
Torpor-like Hypothermia Induced by A1 Adenosine Receptor Agonist: A Novel Approach to Protect against Neuroinflammation
 ,
,
Abstract
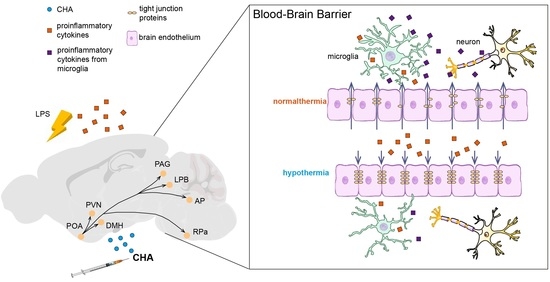
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The (A1AR) Agonist N6-cyclohexyladenosine (CHA) Induces a Stable Torpor State upon LPS Infection in Mice
2.2. CHA Protects against LPS-Induced Neuroinflammation and Microgliosis
2.3. CHA Prevents the LPS-Induced Blood–Brain Barrier Disruption
2.4. CHA Induces Torpor through POA-PVN-DMH-AP Neural Circuitry
2.5. CHA-Induced Hypothermia Mitigates Inflammation and Oxidative Stress in Endothelial Cells
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Torpor Induction and Experimental Groups
4.3. Core Temperature Measurement
4.4. Assessment of Blood–Brain Barrier Permeability
4.5. Cell Culture
4.6. Real-Time Quantitative PCR
4.7. Western Blotting
4.8. Immunohistochemistry and Immunofluorescence
4.9. Image Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Polderman, K.H. Induced hypothermia and fever control for prevention and treatment of neurological injuries. Lancet 2008, 371, 1955–1969. [Google Scholar] [CrossRef] [PubMed]
- Polderman, K.H. Mechanisms of action, physiological effects, and complications of hypothermia. Crit. Care Med. 2009, 37 (Suppl. S7), S186–S202. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.-J.; Cassiere, H.; Bocchieri, K.; DeRosa, S.; Yar, S.; Hartman, A. Hypermetabolism in critically ill patients with COVID-19 and the effects of hypothermia: A case series. Metab. Open 2020, 7, 100046. [Google Scholar] [CrossRef] [PubMed]
- Cruces, P.; Cores, C.; Casanova, D.; Pizarro, F.; Díaz, F. Successful use of mild therapeutic hypothermia as compassionate treatment for severe refractory hypoxemia in COVID-19. J. Crit. Care 2021, 63, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Yenari, M.A.; Han, H.S. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat. Rev. Neurosci. 2012, 13, 267–278. [Google Scholar] [CrossRef]
- Wu, T.-C.; Grotta, J.C. Hypothermia for acute ischaemic stroke. Lancet Neurol. 2013, 12, 275–284. [Google Scholar] [CrossRef]
- Polderman, K.H.; Herold, I. Therapeutic hypothermia and controlled normothermia in the intensive care unit: Practical considerations, side effects, and cooling methods*. Crit. Care Med. 2009, 37, 1101–1120. [Google Scholar] [CrossRef]
- Geiser, F. Hibernation. Curr. Biol. 2013, 23, R188–R193. [Google Scholar] [CrossRef] [Green Version]
- Heldmaier, G.; Ruf, T. Body temperature and metabolic rate during natural hypothermia in endotherms. J. Comp. Physiol. B 2004, 162, 696–706. [Google Scholar] [CrossRef]
- Ruf, T.; Geiser, F. Daily torpor and hibernation in birds and mammals. Biol. Rev. 2015, 90, 891–926. [Google Scholar] [CrossRef] [Green Version]
- Geiser, F. Metabolic Rate and Body Temperature Reduction During Hibernation and Daily Torpor. Annu. Rev. Physiol. 2004, 66, 239–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, J.W.; Scott, I.M. Daily torpor in the laboratory mouse, Mus musculus var. albino. Physiol. Zool. 1979, 52, 205–218. [Google Scholar] [CrossRef]
- Takahashi, T.M.; Sunagawa, G.A.; Soya, S.; Abe, M.; Sakurai, K.; Ishikawa, K.; Yanagisawa, M.; Hama, H.; Hasegawa, E.; Miyawaki, A.; et al. A discrete neuronal circuit induces a hibernation-like state in rodents. Nature 2020, 583, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Hrvatin, S.; Sun, S.; Wilcox, O.F.; Yao, H.; Lavin-Peter, A.J.; Cicconet, M.; Assad, E.G.; Palmer, M.E.; Aronson, S.; Banks, A.S.; et al. Neurons that regulate mouse torpor. Nature 2020, 583, 115–121. [Google Scholar] [CrossRef]
- Bouma, H.R.; Verhaag, E.M.; Otis, J.P.; Heldmaier, G.; Swoap, S.J.; Strijkstra, A.M.; Henning, R.H.; Carey, H.V. Induction of torpor: Mimicking natural metabolic suppression for biomedical applications. J. Cell. Physiol. 2012, 227, 1285–1290. [Google Scholar] [CrossRef]
- Tupone, D.; Madden, C.J.; Morrison, S.F. Central activation of the A1 adenosine receptor (A1AR) induces a hypothermic, torpor-like state in the rat. J. Neurosci. 2013, 33, 14512–14525. [Google Scholar] [CrossRef] [Green Version]
- Carlin, J.L.; Jain, S.; Gizewski, E.; Wan, T.C.; Tosh, D.K.; Xiao, C.; Auchampach, J.A.; Jacobson, K.A.; Gavrilova, O.; Reitman, M.L. Hypothermia in mouse is caused by adenosine A1 and A3 receptor agonists and AMP via three distinct mechanisms. Neuropharmacology 2017, 114, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Li, J.; Wang, R.-m.; Chen, Z.; Zhou, F. Moderate systemic therapeutic hypothermia is insufficient to protect blood-spinal cord barrier in spinal cord injury. Front. Neurol. 2022, 13, 1041099. [Google Scholar] [CrossRef]
- Kadir, R.R.A.; Alwjwaj, M.; McCarthy, Z.Z.S.; Bayraktutan, U. Therapeutic hypothermia augments the restorative effects of PKC-β and Nox2 inhibition on an in vitro model of human blood–brain barrier. Metab. Brain Dis. 2021, 36, 1817–1832. [Google Scholar] [CrossRef]
- Park, Y.H.; Lee, Y.M.; Kim, D.S.; Park, J.; Suk, K.; Kim, J.K.; Han, H.S. Hypothermia enhances induction of protective protein metallothionein under ischemia. J. Neuroinflammation 2013, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Shimizu, K.; Kokubu, Y.; Nishijima, M.; Takeda, S.; Ogura, H.; Kawabata, K. Effect of heat stress on blood-brain barrier integrity in iPS cell-derived microvascular endothelial cell models. PLoS ONE 2019, 14, e0222113. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.J.; Zhang, Z.Y.; Fan, B.; Li, G.Y. Neuroprotection by Therapeutic Hypothermia. Front. Neurosci. 2019, 13, 586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterhout, J.A.; Kapoor, V.; Eichhorn, S.W.; Vaughn, E.; Moore, J.D.; Liu, D.; Lee, D.; DeNardo, L.A.; Luo, L.; Zhuang, X.; et al. A preoptic neuronal population controls fever and appetite during sickness. Nature 2022, 606, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazziotta, C.; Rotondo, J.C.; Lanzillotti, C.; Campione, G.; Martini, F.; Tognon, M. Cancer biology and molecular genetics of A(3) adenosine receptor. Oncogene 2022, 41, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Sebastião, A.M.; Ribeiro, J.A. Adenosine A2 receptor-mediated excitatory actions on the nervous system. Prog. Neurobiol. 1996, 48, 167–189. [Google Scholar] [CrossRef]
- Colella, M.; Zinni, M.; Pansiot, J.; Cassanello, M.; Mairesse, J.; Ramenghi, L.; Baud, O. Modulation of Microglial Activation by Adenosine A2a Receptor in Animal Models of Perinatal Brain Injury. Front. Neurol. 2018, 9, 605. [Google Scholar] [CrossRef]
- Kashio, M.; Sokabe, T.; Shintaku, K.; Uematsu, T.; Fukuta, N.; Kobayashi, N.; Mori, Y.; Tominaga, M. Redox signal-mediated sensitization of transient receptor potential melastatin 2 (TRPM2) to temperature affects macrophage functions. Proc. Natl. Acad. Sci. USA 2012, 109, 6745–6750. [Google Scholar] [CrossRef]
- Socha, M.J.; Hakim, C.H.; Jackson, W.F.; Segal, S.S. Temperature effects on morphological integrity and Ca2⁺ signaling in freshly isolated murine feed artery endothelial cell tubes. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H773–H783. [Google Scholar] [CrossRef] [Green Version]
- Fedinec, A.L.; Liu, J.; Zhang, R.; Harsono, M.; Pourcyrous, M.; Parfenova, H. The cold receptor TRPM8 activation leads to attenuation of endothelium-dependent cerebral vascular functions during head cooling. J. Cereb. Blood Flow Metab. 2021, 41, 2897–2906. [Google Scholar] [CrossRef]
- Layland, J.; Carrick, D.; Lee, M.; Oldroyd, K.; Berry, C. Adenosine: Physiology, pharmacology, and clinical applications. JACC Cardiovasc. Interv. 2014, 7, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Calkins, H. The 2019 ESC Guidelines for the Management of Patients with Supraventricular Tachycardia. Eur. Heart J. 2019, 40, 3812–3813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, C.; Liu, N.; Jacobson, K.A.; Gavrilova, O.; Reitman, M.L. Physiology and effects of nucleosides in mice lacking all four adenosine receptors. PLoS Biol. 2019, 17, e3000161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlin, J.L.; Jain, S.; Duroux, R.; Suresh, R.R.; Xiao, C.; Auchampach, J.A.; Jacobson, K.A.; Gavrilova, O.; Reitman, M.L. Activation of adenosine A2A or A2B receptors causes hypothermia in mice. Neuropharmacology 2018, 139, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, A.; De Simone, R.; Ajmone-Cat, M.A.; Minghetti, L.; Popoli, P. Adenosine Receptors and Neuroinflammation. In The Adenosine Receptors; Borea, P.A., Varani, K., Gessi, S., Merighi, S., Vincenzi, F., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 217–237. [Google Scholar] [CrossRef]
- Pasquini, S.; Contri, C.; Borea, P.A.; Vincenzi, F.; Varani, K. Adenosine and Inflammation: Here, There and Everywhere. Int. J. Mol. Sci. 2021, 22, 7685. [Google Scholar] [CrossRef]
- Ingwersen, J.; Wingerath, B.; Graf, J.; Lepka, K.; Hofrichter, M.; Schröter, F.; Wedekind, F.; Bauer, A.; Schrader, J.; Hartung, H.P.; et al. Dual roles of the adenosine A2a receptor in autoimmune neuroinflammation. J. Neuroinflammation 2016, 13, 48. [Google Scholar] [CrossRef] [Green Version]
- Martí Navia, A.; Dal Ben, D.; Lambertucci, C.; Spinaci, A.; Volpini, R.; Marques-Morgado, I.; Coelho, J.E.; Lopes, L.V.; Marucci, G.; Buccioni, M. Adenosine Receptors as Neuroinflammation Modulators: Role of A1 Agonists and A2A Antagonists. Cells 2020, 9, 1739. [Google Scholar] [CrossRef]
- Song, K.; Wang, H.; Kamm, G.B.; Pohle, J.; Reis, F.C.; Heppenstall, P.; Wende, H.; Siemens, J. The TRPM2 channel is a hypothalamic heat sensor that limits fever and can drive hypothermia. Science 2016, 353, 1393–1398. [Google Scholar] [CrossRef]
- Kollmar, R.; Schellinger, P.D.; Steigleder, T.; Köhrmann, M.; Schwab, S. Ice-cold saline for the induction of mild hypothermia in patients with acute ischemic stroke: A pilot study. Stroke 2009, 40, 1907–1909. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yuan, J.; Field, R.L.; Ye, D.; Hu, Z.; Xu, K.; Xu, L.; Gong, Y.; Yue, Y.; Kravitz, A.V.; et al. Induction of a torpor-like hypothermic and hypometabolic state in rodents by ultrasound. Nat. Metab. 2023, 5, 789–803. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, Y.; Zhou, R.; Li, Y.; Gao, Y.; Tu, D.; Wilson, B.; Song, S.; Feng, J.; Hong, J.S.; et al. A novel role of NLRP3-generated IL-1β in the acute-chronic transition of peripheral lipopolysaccharide-elicited neuroinflammation: Implications for sepsis-associated neurodegeneration. J. Neuroinflammation 2020, 17, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, M.Q.; Wang, Y.J.; Fu, K.; Jiao, H.; Sun, J.; Gao, Y. Orexin receptor type 2 agonism inhibits thermogenesis in brown adipose tissue by attenuating afferent innervation. J. Biomed. Res. 2022, 36, 195–207. [Google Scholar] [CrossRef]
- Jangula, A.; Murphy, E.J. Lipopolysaccharide-induced blood brain barrier permeability is enhanced by alpha-synuclein expression. Neurosci. Lett. 2013, 551, 23–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, C.; Lv, J.; Zhu, Z.; Cong, W.; Bian, H.; Zhang, C.; Gu, R.; Chen, D.; Tan, X.; Su, L.; et al. Regulation of microglia related neuroinflammation contributes to the protective effect of Gelsevirine on ischemic stroke. Front. Immunol. 2023, 14, 1164278. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Vidal-Itriago, A.; Kalsbeek, M.J.; Layritz, C.; García-Cáceres, C.; Tom, R.Z.; Eichmann, T.O.; Vaz, F.M.; Houtkooper, R.H.; van der Wel, N.; et al. Lipoprotein Lipase Maintains Microglial Innate Immunity in Obesity. Cell Rep. 2017, 20, 3034–3042. [Google Scholar] [CrossRef] [Green Version]
- McHugh, M.L. Multiple comparison analysis testing in ANOVA. Biochem. Med. 2011, 21, 203–209. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, K.; Hui, C.; Wang, X.; Ji, T.; Li, X.; Sun, R.; Xing, C.; Fan, X.; Gao, Y.; Su, L. Torpor-like Hypothermia Induced by A1 Adenosine Receptor Agonist: A Novel Approach to Protect against Neuroinflammation. Int. J. Mol. Sci. 2023, 24, 11036. https://doi.org/10.3390/ijms241311036
Fu K, Hui C, Wang X, Ji T, Li X, Sun R, Xing C, Fan X, Gao Y, Su L. Torpor-like Hypothermia Induced by A1 Adenosine Receptor Agonist: A Novel Approach to Protect against Neuroinflammation. International Journal of Molecular Sciences. 2023; 24(13):11036. https://doi.org/10.3390/ijms241311036
Chicago/Turabian StyleFu, Kang, Chunlei Hui, Xinyuan Wang, Tingting Ji, Xiuqing Li, Rui Sun, Chunlei Xing, Xi Fan, Yuanqing Gao, and Li Su. 2023. "Torpor-like Hypothermia Induced by A1 Adenosine Receptor Agonist: A Novel Approach to Protect against Neuroinflammation" International Journal of Molecular Sciences 24, no. 13: 11036. https://doi.org/10.3390/ijms241311036
APA StyleFu, K., Hui, C., Wang, X., Ji, T., Li, X., Sun, R., Xing, C., Fan, X., Gao, Y., & Su, L. (2023). Torpor-like Hypothermia Induced by A1 Adenosine Receptor Agonist: A Novel Approach to Protect against Neuroinflammation. International Journal of Molecular Sciences, 24(13), 11036. https://doi.org/10.3390/ijms241311036