
Opioids Alleviate Oxidative Stress via the Nrf2/HO-1 Pathway in LPS-Stimulated Microglia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Opioid Receptor Agonists Do Not Affect Microglial Viability and Reduce LPS Cytotoxicity
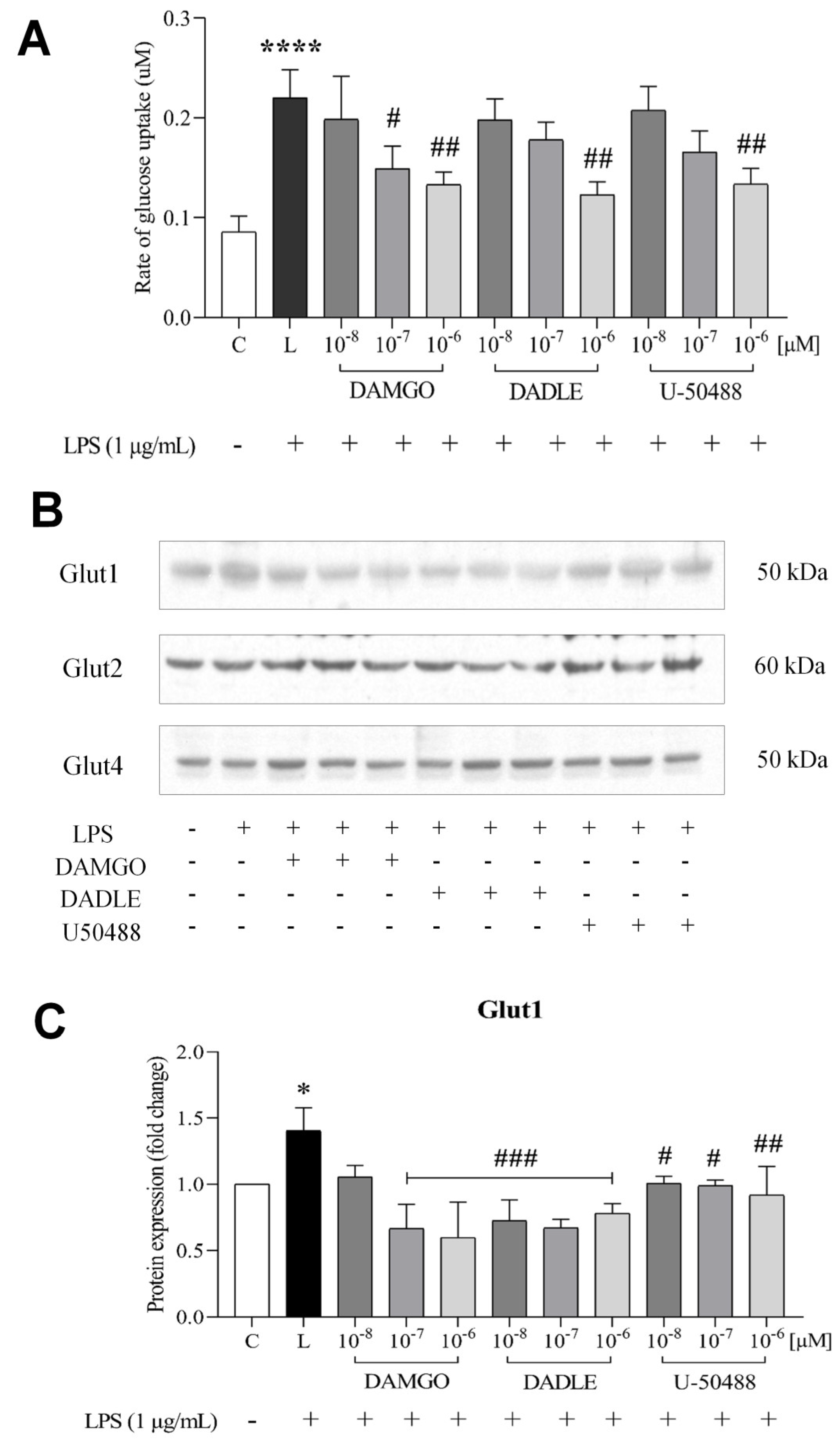
2.2. Opioid Receptor Agonists Prevent the LPS-Induced Increase in Glucose Uptake and Downregulate GLUTs
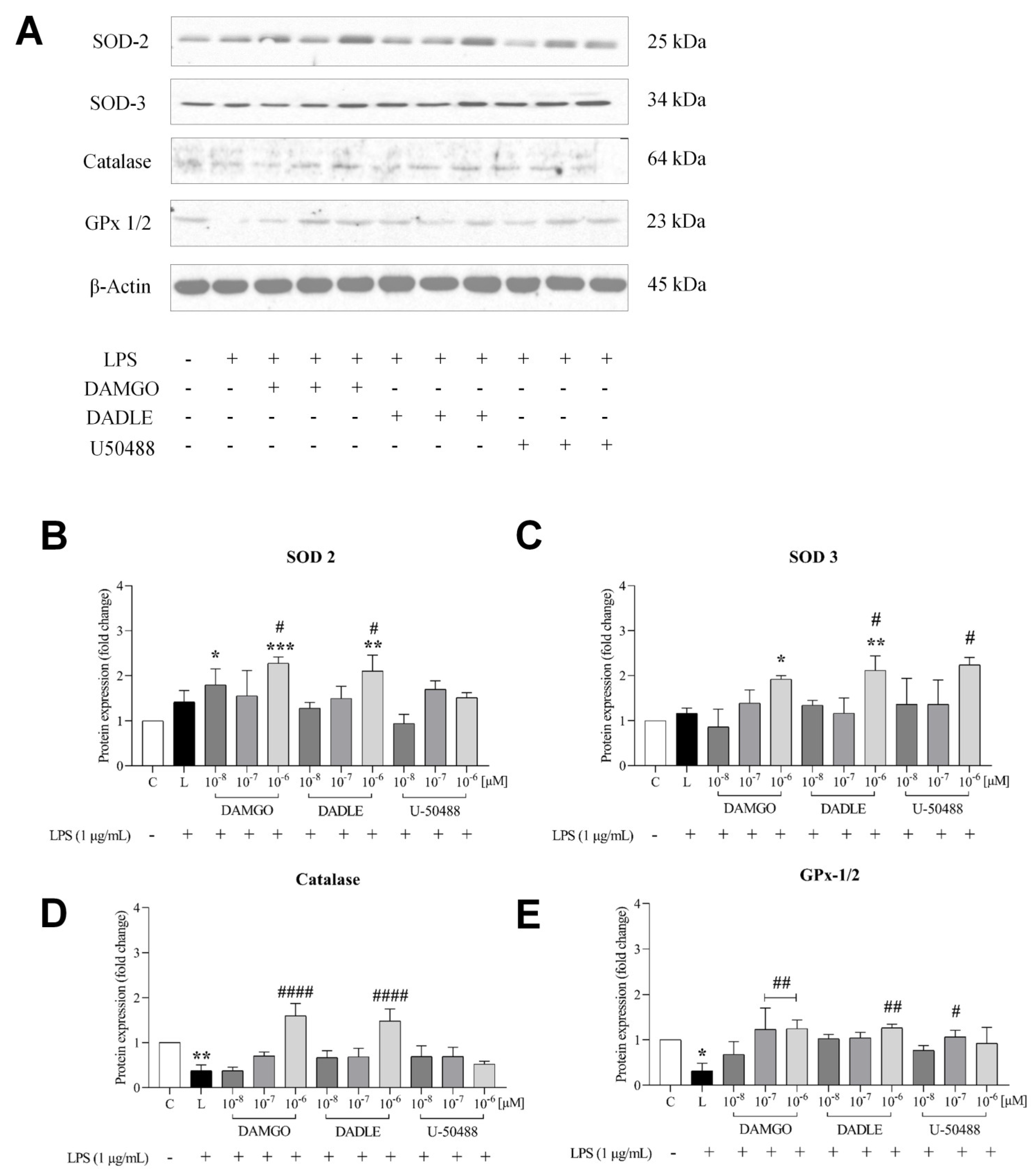
2.3. Opioid Receptor Agonists Inhibit the LPS-Induced Inflammatory Response by Increasing Antioxidant Capacity, Preventing the Increase in NADPH Production, and Promoting GSH Upregulation
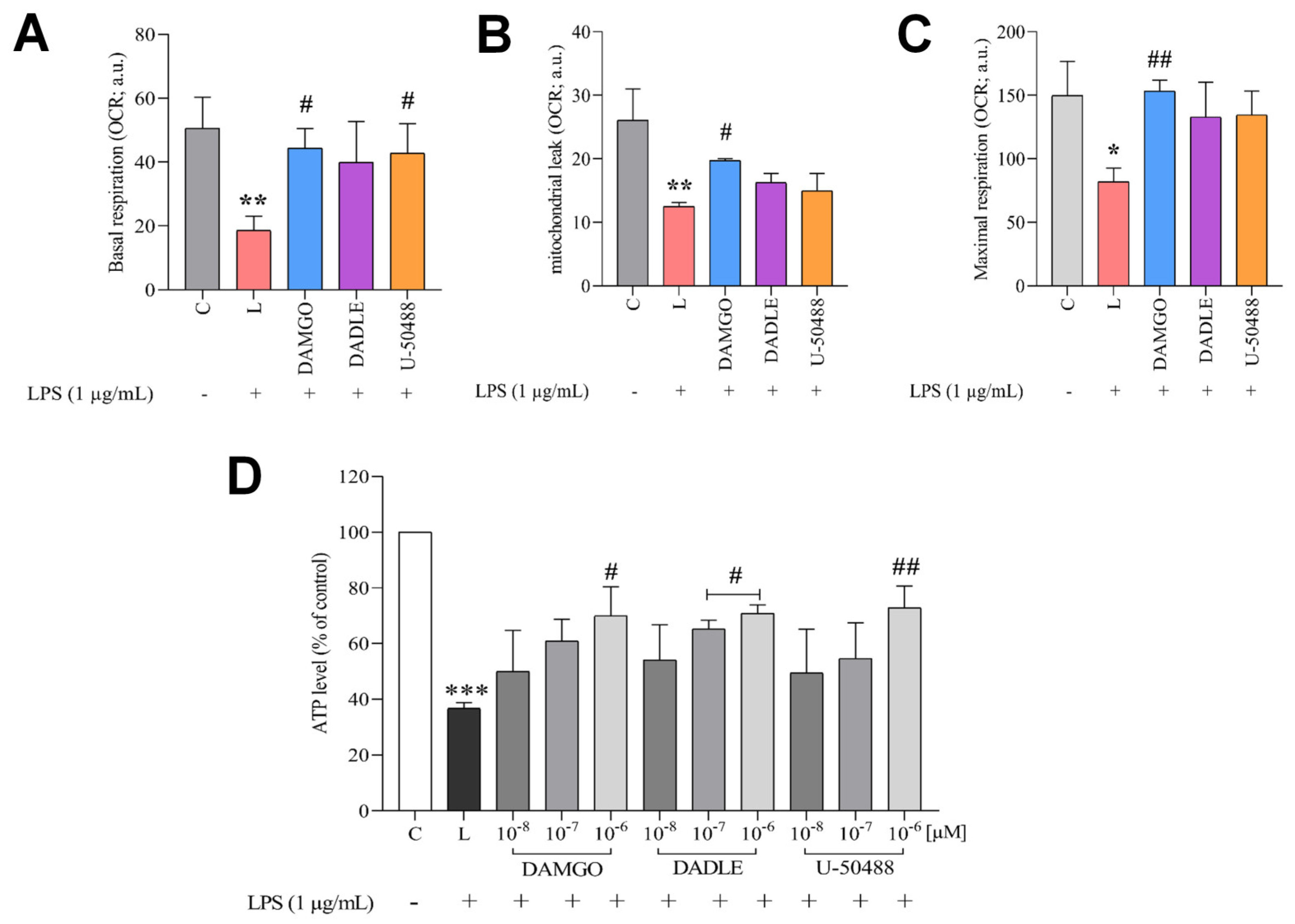
2.4. Opioid Receptor Agonists Reverse LPS-Induced Dysregulation of Cellular Redox Balance and Bioenergetics
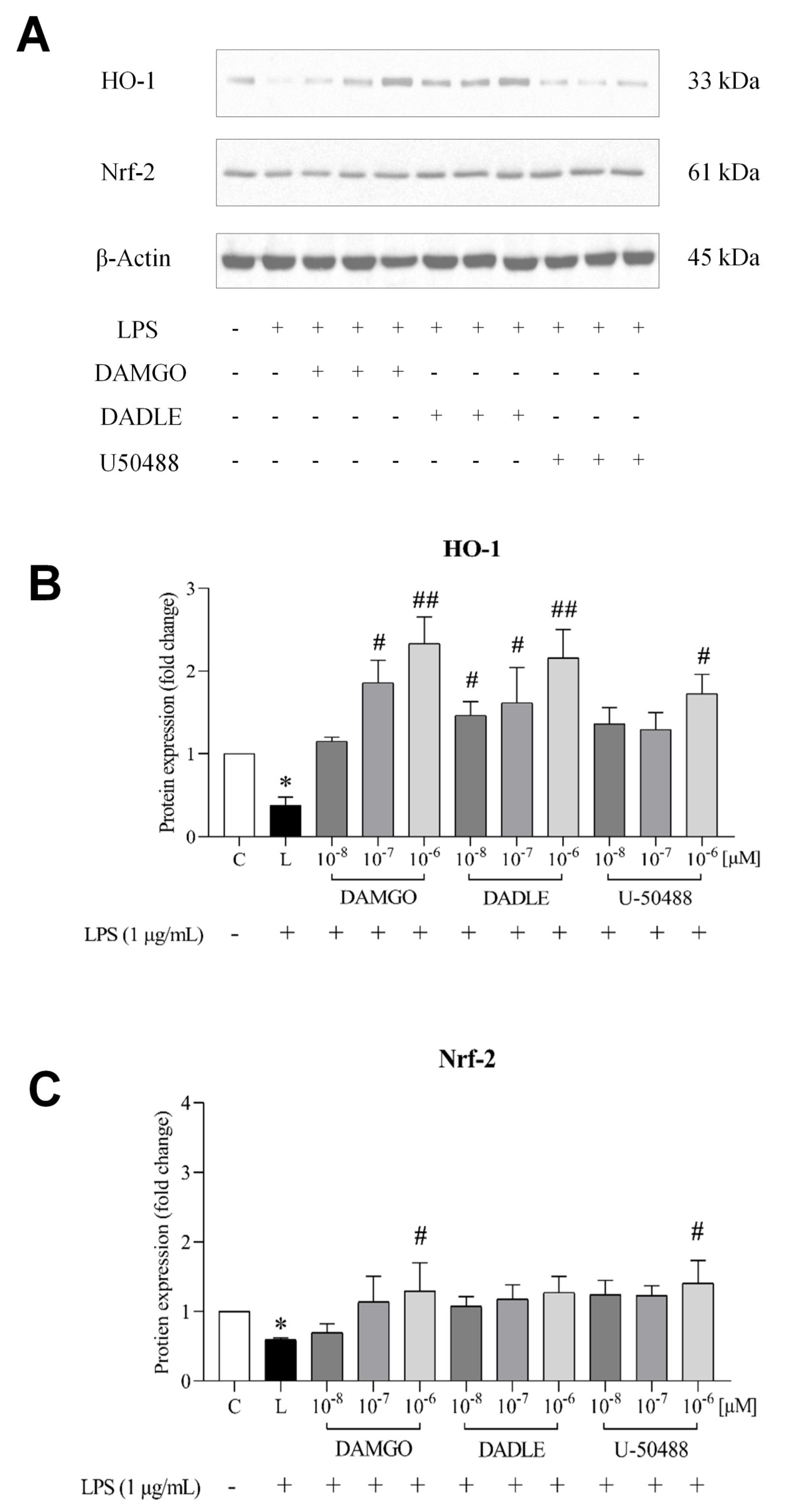
2.5. Opioid Receptor Agonists Activate the Nrf2/HO-1 Pathway in LPS-Treated Microglia
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Treatment
4.3. Cell Viability Assay
4.4. Cytotoxicity Assay
4.5. Measurement of Intracellular ATP Content
4.6. Glucose Uptake Assay
4.7. Reduced Glutathione Assay
4.8. NADPH Assay
4.9. Mitochondrial Superoxide Assay
4.10. Intracellular ROS Measurement
4.11. High-Resolution Respirometry
4.12. Western Blot Analysis
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.J.; Flood, P.M. Oxidative Stress and Microglial Cells in Parkinson’s Disease. Mediat. Inflamm. 2012, 2012, 401264. [Google Scholar] [CrossRef] [Green Version]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated CD4+T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [Green Version]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; De Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell. Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Huang, Z.; Sun, X.; Zhu, X.; Zhou, L.; Li, M.; Cheng, B.; Liu, X.; He, C. Microglia Polarization with M1/M2 Phenotype Changes in Rd1 Mouse Model of Retinal Degeneration. Front. Neuroanat. 2017, 11, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, L.; Lee, J.; Yumnam, S.; Ji, E.; Kim, S. Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-ΚB Inhibition and Nrf2/HO-1 Activation. Cells 2019, 8, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milosevic, K.; Stevanovic, I.; Bozic, I.D.; Milosevic, A.; Janjic, M.M.; Laketa, D.; Bjelobaba, I.; Lavrnja, I.; Savic, D. Agmatine Mitigates Inflammation-Related Oxidative Stress in BV-2 Cells by Inducing a Pre-Adaptive Response. Int. J. Mol. Sci. 2022, 23, 3561. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Tossetta, G.; Fantone, S.; Piani, F.; Crescimanno, C.; Ciavattini, A.; Giannubilo, S.R.; Marzioni, D. Modulation of NRF2/KEAP1 Signaling in Preeclampsia. Cells 2023, 12, 1545. [Google Scholar] [CrossRef] [PubMed]
- Tossetta, G.; Marzioni, D. Targeting the NRF2/KEAP1 Pathway in Cervical and Endometrial Cancers. Eur. J. Pharmacol. 2023, 941, 175503. [Google Scholar] [CrossRef] [PubMed]
- Bukke, V.N.; Moola, A.; Serviddio, G.; Vendemiale, G.; Bellanti, F. Nuclear Factor Erythroid 2-Related Factor 2-Mediated Signaling and Metabolic Associated Fatty Liver Disease. World J. Gastroenterol. 2022, 28, 6909–6921. [Google Scholar] [CrossRef]
- Khan, N.M.; Sandur, S.K.; Checker, R.; Sharma, D.; Poduval, T.B.; Sainis, K.B. Pro-Oxidants Ameliorate Radiation-Induced Apoptosis through Activation of the Calcium–ERK1/2–Nrf2 Pathway. Free Radic. Biol. Med. 2011, 51, 115–128. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 System in Development, Oxidative Stress Response and Diseases: An Evolutionarily Conserved Mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Yang, J.; Yan, K.; Guo, J. Ginsenoside Rk1 Protects Human Melanocytes from H2O2-induced Oxidative Injury via Regulation of the PI3K/AKT/Nrf2/HO-1 Pathway. Mol. Med. Rep. 2021, 24, 821. [Google Scholar] [CrossRef]
- Nair, S.; Sobotka, K.S.; Joshi, P.; Gressens, P.; Fleiss, B.; Thornton, C.; Mallard, C.; Hagberg, H. Lipopolysaccharide-induced Alteration of Mitochondrial Morphology Induces a Metabolic Shift in Microglia Modulating the Inflammatory Response in Vitro and in Vivo. Glia 2019, 67, 1047–1061. [Google Scholar] [CrossRef] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 Polarization and Metabolic States: Microglia Bioenergetics with Acute Polarization. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Liang, X.; Liu, X.; Li, Y.; Wang, Y.; Kong, L.; Tang, M. Induction of Ferroptosis in Response to Graphene Quantum Dots through Mitochondrial Oxidative Stress in Microglia. Part. Fibre Toxicol. 2020, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Chao, C.C.; Hegg, C.C.; Thayer, S.; Peterson, P.K. Morphine Inhibits Human Microglial Cell Production of, and Migration towards, RANTES. J. Psychopharmacol. 2000, 14, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Mali, A.S.; Novotny, J. Opioid Receptor Activation Suppresses the Neuroinflammatory Response by Promoting Microglial M2 Polarization. Mol. Cell. Neurosci. 2022, 121, 103744. [Google Scholar] [CrossRef] [PubMed]
- Burlacu, C.-C.; Neag, M.-A.; Mitre, A.-O.; Sirbu, A.-C.; Badulescu, A.-V.; Buzoianu, A.-D. The Role of MiRNAs in Dexmedetomidine’s Neuroprotective Effects against Brain Disorders. Int. J. Mol. Sci. 2022, 23, 5452. [Google Scholar] [CrossRef]
- Cai, B.; Zhong, L.; Liu, Y.; Xu, Q.; Chen, T. δ-Opioid Receptor Activation Inhibits Ferroptosis by Activating the Nrf2 Pathway in MPTP-Induced Parkinson Disease Models. Evid.-Based Complement. Altern. Med. 2023, 2023, 4130937. [Google Scholar] [CrossRef]
- Chao, D.; Xia, Y. Ionic Storm in Hypoxic/Ischemic Stress: Can Opioid Receptors Subside It? Prog. Neurobiol. 2010, 90, 439–470. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Sandhu, H.K.; Yang, Y.; Hua, F.; Belser, N.; Kim, D.H.; Xia, Y. Neuroprotection against Hypoxia/Ischemia: δ-Opioid Receptor-Mediated Cellular/Molecular Events. Cell. Mol. Life Sci. 2013, 70, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- dos-Santos-Pereira, M.; Guimarães, F.S.; Del-Bel, E.; Raisman-Vozari, R.; Michel, P.P. Cannabidiol Prevents LPS-induced Microglial Inflammation by Inhibiting ROS/NF-κB-dependent Signaling and Glucose Consumption. Glia 2020, 68, 561–573. [Google Scholar] [CrossRef]
- Wang, L.; Pavlou, S.; Du, X.; Bhuckory, M.; Xu, H.; Chen, M. Glucose Transporter 1 Critically Controls Microglial Activation through Facilitating Glycolysis. Mol. Neurodegener. 2019, 14, 2. [Google Scholar] [CrossRef] [Green Version]
- Xiang, X.; Wind, K.; Wiedemann, T.; Blume, T.; Shi, Y.; Briel, N.; Beyer, L.; Biechele, G.; Eckenweber, F.; Zatcepin, A.; et al. Microglial Activation States Drive Glucose Uptake and FDG-PET Alterations in Neurodegenerative Diseases. Sci. Transl. Med. 2021, 13, eabe5640. [Google Scholar] [CrossRef]
- Fu, D.; Liu, H.; Li, S.; Chen, L.; Yao, J. Antioxidative and Antiapoptotic Effects of Delta-Opioid Peptide [D-Ala2, D-Leu5] Enkephalin on Spinal Cord Ischemia-Reperfusion Injury in Rabbits. Front. Neurosci. 2017, 11, 603. [Google Scholar] [CrossRef]
- Reymond, S.; Vujić, T.; Schvartz, D.; Sanchez, J.-C. Morphine-Induced Modulation of Nrf2-Antioxidant Response Element Signaling Pathway in Primary Human Brain Microvascular Endothelial Cells. Sci. Rep. 2022, 12, 4588. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Abiko, Y.; Yamano, S.; Toriba, A.; Matsusue, K.; Kumagai, Y. Activation of the Keap1/Nrf2 Pathway as an Adaptive Response to an Electrophilic Metabolite of Morphine. Biol. Pharm. Bull. 2023, 46, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Gwak, M.-S.; Li, L.; Zuo, Z. Morphine Preconditioning Reduces Lipopolysaccharide and Interferon-γ-Induced Mouse Microglial Cell Injury via Δ1 Opioid Receptor Activation. Neuroscience 2010, 167, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrabalova, J.; Karlovska, I.; Hejnova, L.; Novotny, J. Protective Effect of Morphine Against the Oxidant-Induced Injury in H9c2 Cells. Cardiovasc. Toxicol. 2018, 18, 374–385. [Google Scholar] [CrossRef]
- Sasaki, A.; Yamaguchi, H.; Horikoshi, Y.; Tanaka, G.; Nakazato, Y. Expression of Glucose Transporter 5 by Microglia in Human Gliomas: GLUT5 in Human Gliomas. Neuropathol. Appl. Neurobiol. 2004, 30, 447–455. [Google Scholar] [CrossRef]
- Yang, T.-T.; Liu, I.-M.; Wu, H.-T.; Cheng, J.-T. Mediation of Protein Kinase C Zeta in μ-Opioid Receptor Activation for Increase of Glucose Uptake into Cultured Myoblast C2C12 Cells. Neurosci. Lett. 2009, 465, 177–180. [Google Scholar] [CrossRef]
- Huang, J.-C.; Yue, Z.-P.; Yu, H.-F.; Yang, Z.-Q.; Wang, Y.-S.; Guo, B. TAZ Ameliorates the Microglia-Mediated Inflammatory Response via the Nrf2-ROS-NF-ΚB Pathway. Mol. Ther.-Nucleic Acids 2022, 28, 435–449. [Google Scholar] [CrossRef]
- Huang, S.C.-C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-Intrinsic Lysosomal Lipolysis Is Essential for Alternative Activation of Macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef]
- Pavlou, S.; Wang, L.; Xu, H.; Chen, M. Higher Phagocytic Activity of Thioglycollate-Elicited Peritoneal Macrophages Is Related to Metabolic Status of the Cells. J. Inflamm. 2017, 14, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhen, Y.; Wang, G.; Liu, B. Deconvoluting the Complexity of Reactive Oxygen Species (ROS) in Neurodegenerative Diseases. Front. Neuroanat. 2022, 16, 910427. [Google Scholar] [CrossRef]
- Jang, J.; Hong, A.; Chung, Y.; Jin, B. Interleukin-4 Aggravates LPS-Induced Striatal Neurodegeneration In Vivo via Oxidative Stress and Polarization of Microglia/Macrophages. Int. J. Mol. Sci. 2022, 23, 571. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Ali, T.; Kim, M.W.; Jo, M.H.; Jo, M.G.; Badshah, H.; Kim, M.O. Anthocyanins Protect against LPS-Induced Oxidative Stress-Mediated Neuroinflammation and Neurodegeneration in the Adult Mouse Cortex. Neurochem. Int. 2016, 100, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Jiang, L.; Oyarzabal, E.A.; Wilson, B.; Li, Z.; Shih, Y.-Y.I.; Wang, Q.; Hong, J.-S. Loss of Brain Norepinephrine Elicits Neuroinflammation-Mediated Oxidative Injury and Selective Caudo-Rostral Neurodegeneration. Mol. Neurobiol. 2019, 56, 2653–2669. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. Oxidative Stress and Neurodegeneration: Where Are We Now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef]
- Pei, J.; Pan, X.; Wei, G.; Hua, Y. Research Progress of Glutathione Peroxidase Family (GPX) in Redoxidation. Front. Pharmacol. 2023, 14, 1147414. [Google Scholar] [CrossRef]
- Chinta, S.J.; Rajagopalan, S.; Butterfield, D.A.; Andersen, J.K. In Vitro and in Vivo Neuroprotection by γ-Glutamylcysteine Ethyl Ester against MPTP: Relevance to the Role of Glutathione in Parkinson’s Disease. Neurosci. Lett. 2006, 402, 137–141. [Google Scholar] [CrossRef]
- Mendiola, A.S.; Ryu, J.K.; Bardehle, S.; Meyer-Franke, A.; Ang, K.K.-H.; Wilson, C.; Baeten, K.M.; Hanspers, K.; Merlini, M.; Thomas, S.; et al. Transcriptional Profiling and Therapeutic Targeting of Oxidative Stress in Neuroinflammation. Nat. Immunol. 2020, 21, 513–524. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Marzioni, D.; Mazzucchelli, R. Role of Natural and Synthetic Compounds in Modulating NRF2/KEAP1 Signaling Pathway in Prostate Cancer. Cancers 2023, 15, 3037. [Google Scholar] [CrossRef]
- Liu, T.; Sun, L.; Zhang, Y.; Wang, Y.; Zheng, J. Imbalanced GSH/ROS and Sequential Cell Death. J. Biochem. Mol. Toxicol. 2022, 36, e22942. [Google Scholar] [CrossRef]
- Yang, W.; Wang, Y.; Zhang, C.; Huang, Y.; Yu, J.; Shi, L.; Zhang, P.; Yin, Y.; Li, R.; Tao, K. Maresin1 Protect Against Ferroptosis-Induced Liver Injury Through ROS Inhibition and Nrf2/HO-1/GPX4 Activation. Front. Pharmacol. 2022, 13, 865689. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-ҚB Interplay in Cerebrovascular and Neurodegenerative Disorders: Molecular Mechanisms and Possible Therapeutic Approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 Pathway: Mechanisms of Activation and Dysregulation in Cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.M.; Haseeb, A.; Ansari, M.Y.; Devarapalli, P.; Haynie, S.; Haqqi, T.M. Wogonin, a Plant Derived Small Molecule, Exerts Potent Anti-Inflammatory and Chondroprotective Effects through the Activation of ROS/ERK/Nrf2 Signaling Pathways in Human Osteoarthritis Chondrocytes. Free Radic. Biol. Med. 2017, 106, 288–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.-C.; Ji, J.-A.; Jiang, Z.-Y.; You, Q.-D. The Keap1-Nrf2-ARE Pathway As a Potential Preventive and Therapeutic Target: An Update: THE KEAP1-NRF2-ARE PATHWAY. Med. Res. Rev. 2016, 36, 924–963. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1–Nrf2 System in Cancers: Stress Response and Anabolic Metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef] [Green Version]
- Uruno, A.; Yamamoto, M. The KEAP1-NRF2 System and Neurodegenerative Diseases. Antioxid. Redox Signal. 2023, 38, 974–988. [Google Scholar] [CrossRef]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the Heart of Oxidative Stress and Cardiac Protection. Physiol. Genom. 2018, 50, 77–97. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; Chen, X.-L.; Mackman, N.; Ogborne, R.M.; O’Connell, M.A. Lipopolysaccharide-Induced Heme Oxygenase-1 Expression in Human Monocytic Cells Is Mediated via Nrf2 and Protein Kinase C. J. Immunol. 2005, 175, 4408–4415. [Google Scholar] [CrossRef] [Green Version]
- Immenschuh, S.; Stritzke, J.; Iwahara, S.-I.; Ramadori, G. Up-regulation of heme-binding protein 23 (HBP23) gene expression by lipopolysaccharide is mediated via a nitric oxide-dependent signaling pathway in rat Kupffer cells. Hepatology 1999, 30, 118–127. [Google Scholar] [CrossRef]
- Lee, T.-S.; Tsai, H.-L.; Chau, L.-Y. Induction of Heme Oxygenase-1 Expression in Murine Macrophages Is Essential for the Anti-Inflammatory Effect of Low Dose 15-Deoxy-Δ12,14-Prostaglandin J2. J. Biol. Chem. 2003, 278, 19325–19330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Du, S.-H.; Yu, J.-B.; Zhang, Y.-F.; He, S.-M.; Dong, S.-A.; Zhang, Y.; Wu, L.-L.; Li, C.; Li, H.-B. Hydromorphone Protects against CO2 Pneumoperitoneum-Induced Lung Injury via Heme Oxygenase-1-Regulated Mitochondrial Dynamics. Oxidative Med. Cell. Longev. 2021, 2021, 9034376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J.; An, Z.; Qi, J. Hydromorphone Mitigates Cardiopulmonary Bypass-Induced Acute Lung Injury by Repressing Pyroptosis of Alveolar Macrophages. Shock 2023. Publish Ahead of Print. [Google Scholar] [CrossRef]
- Dai, H.; Wang, P.; Mao, H.; Mao, X.; Tan, S.; Chen, Z. Dynorphin Activation of Kappa Opioid Receptor Protects against Epilepsy and Seizure-Induced Brain Injury via PI3K/Akt/Nrf2/HO-1 Pathway. Cell Cycle 2019, 18, 226–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Yao, H.; Zhang, B.; Zhou, S.; Hou, P.; Chen, K. κ Opioid Receptor Agonist Inhibits Myocardial Injury in Heart Failure Rats through Activating Nrf2/HO-1 Pathway and Regulating Ca2+-SERCA2a. Oxidative Med. Cell. Longev. 2021, 2021, 7328437. [Google Scholar] [CrossRef]
- Kauffman, M.; Kauffman, M.; Traore, K.; Zhu, H.; Trush, M.; Jia, Z.; Li, Y. MitoSOX-Based Flow Cytometry for Detecting Mitochondrial ROS. React. Oxyg. Species 2016, 2, 361. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.; Moghimi, S.M. Determination of Polycation-Mediated Perturbation of Mitochondrial Respiration in Intact Cells by High-Resolution Respirometry (Oxygraph-2k, OROBOROS); Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1943, pp. 313–322. ISBN 978-1-4939-9091-7. [Google Scholar]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shivling Mali, A.; Honc, O.; Hejnova, L.; Novotny, J. Opioids Alleviate Oxidative Stress via the Nrf2/HO-1 Pathway in LPS-Stimulated Microglia. Int. J. Mol. Sci. 2023, 24, 11089. https://doi.org/10.3390/ijms241311089
Shivling Mali A, Honc O, Hejnova L, Novotny J. Opioids Alleviate Oxidative Stress via the Nrf2/HO-1 Pathway in LPS-Stimulated Microglia. International Journal of Molecular Sciences. 2023; 24(13):11089. https://doi.org/10.3390/ijms241311089
Chicago/Turabian StyleShivling Mali, Akash, Ondrej Honc, Lucie Hejnova, and Jiri Novotny. 2023. "Opioids Alleviate Oxidative Stress via the Nrf2/HO-1 Pathway in LPS-Stimulated Microglia" International Journal of Molecular Sciences 24, no. 13: 11089. https://doi.org/10.3390/ijms241311089
APA StyleShivling Mali, A., Honc, O., Hejnova, L., & Novotny, J. (2023). Opioids Alleviate Oxidative Stress via the Nrf2/HO-1 Pathway in LPS-Stimulated Microglia. International Journal of Molecular Sciences, 24(13), 11089. https://doi.org/10.3390/ijms241311089