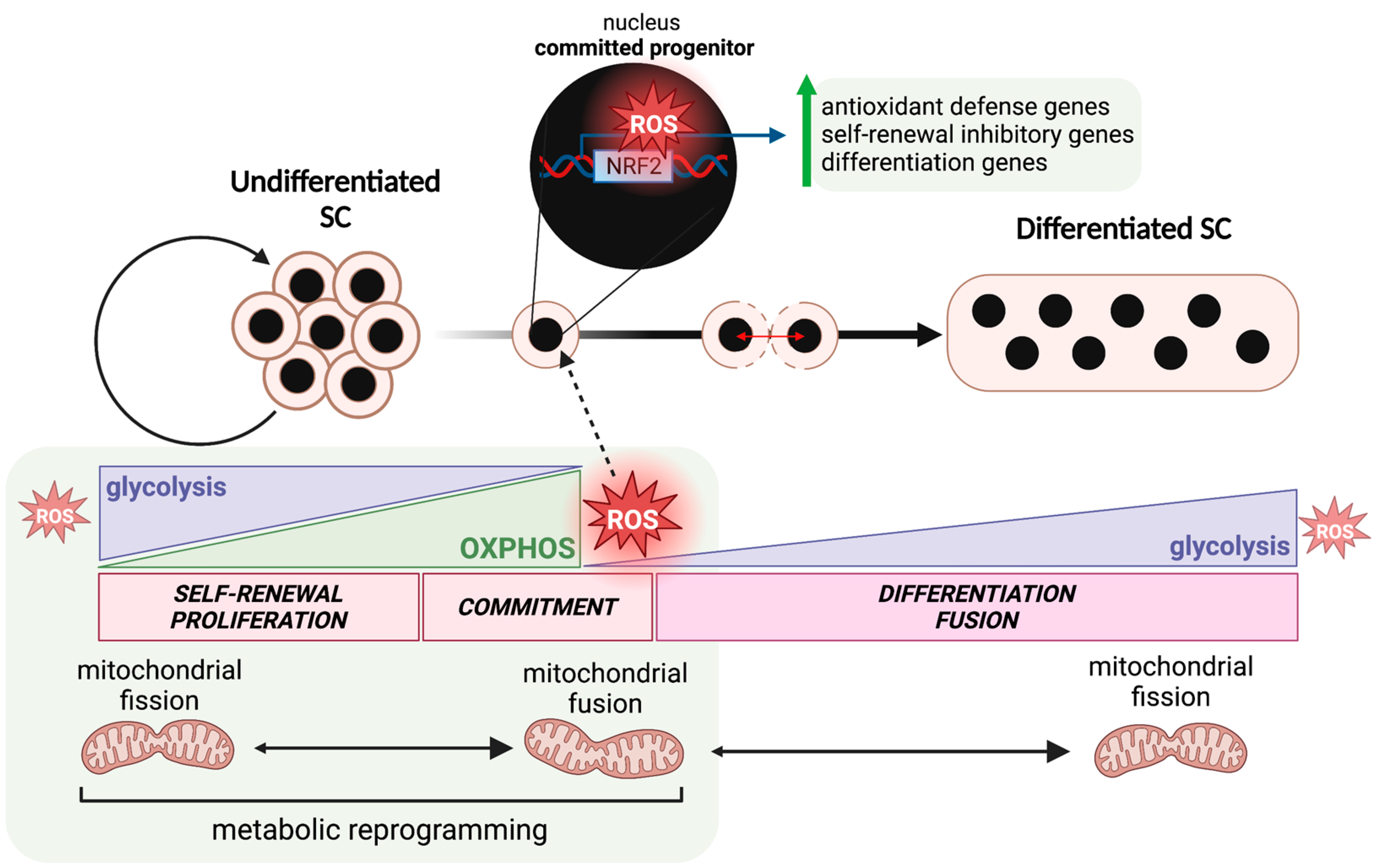
2.1. Stem Cell Characteristics
2.2. Key Intracellular Signaling Pathways Involved in Stem Cell Function and Differentiation
3. Metabolic Regulation of Stem Cell Fate Decisions
3.1. Mitochondrial Dynamics
3.2. Metabolic Switch as a Driver of Stem Cell Differentiation
3.3. Endocannabinoid Signaling at Mitochondrial CB1 (mtCB1)
4. Endoplasmic Reticulum (ER) and the ER Stress Response
4.1. Structure of the ER
4.2. Function of ER in Key Cellular Processes
4.3. ER and Mitochondrial Cross-Talk Mechanisms
4.4. ER-Dependent Unfolded Protein Response
5. Endocannabinoid Signaling and Intracellular Functions
5.1. Regulation of Mitochondrial Function through Endocannabinoid Signaling
5.2. Regulation of ER Function through Endocannabinoid Signaling
6. Potential Impacts of Cannabinoids on Cellular Differentiation
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bisogno, T.; Berrendero, F.; Ambrosino, G.; Cebeira, M.; Ramos, J.; Fernandez-Ruiz, J.; Di Marzo, V. Brain regional distribution of endocannabinoids: Implications for their biosynthesis and biological function. Biochem. Biophys. Res. Commun. 1999, 256, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.C.; Nielsen, A.; Briley, E.M.; Palkovits, M.; Priller, J.; Axelrod, J.; Nguyen, D.N.; Richardson, J.M.; Riggin, R.M.; Koppel, G.A.; et al. Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS Lett. 1996, 393, 231–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Giagnoni, G.; Franke, C.; Trovato, A.E.; Colleoni, M. Vanilloid TRPV1 receptor mediates the antihyperalgesic effect of the nonpsychoactive cannabinoid, cannabidiol, in a rat model of acute inflammation. Br. J. Pharmacol. 2004, 143, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Zhang, J.; Fan, N.; Teng, Z.-Q.; Wu, Y.; Yang, H.; Tang, Y.-P.; Sun, H.; Song, Y.; Chen, C. Δ9-THC-caused synaptic and memory impairments are mediated through COX-2 signaling. Cell 2013, 155, 1154–1165. [Google Scholar] [CrossRef] [Green Version]
- Kirkham, T.C.; Williams, C.M.; Fezza, F.; Di Marzo, V. Endocannabinoid levels in rat limbic forebrain and hypothalamus in relation to fasting, feeding and satiation: Stimulation of eating by 2-arachidonoyl glycerol. Br. J. Pharmacol. 2002, 136, 550–557. [Google Scholar] [CrossRef] [Green Version]
- Xapelli, S.; Agasse, F.; Sardà-Arroyo, L.; Bernardino, L.; Santos, T.; Ribeiro, F.F.; Valero, J.; Bragança, J.; Schitine, C.; de Melo Reis, R.A.; et al. Activation of type 1 cannabinoid receptor (CB1R) promotes neurogenesis in murine subventricular zone cell cultures. PLoS ONE 2013, 8, e63529. [Google Scholar] [CrossRef]
- Bagavandoss, P.; Grimshaw, S. Temporal and spatial distribution of the cannabinoid receptors (CB1, CB2) and fatty acid amide hydroxylase in the rat ovary. Anat. Rec. 2010, 293, 1425–1432. [Google Scholar] [CrossRef]
- Liu, Q.-R.; Pan, C.-H.; Hishimoto, A.; Li, C.-Y.; Xi, Z.-X.; Llorente-Berzal, A.; Viveros, M.-P.; Ishiguro, H.; Arinami, T.; Onaivi, E.S.; et al. Species differences in cannabinoid receptor 2 (CNR2 gene): Identification of novel human and rodent CB2 isoforms, differential tissue expression and regulation by cannabinoid receptor ligands. Genes Brain Behav. 2009, 8, 519–530. [Google Scholar] [CrossRef]
- Jiang, S.; Fu, Y.; Avraham, H.K. Regulation of hematopoietic stem cell trafficking and mobilization by the endocannabinoid system. Transfusion 2011, 51 (Suppl. 4), 65S–71S. [Google Scholar] [CrossRef]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef] [Green Version]
- Fezza, F.; Bari, M.; Florio, R.; Talamonti, E.; Feole, M.; Maccarrone, M. Endocannabinoids, related compounds and their metabolic routes. Molecules 2014, 19, 17078–17106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galve-Roperh, I.; Chiurchiù, V.; Díaz-Alonso, J.; Bari, M.; Guzmán, M.; Maccarrone, M. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog. Lipid Res. 2013, 52, 633–650. [Google Scholar] [CrossRef] [PubMed]
- Ramalho-Santos, M.; Willenbring, H. On the origin of the term “stem cell”. Cell Stem Cell 2007, 1, 35–38. [Google Scholar] [CrossRef] [Green Version]
- Morales, A.V.; Mira, H. Adult Neural Stem Cells: Born to Last. Front. Cell Dev. Biol. 2019, 7, 96. [Google Scholar] [CrossRef] [Green Version]
- Till, J.E.; McCulloch, E.A. Hemopoietic stem cell differentiation. Biochim. Biophys. Acta 1980, 605, 431–459. [Google Scholar] [CrossRef] [PubMed]
- Weissman, I.L. Stem cells: Units of development, units of regeneration, and units in evolution. Cell 2000, 100, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Morrison, S.J.; Shah, N.M.; Anderson, D.J. Regulatory mechanisms in stem cell biology. Cell 1997, 88, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Cavallucci, V.; Fidaleo, M.; Pani, G. Neural Stem Cells and Nutrients: Poised Between Quiescence and Exhaustion. Trends Endocrinol. Metab. 2016, 27, 756–769. [Google Scholar] [CrossRef]
- Takahashi, S.; Kobayashi, S.; Hiratani, I. Epigenetic differences between naïve and primed pluripotent stem cells. Cell. Mol. Life Sci. 2018, 75, 1191–1203. [Google Scholar] [CrossRef] [Green Version]
- Weinberger, L.; Ayyash, M.; Novershtern, N.; Hanna, J.H. Dynamic stem cell states: Naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 2016, 17, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Hasan, F.; Chiu, Y.; Shaw, R.M.; Wang, J.; Yee, C. Hypoxia acts as an environmental cue for the human tissue-resident memory T cell differentiation program. JCI Insight 2021, 6, e138970. [Google Scholar] [CrossRef] [PubMed]
- Podkalicka, P.; Stępniewski, J.; Mucha, O.; Kachamakova-Trojanowska, N.; Dulak, J.; Łoboda, A. Hypoxia as a Driving Force of Pluripotent Stem Cell Reprogramming and Differentiation to Endothelial Cells. Biomolecules 2020, 10, 1614. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Liu, F.; Han, L.; Zhao, L.; Chen, J.; Olopade, O.I.; He, M.; Wei, M. HIF-2α promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J. Exp. Clin. Cancer Res. 2018, 37, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.-M.; Kim, J.; Han, H.-W.; Chae, J.-I.; Son, M.-Y.; Cho, S.; Chung, H.-M.; Han, Y.-M.; Kang, Y.-K. Notch signaling is required for maintaining stem-cell features of neuroprogenitor cells derived from human embryonic stem cells. BMC Neurosci. 2009, 10, 97. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yu, L.; Zhu, L.-Y.; He, H.; Ren, J.; Pan, J.; Xie, X.; Cai, C.; Lu, L.; Tian, H.; et al. Cytokines Induce Monkey Neural Stem Cell Differentiation through Notch Signaling. BioMed Res. Int. 2020, 2020, 1308526. [Google Scholar] [CrossRef]
- Lee, S.; Remark, L.H.; Josephson, A.M.; Leclerc, K.; Lopez, E.M.; Kirby, D.J.; Mehta, D.; Litwa, H.P.; Wong, M.Z.; Shin, S.Y.; et al. Notch-Wnt signal crosstalk regulates proliferation and differentiation of osteoprogenitor cells during intramembranous bone healing. NPJ Regen. Med. 2021, 6, 29. [Google Scholar] [CrossRef]
- Wend, P.; Holland, J.D.; Ziebold, U.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Semin. Cell Dev. Biol. 2010, 21, 855–863. [Google Scholar] [CrossRef]
- Sato, N.; Meijer, L.; Skaltsounis, L.; Greengard, P.; Brivanlou, A.H. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 2004, 10, 55–63. [Google Scholar] [CrossRef]
- Xu, Z.; Robitaille, A.M.; Berndt, J.D.; Davidson, K.C.; Fischer, K.A.; Mathieu, J.; Potter, J.C.; Ruohola-Baker, H.; Moon, R.T. Wnt/β-catenin signaling promotes self-renewal and inhibits the primed state transition in naïve human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6382–E6390. [Google Scholar] [CrossRef]
- Rodesch, F.; Simon, P.; Donner, C.; Jauniaux, E. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet. Gynecol. 1992, 80, 283–285. [Google Scholar]
- Dahl, K.D.C.; Fryer, B.H.; Mack, F.A.; Compernolle, V.; Maltepe, E.; Adelman, D.M.; Carmeliet, P.; Simon, M.C. Hypoxia-Inducible Factors 1α and 2α Regulate Trophoblast Differentiation. Mol. Cell. Biol. 2005, 25, 10479–10491. [Google Scholar] [CrossRef] [Green Version]
- Caniggia, I.; Grisaru-Gravnosky, S.; Kuliszewsky, M.; Post, M.; Lye, S.J. Inhibition of TGF-beta 3 restores the invasive capability of extravillous trophoblasts in preeclamptic pregnancies. J. Clin. Investig. 1999, 103, 1641–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caniggia, I.; Mostachfi, H.; Winter, J.; Gassmann, M.; Lye, S.J.; Kuliszewski, M.; Post, M. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3). J. Clin. Investig. 2000, 105, 577–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kizil, C.; Kyritsis, N.; Brand, M. Effects of inflammation on stem cells: Together they strive? EMBO Rep. 2015, 16, 416–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iosif, R.E.; Ekdahl, C.T.; Ahlenius, H.; Pronk, C.J.H.; Bonde, S.; Kokaia, Z.; Jacobsen, S.-E.W.; Lindvall, O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J. Neurosci. 2006, 26, 9703–9712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory blockade restores adult hippocampal neurogenesis. Science 2003, 302, 1760–1765. [Google Scholar] [CrossRef]
- Stolp, H.B.; Turnquist, C.; Dziegielewska, K.M.; Saunders, N.R.; Anthony, D.C.; Molnár, Z. Reduced ventricular proliferation in the foetal cortex following maternal inflammation in the mouse. Brain 2011, 134 Pt 11, 3236–3248. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.A.; Steiner, B.; Wengner, A.; Lipp, M.; Kammertoens, T.; Kempermann, G. Adaptive peripheral immune response increases proliferation of neural precursor cells in the adult hippocampus. FASEB J. 2009, 23, 3121–3128. [Google Scholar] [CrossRef]
- Wang, X.; Fu, S.; Wang, Y.; Yu, P.; Hu, J.; Gu, W.; Xu, X.-M.; Lu, P. Interleukin-1beta mediates proliferation and differentiation of multipotent neural precursor cells through the activation of SAPK/JNK pathway. Mol. Cell. Neurosci. 2007, 36, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Widera, D.; Mikenberg, I.; Elvers, M.; Kaltschmidt, C.; Kaltschmidt, B. Tumor necrosis factor alpha triggers proliferation of adult neural stem cells via IKK/NF-kappaB signaling. BMC Neurosci. 2006, 7, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Ji, J.; Yan, X.H. Cross-talk between AMPK and mTOR in regulating energy balance. Crit. Rev. Food Sci. Nutr. 2012, 52, 373–381. [Google Scholar] [CrossRef]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.-E.W.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.; Sherman, N.; Pagano, M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef]
- Gingras, A.-C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Meng, D.; Frank, A.R.; Jewell, J.L. mTOR signaling in stem and progenitor cells. Development 2018, 145, dev152595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, R.M.; La, H.M.; Mäkelä, J.; Kobayashi, T.; Noda, T.; Pandolfi, P.P. Distinct germline progenitor subsets defined through Tsc2-mTORC1 signaling. EMBO Rep. 2015, 16, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.K.; Fitter, S.; Dutta, A.K.; Matthews, M.P.; Walkley, C.R.; Hall, M.N.; Ruegg, M.A.; Gronthos, S.; Zannettino, A.C.W. Brief report: The differential roles of mTORC1 and mTORC2 in mesenchymal stem cell differentiation. Stem Cells 2015, 33, 1359–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohapatra, B.; Zutshi, N.; An, W.; Goetz, B.; Arya, P.; Bielecki, T.A.; Mushtaq, I.; Storck, M.D.; Meza, J.L.; Band, V.; et al. An essential role of CBL and CBL-B ubiquitin ligases in mammary stem cell maintenance. Development 2017, 144, 1072–1086. [Google Scholar] [PubMed] [Green Version]
- Otaegi, G.; Yusta-Boyo, M.J.; Vergaño-Vera, E.; Méndez-Gómez, H.R.; Carrera, A.C.; Abad, J.L.; González, M.; de la Rosa, E.J.; Vicario-Abejón, C.; de Pablo, F. Modulation of the PI 3-kinase-Akt signalling pathway by IGF-I and PTEN regulates the differentiation of neural stem/precursor cells. J. Cell Sci. 2006, 119 Pt 13, 2739–2748. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Easley, C.A.; Ben-Yehudah, A.; Redinger, C.J.; Oliver, S.L.; Varum, S.T.; Eisinger, V.M.; Carlisle, D.L.; Donovan, P.J.; Schatten, G.P. mTOR-mediated activation of p70 S6K induces differentiation of pluripotent human embryonic stem cells. Cell Reprogram. 2010, 12, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Lim, M.S.; Park, J.H.; Park, C.H.; Koh, H.C. PTEN Promotes Dopaminergic Neuronal Differentiation Through Regulation of ERK-Dependent Inhibition of S6K Signaling in Human Neural Stem Cells. Stem Cells Transl. Med. 2016, 5, 1319–1329. [Google Scholar] [CrossRef] [Green Version]
- Schaub, T.; Gürgen, D.; Maus, D.; Lange, C.; Tarabykin, V.; Dragun, D.; Hegner, B. Publisher Correction: mTORC1 and mTORC2 Differentially Regulate Cell Fate Programs to Coordinate Osteoblastic Differentiation in Mesenchymal Stromal Cells. Sci. Rep. 2020, 10, 3740. [Google Scholar] [CrossRef] [Green Version]
- Cipolat, S.; De Brito, O.M.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117 Pt 26, 6535–6546. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C.; Cipolat, S.; de Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [Green Version]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C.; Cherok, E.; Xu, S.; Li, S.; Das, S.; Meltzer, W.A.; Zalzman, M.; et al. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [Green Version]
- Sugioka, K.; Nakano, M.; Totsune-Nakano, H.; Minakami, H.; Tero-Kubota, S.; Ikegami, Y. Mechanism of O−2 generation in reduction and oxidation cycle of ubiquinones in a model of mitochondrial electron transport systems. Biochim. Biophys. Acta 1988, 936, 377–385. [Google Scholar] [CrossRef]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Heiden, M.G.V. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Folmes, C.D.L.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Nuebel, E.; Daley, G.Q.; Koehler, C.M.; Teitell, M.A. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 2012, 11, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.; Patel, J.; Khacho, M. Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: How mitochondrial structure can regulate bioenergetics. Mitochondrion 2019, 49, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-M.; Liu, X.; Shen, J.; Jovanovic, O.; Pohl, E.E.; Gerson, S.L.; Finkel, T.; Broxmeyer, H.E.; Qu, C.-K. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell 2013, 12, 62–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.M.; Kwon, S.; Pak, Y.K.; Seol, H.W.; Choi, Y.M.; Park, D.J.; Park, K.S.; Lee, H.K. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem. Biophys. Res. Commun. 2006, 348, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.-H.; Wang, R.; Wang, Y.; Kung, C.-P.; Weber, J.; Patti, G.J. Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife 2019, 8, e41351. [Google Scholar] [CrossRef]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.-E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 2016, 19, 232–247. [Google Scholar] [CrossRef] [Green Version]
- Luchsinger, L.L.; de Almeida, M.J.; Corrigan, D.J.; Mumau, M.; Snoeck, H.-W. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 2016, 529, 528–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, W.; Hegde, V.L.; Singh, N.P.; Sisco, D.; Grant, S.; Nagarkatti, M.; Nagarkatti, P.S. Delta9-tetrahydrocannabinol-induced apoptosis in Jurkat leukemia T cells is regulated by translocation of Bad to mitochondria. Mol Cancer Res. 2006, 4, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, O.S.; Ragos, R.; Gurm, H.; Lapierre, M.; May, L.L.; Raha, S. Delta-9-tetrahydrocannabinol disrupts mitochondrial function and attenuates syncytialization in human placental BeWo cells. Physiol. Rep. 2020, 8, e14476. [Google Scholar] [CrossRef] [PubMed]
- Walker, O.S.; Ragos, R.; Wong, M.K.; Adam, M.; Cheung, A.; Raha, S. Reactive oxygen species from mitochondria impacts trophoblast fusion and the production of endocrine hormones by syncytiotrophoblasts. PLoS ONE 2020, 15, e0229332. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Torres-López, L.; Valle-Reyes, J.S.; Hernández-Cruz, A.; Pottosin, I.; Dobrovinskaya, O. Cannabidiol directly targets mitochondria and disturbs calcium homeostasis in acute lymphoblastic leukemia. Cell Death Dis. 2019, 10, 779. [Google Scholar] [CrossRef] [Green Version]
- Gkoumassi, E.; Dekkers, B.G.J.; Dröge, M.J.; Elzinga, C.R.S.; Hasenbosch, R.E.; Meurs, H.; Nelemans, S.A.; Schmidt, M.; Zaagsma, J. (Endo)cannabinoids mediate different Ca2+ entry mechanisms in human bronchial epithelial cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2009, 380, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Gross, C.; Ramirez, D.A.; McGrath, S.; Gustafson, D.L. Cannabidiol Induces Apoptosis and Perturbs Mitochondrial Function in Human and Canine Glioma Cells. Front. Pharmacol. 2021, 12, 725136. [Google Scholar] [CrossRef]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB₁ receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef]
- Jimenez-Blasco, D.; Busquets-Garcia, A.; Hebert-Chatelain, E.; Serrat, R.; Vicente-Gutierrez, C.; Ioannidou, C.; Gómez-Sotres, P.; Lopez-Fabuel, I.; Resch-Beusher, M.; Resel, E.; et al. Glucose metabolism links astroglial mitochondria to cannabinoid effects. Nature 2020, 583, 603–608. [Google Scholar] [CrossRef]
- Hebert-Chatelain, E.; Desprez, T.; Serrat, R.; Bellocchio, L.; Soria-Gomez, E.; Busquets-Garcia, A.; Zottola, A.C.P.; Delamarre, A.; Cannich, A.; Vincent, P.; et al. A cannabinoid link between mitochondria and memory. Nature 2016, 539, 555–559. [Google Scholar] [CrossRef]
- Ma, L.; Jia, J.; Niu, W.; Jiang, T.; Zhai, Q.; Yang, L.; Bai, F.; Wang, Q.; Xiong, L. Mitochondrial CB1 receptor is involved in ACEA-induced protective effects on neurons and mitochondrial functions. Sci. Rep. 2015, 5, 12440. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.; Varela, L.; Kim, J.G.; Kim, J.D.; Hernández-Nuño, F.; Simonds, S.E.; Castorena, C.M.; Vianna, C.R.; Elmquist, J.K.; Morozov, Y.M.; et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature 2015, 519, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamnate, A.; Sirisin, J.; Watanabe, M.; Kondo, H.; Hipkaeo, W.; Chomphoo, S. Mitochondrial Localization of CB1 in Progesterone-producing Cells of Ovarian Interstitial Glands of Adult Mice. J. Histochem. Cytochem. 2022, 70, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Aquila, S.; Guido, C.; Santoro, A.; Perrotta, I.; Laezza, C.; Bifulco, M.; Sebastiano, A. Human sperm anatomy: Ultrastructural localization of the cannabinoid1 receptor and a potential role of anandamide in sperm survival and acrosome reaction. Anat. Rec. 2010, 293, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Mendizabal-Zubiaga, J.; Melser, S.; Bénard, G.; Ramos, A.; Reguero, L.; Arrabal, S.; Elezgarai, I.; Gerrikagoitia, I.; Suarez, J.; De Fonseca, F.R.; et al. Cannabinoid CB1 Receptors Are Localized in Striated Muscle Mitochondria and Regulate Mitochondrial Respiration. Front. Physiol. 2016, 7, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westrate, L.M.; Lee, J.E.; Prinz, W.A.; Voeltz, G.K. Form follows function: The importance of endoplasmic reticulum shape. Annu. Rev. Biochem. 2015, 84, 791–811. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Ruggiano, A.; Foresti, O.; Carvalho, P. Quality control: ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippincott-Schwartz, J.; Bonifacino, J.S.; Yuan, L.C.; Klausner, R.D. Degradation from the endoplasmic reticulum: Disposing of newly synthesized proteins. Cell 1988, 54, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Sommer, T.; Jentsch, S. A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature 1993, 365, 176–179. [Google Scholar] [CrossRef]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Saido, T.C. Critical review: Involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol. 2018, 8, 180024. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Jiang, L.; Huang, T.; Zhao, Y.; Liu, T.; Zhong, Y.; Li, X.; Campos, A.; Pomeroy, K.; Masliah, E.; et al. ER-associated degradation regulates Alzheimer’s amyloid pathology and memory function by modulating γ-secretase activity. Nat. Commun. 2017, 8, 1472. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Bygrave, F.L.; Benedetti, A. What is the concentration of calcium ions in the endoplasmic reticulum? Cell Calcium 1996, 19, 547–551. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 2009, 1793, 933–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Xie, S.Z.; Garcia-Prat, L.; Voisin, V.; Ferrari, R.; Gan, O.I.; Wagenblast, E.; Kaufmann, K.B.; Zeng, A.G.; Takayanagi, S.-I.; Patel, I.; et al. Sphingolipid Modulation Activates Proteostasis Programs to Govern Human Hematopoietic Stem Cell Self-Renewal. Cell Stem Cell 2019, 25, 639–653.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innamorati, G.; Fontana, E.; Steccanella, F.; Gandhi, K.; Bassi, G.; Zandonà, V.; Giacomello, L. Pleiotropic effects of sphingosine-1-phosphate signaling to control human chorionic mesenchymal stem cell physiology. Cell Death Dis. 2017, 8, e2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inniss, K.; Moore, H. Mediation of apoptosis and proliferation of human embryonic stem cells by sphingosine-1-phosphate. Stem Cells Dev. 2006, 15, 789–796. [Google Scholar] [CrossRef]
- Romani, R.; Manni, G.; Donati, C.; Pirisinu, I.; Bernacchioni, C.; Gargaro, M.; Pirro, M.; Calvitti, M.; Bagaglia, F.; Sahebkar, A.; et al. S1P promotes migration, differentiation and immune regulatory activity in amniotic-fluid-derived stem cells. Eur. J. Pharmacol. 2018, 833, 173–182. [Google Scholar] [CrossRef]
- Siddique, M.M.; Li, Y.; Wang, L.; Ching, J.; Mal, M.; Ilkayeva, O.; Wu, Y.J.; Bay, B.H.; Summers, S.A. Ablation of dihydroceramide desaturase 1, a therapeutic target for the treatment of metabolic diseases, simultaneously stimulates anabolic and catabolic signaling. Mol. Cell. Biol. 2013, 33, 2353–2369. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Khacho, M.; Slack, R.S. Mitochondrial and Reactive Oxygen Species Signaling Coordinate Stem Cell Fate Decisions and Life Long Maintenance. Antioxid. Redox Signal. 2017, 28, 1090–1101. [Google Scholar] [CrossRef]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef] [PubMed]
- Pitts, K.; Yoon, Y.; Krueger, E.; McNiven, M. The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Mol. Biol. Cell 1999, 10, 4403–4417. [Google Scholar] [CrossRef] [Green Version]
- Iwasawa, R.; Mahul-Mellier, A.-L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keinan, N.; Pahima, H.; Ben-Hail, D.; Shoshan-Barmatz, V. The role of calcium in VDAC1 oligomerization and mitochondria-mediated apoptosis. Biochim. Biophys. Acta 2013, 1833, 1745–1754. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Szabadkai, G.; Simoni, A.M.; Chami, M.; Wieckowski, M.R.; Youle, R.J.; Rizzuto, R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell 2004, 16, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Liu, C.Y.; Schröder, M.; Kaufman, R.J. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J. Biol. Chem. 2000, 275, 24881–24885. [Google Scholar] [CrossRef] [Green Version]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.; Newbatt, Y.; McAndrew, P.C.; Stubbs, M.; Burke, R.; Richards, M.W.; Bhatia, C.; Caldwell, J.J.; McHardy, T.; Collins, I.; et al. Molecular mechanisms of human IRE1 activation through dimerization and ligand binding. Oncotarget 2015, 6, 13019–13035. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpędek-Kamińska, W.; Wawrzynkiewicz, A.; Pytel, D.; Diehl, J.A.; Majsterek, I. The Structure, Activation and Signaling of IRE1 and Its Role in Determining Cell Fate. Biomedicines 2021, 9, 156. [Google Scholar] [CrossRef]
- Karagöz, G.E.; Acosta-Alvear, D.; Nguyen, H.T.; Lee, C.P.; Chu, F.; Walter, P. An unfolded protein-induced conformational switch activates mammalian IRE1. Elife 2017, 6, e30700. [Google Scholar] [CrossRef]
- Wang, P.; Li, J.; Tao, J.; Sha, B. The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J. Biol. Chem. 2018, 293, 4110–4121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarafian, T.A.; Habib, N.; Oldham, M.; Seeram, N.; Lee, R.-P.; Lin, L.; Tashkin, D.P.; Roth, M.D. Inhaled marijuana smoke disrupts mitochondrial energetics in pulmonary epithelial cells in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L1202–L1209. [Google Scholar] [CrossRef]
- Sarafian, T.A.; Kouyoumjian, S.; Khoshaghideh, F.; Tashkin, D.P.; Roth, M.D. Delta 9-tetrahydrocannabinol disrupts mitochondrial function and cell energetics. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L298–L306. [Google Scholar] [CrossRef] [Green Version]
- Athanasiou, A.; Clarke, A.B.; Turner, A.E.; Kumaran, N.M.; Vakilpour, S.; Smith, P.A.; Bagiokou, D.; Bradshaw, T.D.; Westwell, A.D.; Fang, L.; et al. Cannabinoid receptor agonists are mitochondrial inhibitors: A unified hypothesis of how cannabinoids modulate mitochondrial function and induce cell death. Biochem. Biophys. Res. Commun. 2007, 364, 131–137. [Google Scholar] [CrossRef]
- Catanzaro, G.; Rapino, C.; Oddi, S.; Maccarrone, M. Anandamide increases swelling and reduces calcium sensitivity of mitochondria. Biochem. Biophys. Res. Commun. 2009, 388, 439–442. [Google Scholar] [CrossRef]
- Zaccagnino, P.; D’oria, S.; Romano, L.L.; Di Venere, A.; Sardanelli, A.M.; Lorusso, M. The endocannabinoid 2-arachidonoylglicerol decreases calcium induced cytochrome c release from liver mitochondria. J. Bioenerg. Biomembr. 2012, 44, 273–280. [Google Scholar] [CrossRef]
- Pfaffenbach, K.T.; Lee, A.S. The critical role of GRP78 in physiologic and pathologic stress. Curr. Opin. Cell Biol. 2011, 23, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Pasquariello, N.; Catanzaro, G.; Marzano, V.; Amadio, D.; Barcaroli, D.; Oddi, S.; Federici, G.; Urbani, A.; Agrò, A.F.; Maccarrone, M. Characterization of the endocannabinoid system in human neuronal cells and proteomic analysis of anandamide-induced apoptosis. J. Biol. Chem. 2009, 284, 29413–29426. [Google Scholar] [CrossRef] [Green Version]
- Soliman, E.; Henderson, K.L.; Danell, A.S.; Van Dross, R. Arachidonoyl-ethanolamide activates endoplasmic reticulum stress-apoptosis in tumorigenic keratinocytes: Role of cyclooxygenase-2 and novel J-series prostamides. Mol. Carcinog. 2016, 55, 117–130. [Google Scholar] [CrossRef]
- Soliman, E.; Van Dross, R. Anandamide-induced endoplasmic reticulum stress and apoptosis are mediated by oxidative stress in non-melanoma skin cancer: Receptor-independent endocannabinoid signaling. Mol. Carcinog. 2016, 55, 1807–1821. [Google Scholar] [CrossRef]
- Almada, M.; Costa, L.; Fonseca, B.; Alves, P.; Braga, J.; Gonçalves, D.; Teixeira, N.; Correia-Da-Silva, G. The endocannabinoid 2-arachidonoylglycerol promotes endoplasmic reticulum stress in placental cells. Reproduction 2020, 160, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Lorente, M.; Egia, A.; Blázquez, C.; García, S.; Giroux, V.; Malicet, C.; Villuendas, R.; Gironella, M.; González-Feria, L.; et al. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell 2006, 9, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernández-Tiedra, S.; Lorente, M.; Egia, A.; Vázquez, P.; Blázquez, C.; Torres, S.; García, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [Green Version]
- De Jaime-Soguero, A.; Aulicino, F.; Ertaylan, G.; Griego, A.; Cerrato, A.; Tallam, A.; Del Sol, A.; Cosma, M.P.; Lluis, F. Wnt/Tcf1 pathway restricts embryonic stem cell cycle through activation of the Ink4/Arf locus. PLoS Genet. 2017, 13, e1006682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, K.; Nishinakamura, R.; Iwamatsu, Y.; Shimosato, D.; Niwa, H. Synergistic action of Wnt and LIF in maintaining pluripotency of mouse ES cells. Biochem. Biophys. Res. Commun. 2006, 343, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Kurek, D.; Neagu, A.; Tastemel, M.; Tüysüz, N.; Lehmann, J.; van de Werken, H.J.; Philipsen, S.; van der Linden, R.; Maas, A.; van Ijcken, W.F.; et al. Endogenous WNT signals mediate BMP-induced and spontaneous differentiation of epiblast stem cells and human embryonic stem cells. Stem Cell Rep. 2015, 4, 114–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Wu, Q.; Lin, X.; Ling, X.; Miao, J.; Liu, X.; Hu, C.; Zhang, Y.; Jia, N.; Hou, F.F.; et al. Cannabinoid receptor type 2 promotes kidney fibrosis through orchestrating β-catenin signaling. Kidney Int. 2021, 99, 364–381. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y. Alzheimer Disease: Crosstalk between the Canonical Wnt/Beta-Catenin Pathway and PPARs Alpha and Gamma. Front. Neurosci. 2016, 10, 459. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; De Filippis, D.; Carnuccio, R.; Izzo, A.; Iuvone, T. The marijuana component cannabidiol inhibits beta-amyloid-induced tau protein hyperphosphorylation through Wnt/beta-catenin pathway rescue in PC12 cells. J. Mol. Med. 2006, 84, 253–258. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.N. Cannabidiol and the Canonical WNT/β-Catenin Pathway in Glaucoma. Int. J. Mol. Sci. 2021, 22, 3798. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; De Filippis, D.; Cipriano, M.; Carratù, M.R.; Iuvone, T.; Steardo, L. Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.C.; Thangavelu, C.S.; Joloya, E.M.; Kuo, A.; Li, Z.; Blumberg, B. Cannabidiol promotes adipogenesis of human and mouse mesenchymal stem cells via PPARγ by inducing lipogenesis but not lipolysis. Biochem. Pharmacol. 2022, 197, 114910. [Google Scholar] [CrossRef]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; MacDougald, O.A. Inhibition of adipogenesis by Wnt signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef]
- Moldes, M.; Zuo, Y.; Morrison, R.F.; Silva, D.; Park, B.-H.; Liu, J.; Farmer, S.R. Peroxisome-proliferator-activated receptor gamma suppresses Wnt/beta-catenin signalling during adipogenesis. Biochem. J. 2003, 376 Pt 3, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Vrechi, T.A.M.; Leão, A.H.F.F.; Morais, I.B.M.; Abílio, V.C.; Zuardi, A.W.; Hallak, J.E.C.; Crippa, J.A.; Bincoletto, C.; Ureshino, R.P.; Smaili, S.S.; et al. Cannabidiol induces autophagy via ERK1/2 activation in neural cells. Sci. Rep. 2021, 11, 5434. [Google Scholar] [CrossRef]
- Heijmans, J.; Jeude, J.F.v.L.d.; Koo, B.-K.; Rosekrans, S.L.; Wielenga, M.C.; van de Wetering, M.; Ferrante, M.; Lee, A.S.; Onderwater, J.J.; Paton, J.C.; et al. ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Rep. 2013, 3, 1128–1139. [Google Scholar] [CrossRef] [Green Version]
- Muncan, V.; Sansom, O.J.; Tertoolen, L.; Phesse, T.J.; Begthel, H.; Sancho, E.; Cole, A.M.; Gregorieff, A.; de Alboran, I.M.; Clevers, H.; et al. Rapid loss of intestinal crypts upon conditional deletion of the Wnt/Tcf-4 target gene c-Myc. Mol. Cell. Biol. 2006, 26, 8418–8426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, B.J.; Smit, W.L.; Koelink, P.J.; Westendorp, B.F.; de Boer, R.J.; van der Meer, J.H.M.; Vermeulen, J.L.M.; Paton, J.C.; Paton, A.W.; Qin, J.; et al. Endoplasmic reticulum stress regulates the intestinal stem cell state through CtBP2. Sci. Rep. 2021, 11, 9892. [Google Scholar] [CrossRef] [PubMed]
- Folmes, C.D.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Laezza, C.; D’alessandro, A.; Paladino, S.; Malfitano, A.M.; Proto, M.C.; Gazzerro, P.; Pisanti, S.; Santoro, A.; Ciaglia, E.; Bifulco, M. Anandamide inhibits the Wnt/β-catenin signalling pathway in human breast cancer MDA MB 231 cells. Eur. J. Cancer 2012, 48, 3112–3122. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, B.; Holovac, K.; Schuebel, K.; Mukhopadhyay, P.; Cinar, R.; Iyer, S.; Marietta, C.; Goldman, D.; Kunos, G. The endocannabinoid system promotes hepatocyte progenitor cell proliferation and maturation by modulating cellular energetics. Cell Death Discov. 2023, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Nalli, Y.; Dar, M.S.; Bano, N.; Rasool, J.U.; Sarkar, A.R.; Banday, J.; Bhat, A.Q.; Rafia, B.; Vishwakarma, R.A.; Ali, A. Analyzing the role of cannabinoids as modulators of Wnt/β-catenin signaling pathway for their use in the management of neuropathic pain. Bioorganic Med. Chem. Lett. 2019, 29, 1043–1046. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Podinić, T.; Werstuck, G.; Raha, S. The Implications of Cannabinoid-Induced Metabolic Dysregulation for Cellular Differentiation and Growth. Int. J. Mol. Sci. 2023, 24, 11003. https://doi.org/10.3390/ijms241311003
Podinić T, Werstuck G, Raha S. The Implications of Cannabinoid-Induced Metabolic Dysregulation for Cellular Differentiation and Growth. International Journal of Molecular Sciences. 2023; 24(13):11003. https://doi.org/10.3390/ijms241311003
Chicago/Turabian StylePodinić, Tina, Geoff Werstuck, and Sandeep Raha. 2023. "The Implications of Cannabinoid-Induced Metabolic Dysregulation for Cellular Differentiation and Growth" International Journal of Molecular Sciences 24, no. 13: 11003. https://doi.org/10.3390/ijms241311003
APA StylePodinić, T., Werstuck, G., & Raha, S. (2023). The Implications of Cannabinoid-Induced Metabolic Dysregulation for Cellular Differentiation and Growth. International Journal of Molecular Sciences, 24(13), 11003. https://doi.org/10.3390/ijms241311003