

Complete Mitochondrial Genome Sequence and Phylogenetic Analysis of Procambarus clarkii and Cambaroides dauricus from China


Abstract
:1. Introduction
2. Results
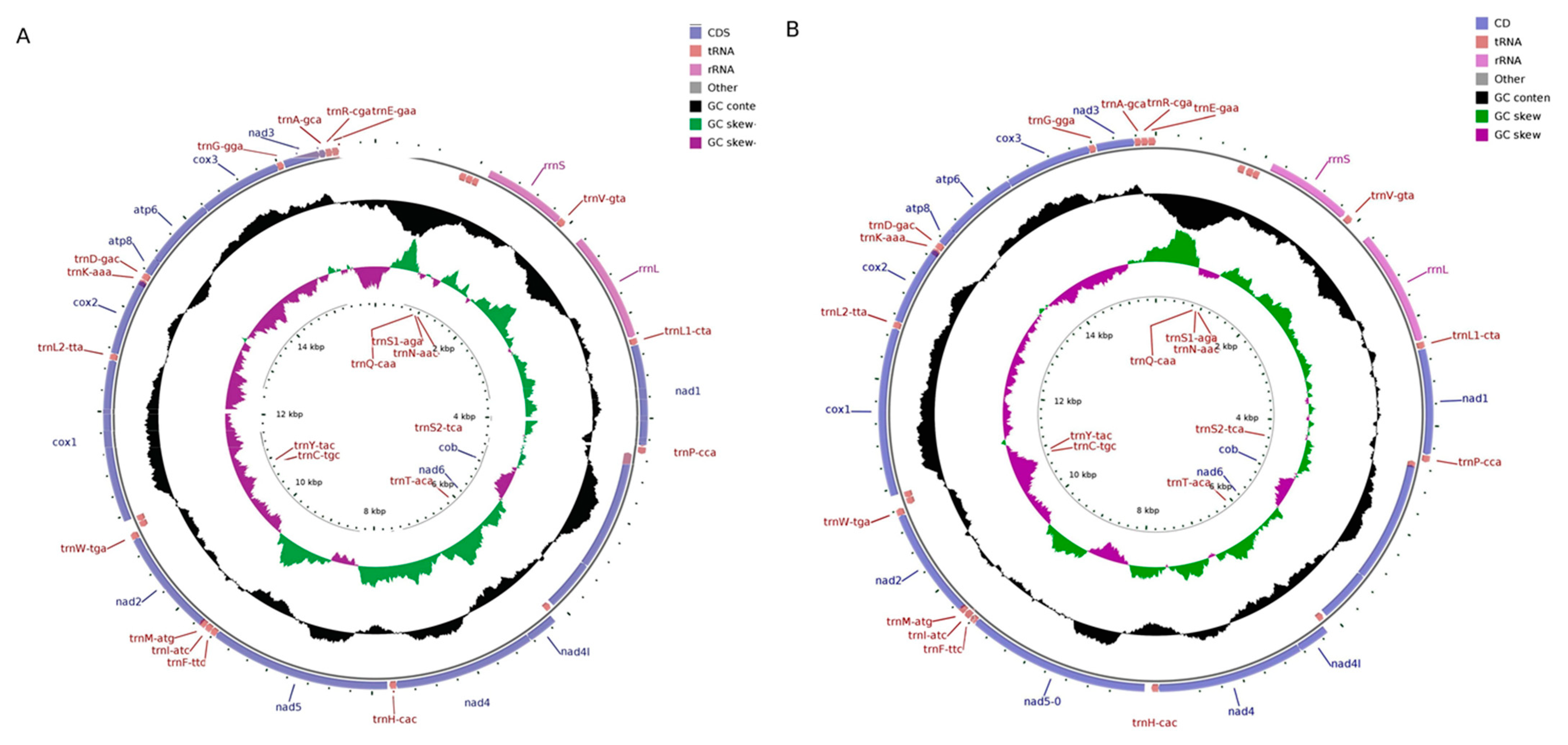
2.1. Structural Characteristics of the Mitochondrial Genomes
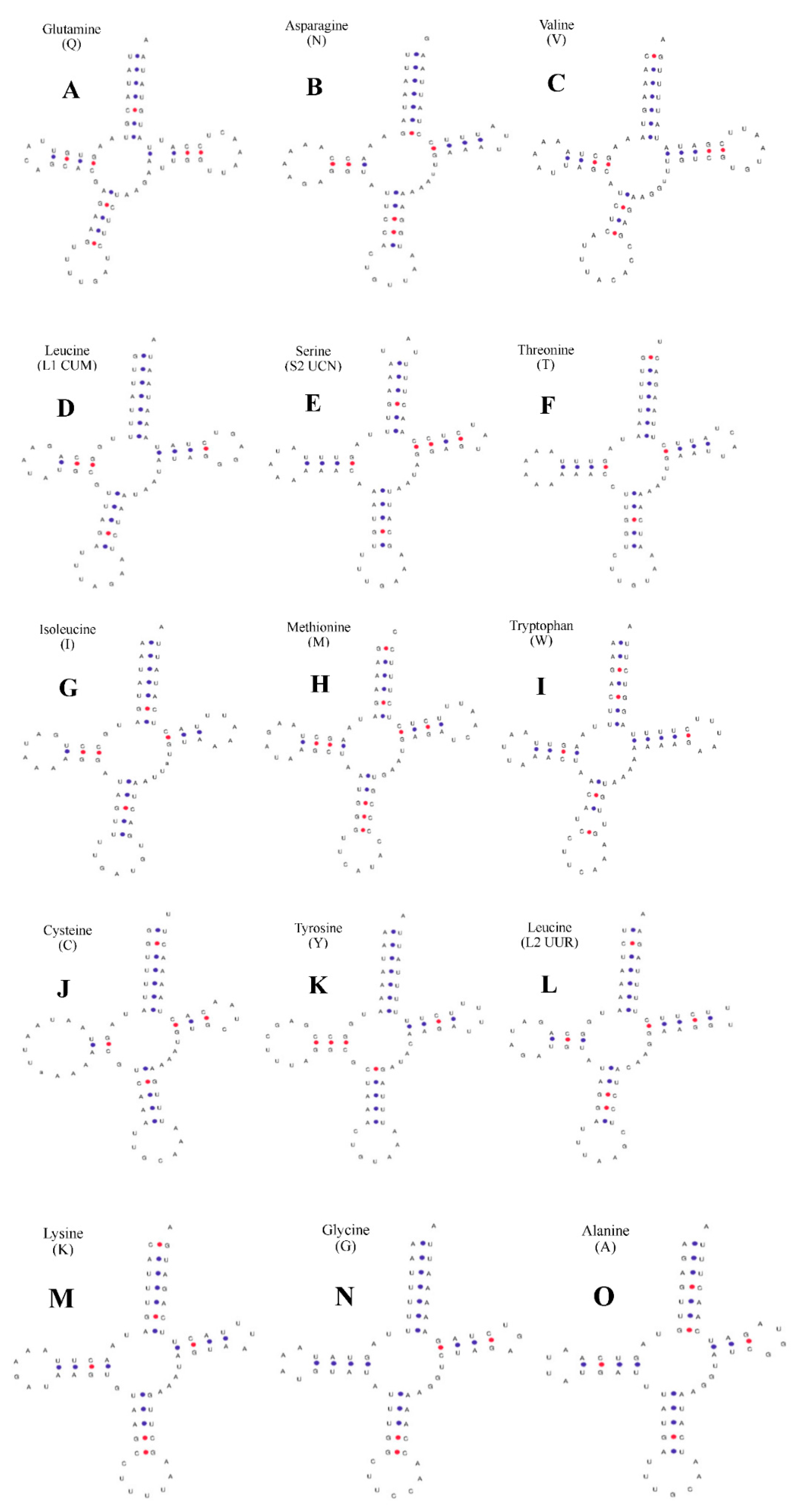
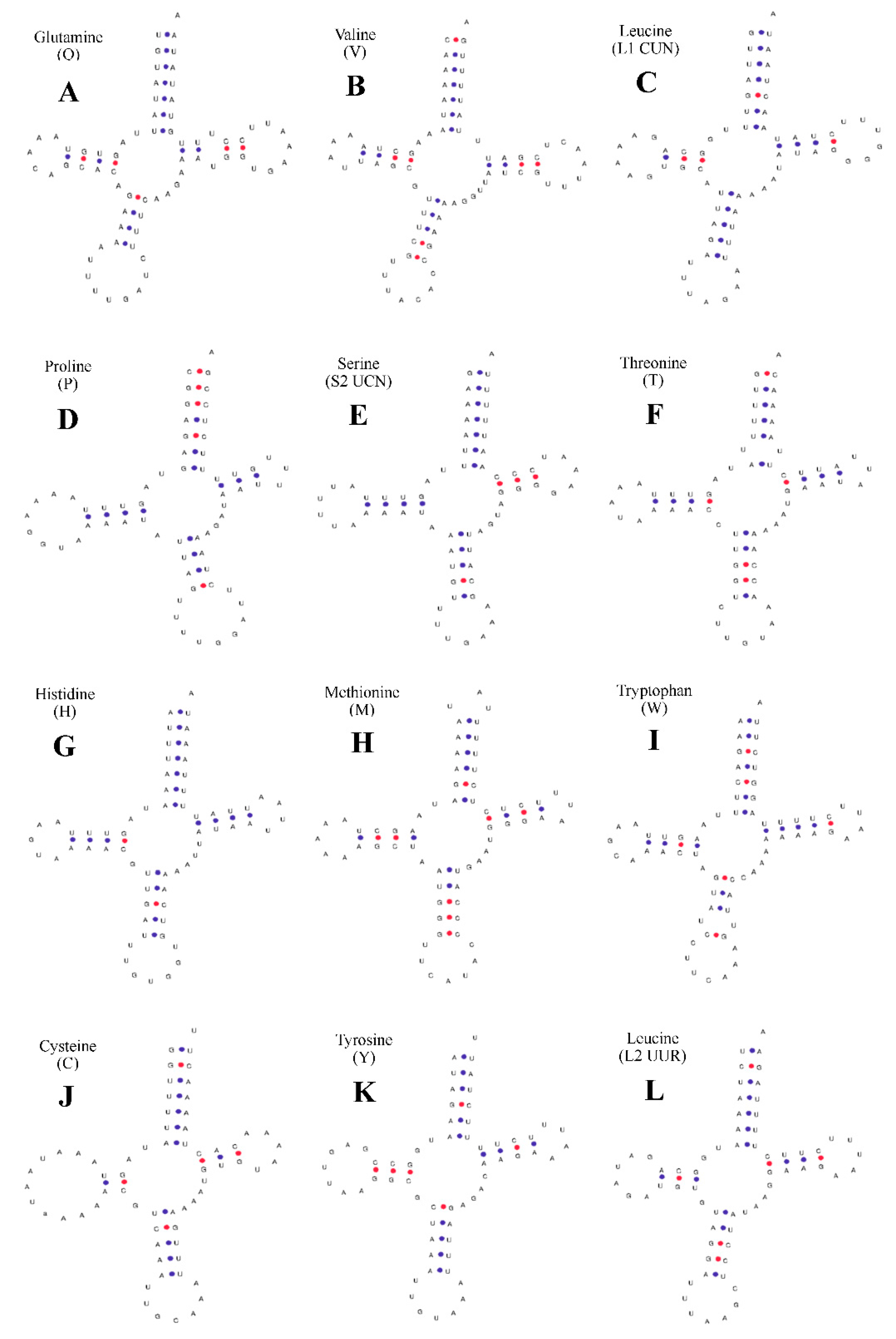
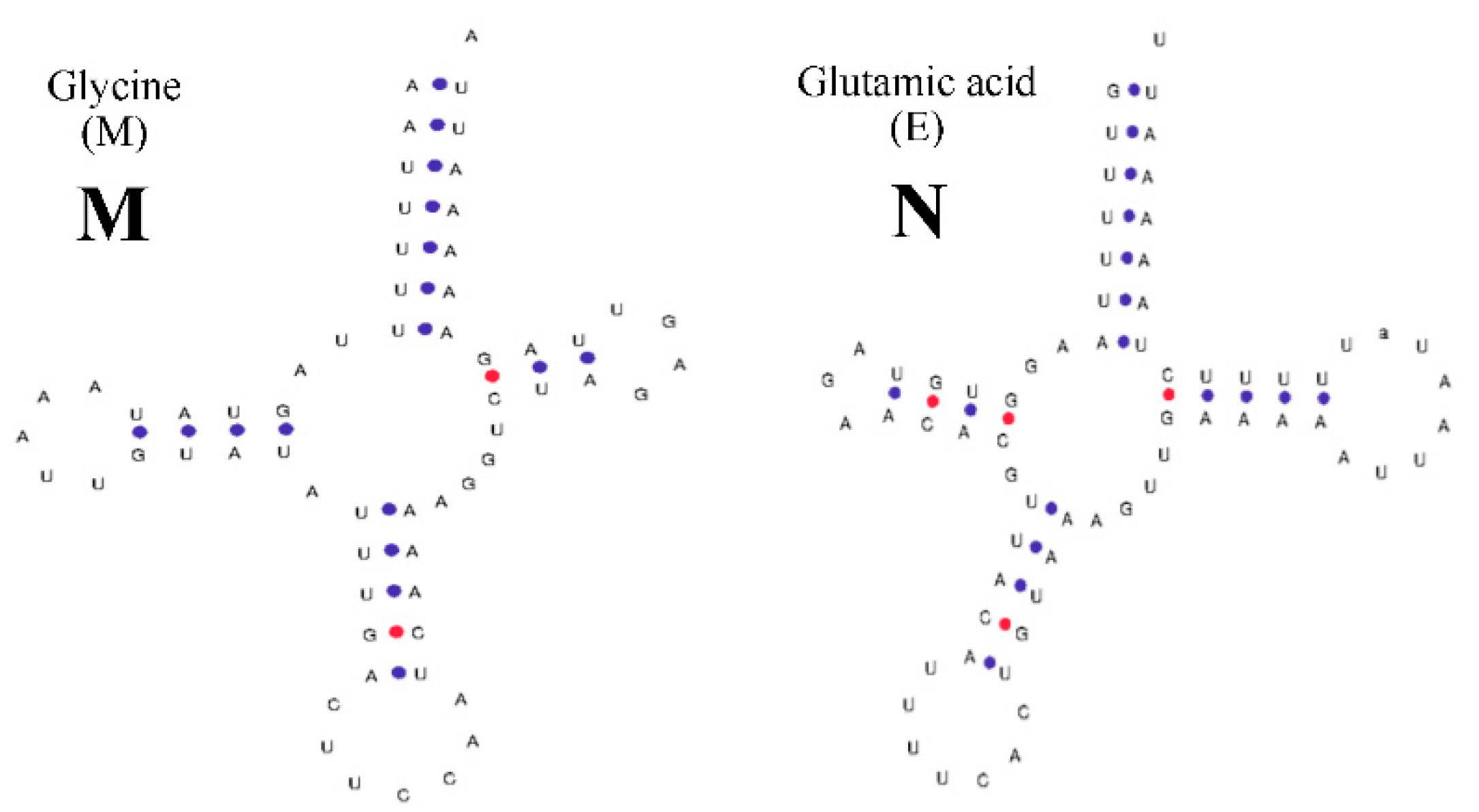
2.2. Structural Analysis of rRNA, tRNA, and D-Loop Regions
2.3. Predicted Structures of rRNA, tRNA, and D-Loop Regions
2.4. Analysis of the Codon Preference Profiles in Protein-Encoding Genes
2.5. The Genetic Variation of the Mitochondrial Genome
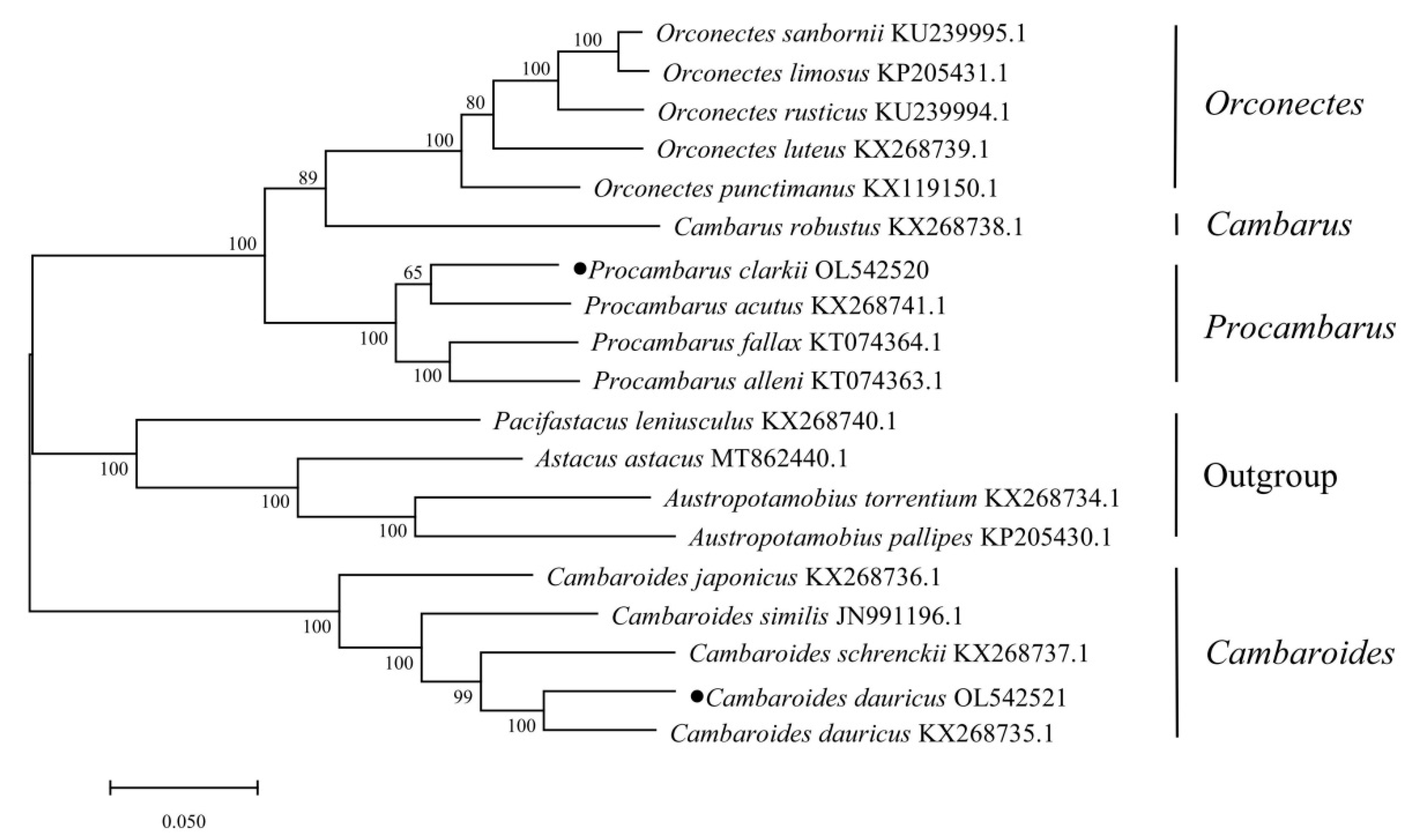
2.6. The Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Identification and DNA Extraction
4.2. Sequence Assembly
4.3. Gene Annotation
4.4. Sequence Feature Analysis
4.5. The Phylogenetic Analysis of Genetic Variation in the Mitochondrial Genome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ding, J.; Kang, J.; Xu, J. Comparison of nutritional compositions in muscle of Cambaroides dauricus and Procambarus clarkii. Food Sci. 2020, 31, 427–431. [Google Scholar]
- Gao, R.; Chen, L.; Zhang, W.; Zhang, S.; Rao, J.; Hu, J. Effect of dietary antarctic krill Euphausia Superba on the growth performance and nonspecific immunity of red swamp crayfish Procambarus clarkia. Fish Shellfish Immunol. 2020, 96, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Xiong, G.; Ling, J.; Hu, Y.; Shi, L.; Qiao, Y.; Yu, J.; Cui, Y.; Liao, L.; Wu, W.; et al. Effect of Ultra-High pressure treatment on shucking and meat properties of red swamp crayfish (Procambarus clarkia). LWT—Food Sci. Technol. 2018, 87, 234–240. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, X.; Gao, X.; Jiang, Q.; Chen, Q.; Zhang, S.; Tong, S.; Liu, X.; Zhu, J.; Zhang, X. Pathogenicity of Aeromonas Hydrophila causing mass mortalities of Procambarus clarkia and its induced host immune response. Microb. Pathog 2020, 147, 104376. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Shui, Y.; Liao, X.; Xu, Z.; Zhou, X. Primary structure of a novel gonadotropin-releasing hormone (Gnrh) in the ovary of red swamp crayfish Procambarus clarkii. Aquaculture 2012, 418, 67–71. [Google Scholar] [CrossRef]
- Siangcham, T.; Tinikul, Y.; Poljaroen, J.; Sroyraya, M.; Changklungmoa, N.; Phoungpetchara, I.; Wilairat, K.; Chanudporn, S.; Chaitip, W.; Peter, J. The effects of serotonin, dopamine, gonadotropin-releasing hormones, and corazonin, on the androgenic gland of the giant freshwater prawn, Macrobrachium rosenbergii. Gen. Comp. Endocrinol 2013, 193, 10–18. [Google Scholar] [CrossRef]
- Cruz, M.J.; Rebelo, R.; Eduardo, G.; Crespo, E. Effects of an introduced crayfish, Procambarus clarkii, on the distribution of south-western iberian amphibians in their breeding habitats. Ecography 2006, 29, 329–338. [Google Scholar] [CrossRef]
- Gutiérrez-Yurrita, P.J.; Montes, C. Bioenergetics and phenology of reproduction of the introduced red swamp crayfish, Procambarus clarkii, in Do· ana National Park, Spain, and implications for species management. Freshw. Biol. 1999, 42, 561–574. [Google Scholar] [CrossRef]
- Cruz, M.; Rui, R. Vulnerability of southwest iberian amphibians to an introduced crayfish, Procambarus clarkii. Amphib. Reptil. 2005, 26, 293–303. [Google Scholar]
- Gherardi, F.; Renai, B.; Corti, C. Crayfish predation on tadpoles: A comparison between a native (Austropotamobius pallipes) and an alien species (Procambarus clarkii). Bull. Français Pêche Piscic. 2001, 361, 659–668. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, J.; Dong, H.; Liu, S.; Zou, Z.; Yuan, M.; Chen, S. Biological characteristics and analysis on current situation of Cambaroides dauricus. Fish Sci. Technol. Inf. 2020, 47, 41–43. [Google Scholar]
- Bao, J.; Xing, Y.; Feng, C.; Kou, S.; Jiang, H.; Li, X. Acute and sub-chronic effects of copper on survival, respiratory metabolism, and metal accumulation in Cambaroides dauricus. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Tadashi, K. Morphology of the mandible and gill of the asian freshwater crayfish cambaroides (Decapoda: Cambaridae) with implications for their phylogeny. J. Crustac. Biol. 2012, 32, 15–23. [Google Scholar]
- Liao, R.; Qu, Y.; Gou, X.; Li, J. Morphological variations and discrimination of four fish species Chinese Bahaba Bahaba flavolabiata, large yellow croaker Pseudosciaena crocea, Ting’wak wak tingi and spinyhead croaker Collichthys lucidus. J. Dalian Fish. Univ. 2009, 24, 305–310. [Google Scholar]
- Çiftci, Y.; İbrahim, O. Fish population genetics and applications of molecular markers to fisheries and aquaculture: I-basic principles of fish population genetics. Turk. J. Fish. Aquat. Sci. 2002, 2, 2. [Google Scholar]
- Nwafili, S.A.; Gao, T.X. Genetic Diversity in the Mtdna Control Region and Population Structure of Chrysichthys Nigrodigitatus From Selected Nigerian Rivers: Implications for Conservation and Aquaculture. Fish. Aquat. Life 2016, 24, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Presti, R.L.; Lisa, C.; Stasio, L.D. Molecular genetics in aquaculture. Ital. J. Anim. Sci. 2009, 8, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Salzburger, W. Meyer a the species flocks of East African cichlid fishes: Recent advances in molecular phylogenetics and population genetics. Naturwissenschaften 2004, 91, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Zhang, Y.; Gao, T.; Yanagimoto, T.; Yabe, M.; Sakurai, Y. Genetic diversity in the mtDNA control region and population structure in the small yellow croaker Larimichthys polyactis. Environ. Biol. Fishes 2009, 85, 303–314. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, X.; Yang, T.; Han, Z.; Yanagimoto, T.; Gao, T. Genetic diversity in the mtDNA control region and population structure in the Sardinella zunasi Bleeker. Afr. J. Biotechnol. 2008, 7, 24. [Google Scholar]
- Moreira, C.; Correia, A.T.; Vaz-Pires, P.; Froufe, E. Genetic diversity and population structure of the blue jack mackerel Trachurus picturatus across its western distribution. J. Fish Biol. 2019, 94, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Huang, Y.; Wang, J.; Huang, J.; Wang, Z.; Peng, Z. The complete mitochondrial genome sequence of Triplophysa bleekeri (Teleostei, Balitoridae, Nemacheilinae). Mitochondrial DNA 2013, 24, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Lüssen, A.; Falk, T.M.; Villwock, W. Phylogenetic patterns in populations of chilean species of the Genus orestias (Teleostei: Cyprinodontidae): Results of mitochondrial DNA analysis. Mol. Phylogenet. Evol. 2003, 29, 151–160. [Google Scholar] [CrossRef]
- Satoh, T.P.; Miya, M.; Mabuchi, K.; Nishida, M. Structure and variation of the mitochondrial genome of fishes. BMC Genom. 2016, 17, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Mi, Z. Advance in molecular biology of animal mitochondrial DNA. China Biotechnol. 1998, 18, 25–31. [Google Scholar]
- Shi, B.; Liu, X.; Liu, Y.; Zhang, Y.; Gao, Q.; Xu, Y.; Wang, B.; Jiang, Y.; Song, X. Complete sequence and gene organization of the mitochondrial genome of Seriola aureovittata. J. Fish. Sci. China 2019, 26, 405–415. [Google Scholar] [CrossRef]
- Xiao, J. The Complete Mitochondrial Genomes and Phylogenetic Analysis of Sillago species. Master’s Thesis, Ocean University of China, Qingdao, China, 2015. [Google Scholar]
- Ma, J. The Complete Mitochondrial Genome Sequence of Oryzias celebensis and the Genetic Structure of Oryzias sinensis. Master’s Thesis, Guangdong Ocean University, Zhanjiang, China, 2020. [Google Scholar]
- Broughton, R.E.; Milam, J.E.; Roe, B.A. The complete sequence of the Zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA. Genome Res. 2001, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Chen, Y.; Cheng, Q.; Qiao, H.; Chen, W. The Complete mitochondrial genome sequence of Schizothorax macropogon (Cypriniformes: Cyprinidae). Mitochondrial DNA 2013, 24, 5. [Google Scholar] [CrossRef]
- Shen, P.; Zhang, Y.; Li, X.; Gao, Q.; Xu, Y.; Huang, Z.; Jiang, W.; Chen, X. Analysis of codon usage bias based on Macrobrachium rosenbergii transcriptome. Mod. Agric. Sci. Technol. 2020, 16, 175–178, 184. [Google Scholar]
- Fang, X. Research on sequencing of mitogenome and phylogenetic analysis within Haliotis discus hannai Ino, Haliotis iris Martyn and Hybrid F1. Master’s Thesis, Shandong University, Jinan, China, 2016. [Google Scholar]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 12. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Zheng, W.; Zhu, S.; Zou, J.; Yang, Y.; Shen, X. Molecular phylogenetic relationship of Carangidae based on partial sequence of mitochondrial 16S ribosomal RNA gene. J. Fish. China 2008, 32, 847–854. [Google Scholar]
- Grandjean, F.; Tan, M.H.; Gan, H.M.; Lee, Y.P.; Kawai, T.; Distefano, R.J.; Blaha, M.; Roles, A.J.; Austin, C.M. Rapid recovery of nuclear and mitochondrial genes by genome skimming from Northern Hemisphere freshwater crayfish. Zool. Scr. 2017, 46, 718–728. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Sudhir, K.; Glen, S.; Koichiro, T. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar]
- Perna, N.T.; Kocher, T.D. Kocher. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Hall, T.A. Bioedit: A User-friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Position (bp) | Size (bp) | Direction | Intergenic Nucleotides | Anti- or Start/ Stop Codons | A + T% |
|---|---|---|---|---|---|---|
| D-loop | 1–844 | 844 | − | - | 82.9 | |
| trnGln(Q) | 845–912 | 68 | + | − | - | 69.1 |
| trnSer1(AGN) | 914–980 | 67 | + | 1 | - | 71.6 |
| trnAsn(N) | 981–1044 | 64 | + | 0 | - | 75.0 |
| rrnS | 1111–1903 | 793 | − | 66 | - | 75.4 |
| trnVal (V) | 1903–1972 | 70 | − | −1 | - | 68.6 |
| rrnL | 2174–3223 | 1050 | − | 201 | - | 74.8 |
| trnLeu1(CUN) | 3238–3300 | 63 | − | 14 | - | 74.6 |
| ND1 | 3304–4266 | 963 | − | 3 | ATG/ATG | 71.5 |
| trnPro (P) | 4275–4339 | 65 | − | 8 | - | 76.9 |
| trnSer2(UCN) | 4342–4404 | 63 | + | 2 | - | 77.8 |
| CYTB | 4388–5539 | 1152 | + | −17 | ATG/TAA | 68.3 |
| ND6 | 5539–6051 | 513 | + | −1 | ATT/TAA | 72.1 |
| trnThr(T) | 6073–6137 | 65 | + | 21 | - | 78.5 |
| ND4L | 6140–6433 | 294 | − | 2 | ATG/TAA | 75.9 |
| ND4 | 6433–7776 | 1344 | − | −1 | ATG/TAA | 71.9 |
| trnHis(H) | 7776–7840 | 65 | − | −1 | - | 84.6 |
| ND5 | 7859–9574 | 1716 | − | 18 | ATG/TAA | 73.4 |
| trnPhe(F) | 9574–9636 | 63 | − | −1 | - | 73.0 |
| trnIle(I) | 9637–9699 | 63 | − | 0 | - | 76.2 |
| trnMet(M) | 9701–9767 | 67 | − | 1 | - | 61.2 |
| ND2 | 9744–10,760 | 1017 | − | −24 | ATG/TAA | 74.5 |
| trnTrp(W) | 10,759–10,825 | 67 | − | −2 | - | 77.6 |
| trnCys(C) | 10,827–10,890 | 64 | + | 1 | - | 76.6 |
| trnTyr(Y) | 10,891–10,955 | 65 | + | 0 | - | 72.3 |
| COX1 | 10,948–12,492 | 1545 | − | −8 | ATT/TAA | 68.4 |
| trnL2 (UUR) | 12,494–12,556 | 63 | − | 1 | - | 66.7 |
| COX2 | 12,557–13,291 | 735 | − | 0 | ATG/TAG | 71.2 |
| trnLys(K) | 13,245–13,308 | 64 | − | −47 | - | 87.5 |
| trnAsp(D) | 13,311–13,377 | 67 | − | 2 | - | 86.6 |
| ATP8 | 13,378–13,536 | 159 | − | 0 | ATG/TAA | 75.5 |
| ATP6 | 13,530–14,204 | 675 | − | −7 | ATG/TAA | 72.9 |
| COX3 | 14,204–14,992 | 789 | − | −1 | ATG/TAA | 67.0 |
| trnGly(G) | 14,991–15,052 | 62 | − | −2 | - | 74.2 |
| ND3 | 15,059–15,406 | 348 | − | 6 | ATT/TAG | 69.5 |
| trnAla(A) | 15,405–15,465 | 61 | − | −2 | - | 70.5 |
| trnArg(R) | 15,468–15,529 | 62 | − | 2 | - | 67.7 |
| trnGlu(E) | 15,530–15,593 | 64 | − | 0 | - | 71.9 |
| Gene | Position (bp) | Size (bp) | Direction | Intergenic Nucleotides | Anti- or Start/Stop Codons | A + T% |
|---|---|---|---|---|---|---|
| D-loop | 1–791 | 791 | − | − | 84.5 | |
| trnGln(Q) | 792–860 | 69 | + | − | − | 73.9 |
| trnSer1(AGN) | 877–943 | 67 | + | 16 | − | 67.2 |
| trnAsn(N) | 944–1007 | 64 | + | 0 | − | 75.0 |
| rrnS | 1084–1874 | 791 | − | 76 | − | 75.7 |
| trnVal (V) | 1897–1964 | 68 | − | 22 | − | 70.6 |
| rrnL | 2166–3219 | 1054 | − | 201 | − | 74.6 |
| trnLeu1(CUN) | 3233–3297 | 65 | − | 13 | − | 72.3 |
| ND1 | 3301–4263 | 963 | − | 3 | ATG/TAG | 72.5 |
| trnPro (P) | 4271–4334 | 64 | − | 7 | − | 71.9 |
| trnSer2(UCN) | 4338–4401 | 64 | + | 3 | − | 78.1 |
| CYTB | 4382–5536 | 1155 | + | −20 | ATG/TAA | 68.8 |
| ND6 | 5536–6048 | 513 | + | −1 | ATT/TAA | 75.0 |
| trnThr(T) | 6072–6134 | 63 | + | 23 | − | 79.4 |
| ND4L | 6137–6430 | 294 | − | 2 | ATG/TAA | 74.1 |
| ND4 | 6430–7770 | 1341 | − | −1 | ATG/TAA | 73.6 |
| trnHis(H) | 7770–7833 | 64 | − | −1 | − | 84.4 |
| ND5 | 7894–9564 | 1671 | − | 60 | − | 71.9 |
| trnPhe(F) | 9564–9624 | 61 | − | −1 | − | 72.1 |
| trnIle(I) | 9628–9691 | 64 | − | 3 | − | 73.4 |
| trnMet(M) | 9695–9757 | 63 | − | 3 | − | 68.3 |
| ND2 | 9737–10,750 | 1014 | − | −1 | ATG/TAA | 83.3 |
| trnTrp(W) | 10,750–10,815 | 66 | − | −1 | − | 72.7 |
| trnCys(C) | 10,815–10,879 | 65 | + | −1 | − | 78.5 |
| trnTyr(Y) | 10,880–10,942 | 63 | + | 0 | − | 68.3 |
| COX1 | 10,940–12,478 | 1539 | − | −3 | ATC/TAG | 66.8 |
| trnL2 (UUR) | 12,481–12,544 | 64 | − | 2 | − | 68.8 |
| COX2 | 12,545–13,276 | 732 | − | 0 | ATG/TAG | 68.0 |
| trnLys(K) | 13,230–13,293 | 64 | − | −47 | − | 75.0 |
| trnAsp(D) | 13,295–13,358 | 64 | − | 1 | − | 87.5 |
| ATP8 | 13,359–13,517 | 159 | − | 0 | ATG/TAA | 75.5 |
| ATP6 | 13,511–14,185 | 675 | − | −7 | ATG/TAA | 69.8 |
| COX3 | 14,185–14,973 | 789 | − | −1 | ATG/TAA | 66.5 |
| trnGly(G) | 14,972–15,033 | 62 | − | −2 | − | 79.0 |
| ND3 | 15,040–15,387 | 348 | − | 6 | ATT/TAA | 70.7 |
| trnAla(A) | 15,389–15,449 | 61 | − | 1 | − | 68.9 |
| trnArg(R) | 15,450–15,512 | 63 | − | 0 | − | 68.3 |
| trnGlu(E) | 15,513–15,580 | 68 | − | 0 | − | 77.9 |
| Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU (F) | 299 | 1.84 | UCU (S) | 140 | 2.89 | UAU (Y) | 144 | 1.88 | UGU (C) | 40 | 1.69 |
| UUC (F) | 27 | 0.16 | UCC (S) | 9 | 0.40 | UAC (Y) | 12 | 0.12 | UGC (C) | 0 | 0.00 |
| UUA (L) | 419 | 4.29 | UCA (S) | 58 | 1.20 | UAA (*) | 10 | 0.46 | UGA (*) | 79 | 2.38 |
| UUG (L) | 76 | 0.81 | UCG (S) | 6 | 0.09 | UAG (*) | 3 | 0.16 | UGG (W) | 25 | 0.69 |
| CUU (L) | 45 | 0.55 | CCU (P) | 97 | 2.82 | CAU (H) | 65 | 1.25 | CGU (R) | 23 | 0.70 |
| CUC (L) | 6 | 0.05 | CCC (P) | 12 | 0.32 | CAC (H) | 12 | 0.44 | CGC (R) | 2 | 0.06 |
| CUA (L) | 25 | 0.27 | CCA (P) | 22 | 0.48 | CAA (Q) | 40 | 1.10 | CGA (R) | 26 | 0.92 |
| CUG (L) | 4 | 0.04 | CCG (P) | 3 | 0.07 | CAG (Q) | 24 | 0.59 | CGG (R) | 8 | 0.68 |
| AUU (I) | 302 | 1.85 | ACU (T) | 101 | 2.47 | AAU (N) | 117 | 1.75 | AGU (S) | 62 | 1.33 |
| AUC (I) | 11 | 0.07 | ACC (T) | 14 | 0.22 | AAC (N) | 21 | 0.25 | AGC (S) | 5 | 0.10 |
| AUA (I) | 179 | 1.08 | ACA (T) | 42 | 0.85 | AAA (K) | 61 | 1.43 | AGA (R) | 65 | 2.13 |
| AUG (M) | 50 | 0.85 | ACG (T) | 8 | 0.15 | AAG (K) | 30 | 0.57 | AGG (R) | 53 | 1.51 |
| GUU (V) | 104 | 1.28 | GCU (A) | 106 | 2.43 | GAU (D) | 71 | 1.77 | GGU (G) | 86 | 1.27 |
| GUC (V) | 11 | 0.19 | GCC (A) | 18 | 0.59 | GAC (D) | 6 | 0.23 | GGC (G) | 15 | 0.22 |
| GUA (V) | 131 | 1.97 | GCA (A) | 35 | 0.78 | GAA (E) | 46 | 1.11 | GGA (G) | 70 | 1.40 |
| GUG (V) | 47 | 0.56 | GCG (A) | 10 | 0.19 | GAG (E) | 40 | 0.89 | GGG (G) | 72 | 1.10 |
| Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU (F) | 306 | 1.82 | UCU (S) | 140 | 2.81 | UAU (Y) | 121 | 1.60 | UGU (C) | 36 | 1.40 |
| UUC (F) | 29 | 0.18 | UCC (S) | 22 | 0.47 | UAC (Y) | 30 | 0.40 | UGC (C) | 5 | 0.13 |
| UUA (L) | 394 | 4.01 | UCA (S) | 64 | 1.69 | UAA (*) | 10 | 0.67 | UGA (*) | 69 | 2.20 |
| UUG (L) | 58 | 0.59 | UCG (S) | 9 | 0.18 | UAG (*) | 3 | 0.13 | UGG (W) | 32 | 0.85 |
| CUU (L) | 83 | 0.82 | CCU (P) | 74 | 2.25 | CAU (H) | 65 | 1.32 | CGU (R) | 16 | 0.45 |
| CUC (L) | 10 | 0.08 | CCC (P) | 15 | 0.45 | CAC (H) | 13 | 0.37 | CGC (R) | 2 | 0.12 |
| CUA (L) | 44 | 0.45 | CCA (P) | 34 | 0.75 | CAA (Q) | 51 | 1.23 | CGA (R) | 28 | 0.89 |
| CUG (L) | 6 | 0.05 | CCG (P) | 7 | 0.25 | CAG (Q) | 16 | 0.62 | CGG (R) | 13 | 0.45 |
| AUU (I) | 283 | 1.86 | ACU (T) | 91 | 2.28 | AAU (N) | 121 | 1.48 | AGU (S) | 34 | 0.72 |
| AUC (I) | 24 | 0.15 | ACC (T) | 14 | 0.24 | AAC (N) | 27 | 0.52 | AGC (S) | 9 | 0.14 |
| AUA (I) | 182 | 0.99 | ACA (T) | 46 | 1.01 | AAA (K) | 65 | 1.36 | AGA (R) | 90 | 2.80 |
| AUG (M) | 28 | 1.00 | ACG (T) | 9 | 0.15 | AAG (K) | 25 | 0.64 | AGG (R) | 41 | 1.29 |
| GUU (V) | 124 | 1.81 | GCU (A) | 101 | 2.13 | GAU (D) | 45 | 1.28 | GGU (G) | 52 | 1.13 |
| GUC (V) | 10 | 0.18 | GCC (A) | 10 | 0.32 | GAC (D) | 21 | 0.41 | GGC (G) | 17 | 0.27 |
| GUA (V) | 106 | 1.68 | GCA (A) | 60 | 1.40 | GAA (E) | 49 | 1.20 | GGA (G) | 111 | 1.57 |
| GUG (V) | 22 | 0.32 | GCG (A) | 10 | 0.16 | GAG (E) | 35 | 0.80 | GGG (G) | 64 | 1.03 |
| Gene | Total Number of Sites | Non-Synonymous Substitution Rate (Ka) | Synonymous Substitution Rate (Ks) | Ka/Ks Ratio |
|---|---|---|---|---|
| nad1 | 963 | 0.1640 | 0.7698 | 0.2130 |
| cob | 1152 | 0.1204 | 1.3004 | 0.0926 |
| nad6 | 513 | 0.9050 | 1.0257 | 0.8823 |
| nad4l | 294 | 0.1541 | 0.7320 | 0.2105 |
| nad4 | 1341 | 0.7720 | 0.8137 | 0.9488 |
| nad5 | 1671 | 1.9679 | 0.8773 | 2.2431 |
| nad2 | 1014 | 1.5361 | 1.1646 | 1.3190 |
| cox1 | 1539 | 2.2137 | 1.0517 | 2.1049 |
| cox2 | 732 | 0.5639 | 1.3191 | 0.4275 |
| atp8 | 159 | 0.3522 | 0.8095 | 0.4351 |
| atp6 | 675 | 0.1645 | 0.6540 | 0.2515 |
| cox3 | 789 | 0.0758 | 0.8173 | 0.0927 |
| nad3 | 348 | 0.2033 | 1.4072 | 0.1445 |
| Family | Subfamily | Genus | Species | Accession Number |
|---|---|---|---|---|
| Astacidae | Cambarinas | Cambaroides | Cambaroides dauricus | OL542521 |
| Cambaroides dauricus | KX268735.1 | |||
| Cambaroides similis | JN991196.1 | |||
| Cambaroides japonicas | KX268736.1 | |||
| Cambaroides schrenckii | KX268737.1 | |||
| Cambarus | Cambarus robustus | KX268738.1 | ||
| Orconectes | Orconectes punctimanus | KX119150.1 | ||
| Orconectes luteus | KX268739.1 | |||
| Orconectes rusticus | KU239994.1 | |||
| Orconectes limosus | KP205431.1 | |||
| Orconectes sanbornii | KU239995.1 | |||
| pacifastacus | pacifastacus leniusculus | KX268740.1 | ||
| procambarus | Procambarus clarkii | OL542520 | ||
| procambarus alleni | KT074363.1 | |||
| Procambarus fallax | KT074364.1 | |||
| Procambarus acutus | KX268741.1 | |||
| Astacus | Astacus astacus | MT862440.1 | ||
| Austropotamobius | Austropotamobius torrentium | KX268734.1 | ||
| Austropotamobius pallipes | KP205430.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, L.; Xu, Y.; Wang, S.; Zhang, R.; Guo, K.; Xu, W.; Zhao, Z. Complete Mitochondrial Genome Sequence and Phylogenetic Analysis of Procambarus clarkii and Cambaroides dauricus from China. Int. J. Mol. Sci. 2023, 24, 11282. https://doi.org/10.3390/ijms241411282
Luo L, Xu Y, Wang S, Zhang R, Guo K, Xu W, Zhao Z. Complete Mitochondrial Genome Sequence and Phylogenetic Analysis of Procambarus clarkii and Cambaroides dauricus from China. International Journal of Molecular Sciences. 2023; 24(14):11282. https://doi.org/10.3390/ijms241411282
Chicago/Turabian StyleLuo, Liang, Yue Xu, Shihui Wang, Rui Zhang, Kun Guo, Wei Xu, and Zhigang Zhao. 2023. "Complete Mitochondrial Genome Sequence and Phylogenetic Analysis of Procambarus clarkii and Cambaroides dauricus from China" International Journal of Molecular Sciences 24, no. 14: 11282. https://doi.org/10.3390/ijms241411282
APA StyleLuo, L., Xu, Y., Wang, S., Zhang, R., Guo, K., Xu, W., & Zhao, Z. (2023). Complete Mitochondrial Genome Sequence and Phylogenetic Analysis of Procambarus clarkii and Cambaroides dauricus from China. International Journal of Molecular Sciences, 24(14), 11282. https://doi.org/10.3390/ijms241411282