
Transcriptome and Metabolome Analyses Reveal Mechanisms Underlying the Response of Quinoa Seedlings to Nitrogen Fertilizers

Abstract
:1. Introduction
2. Results
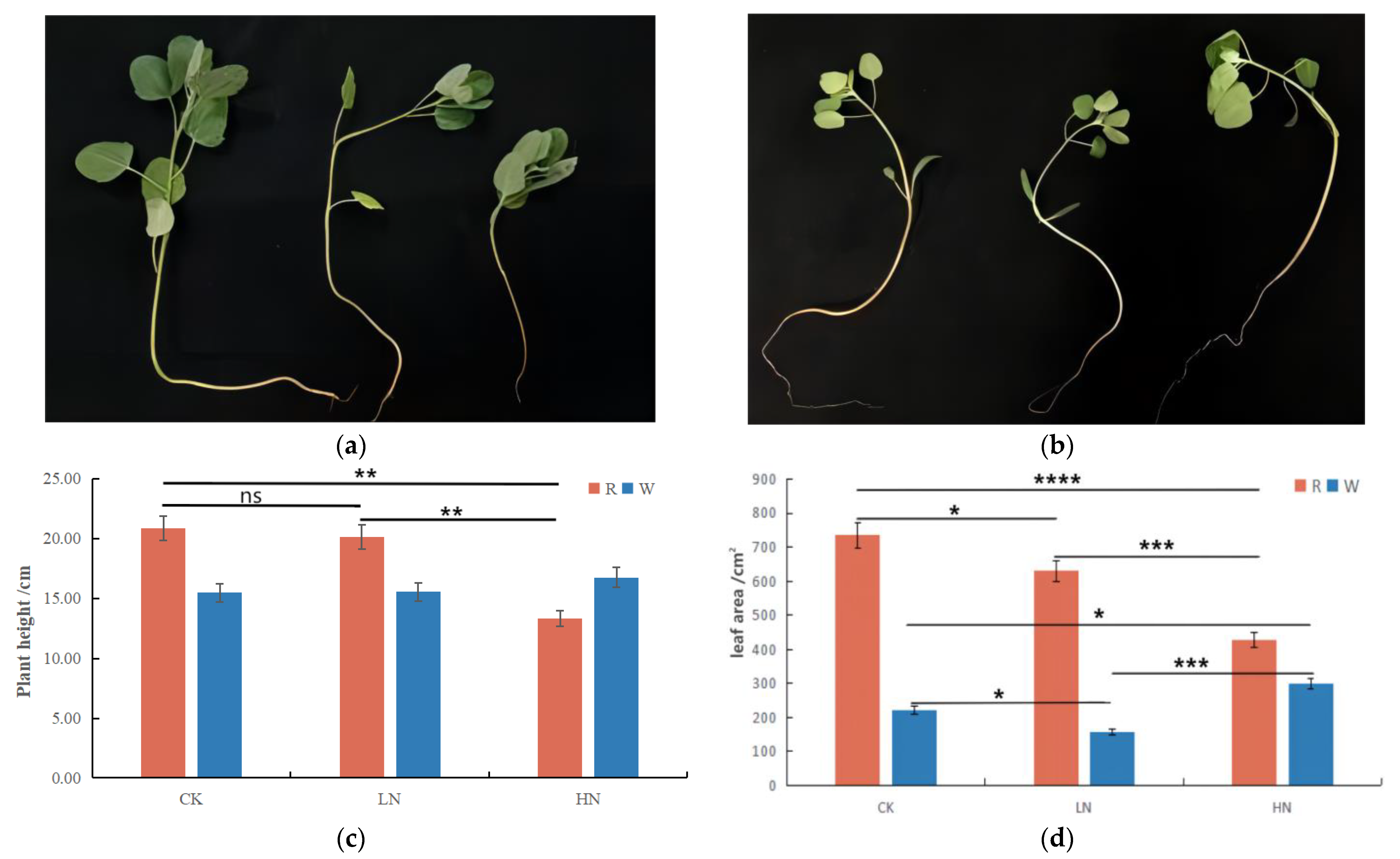
2.1. Changes in Agronomic Characteristics of Quinoa Seedlings under Different Treatments
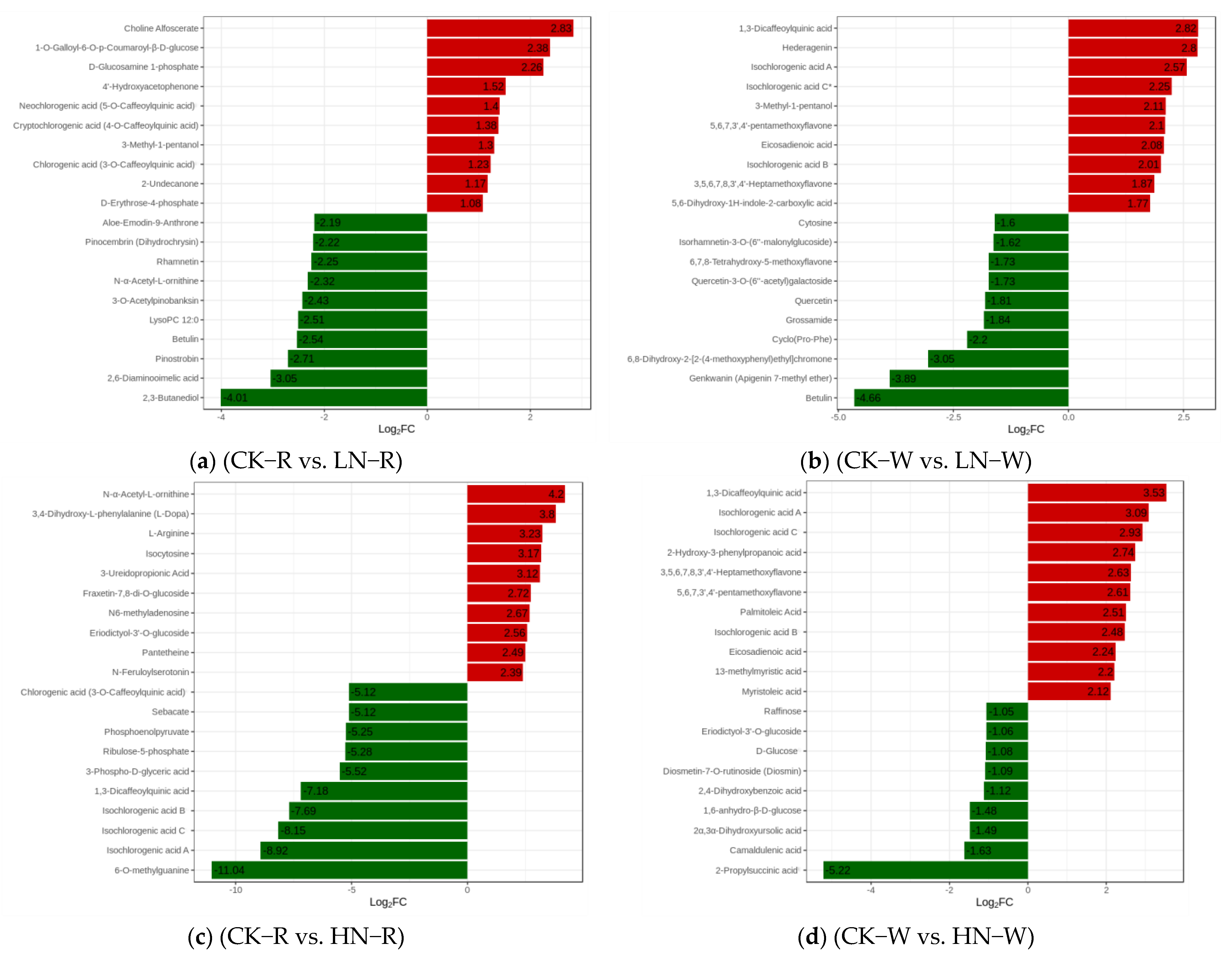
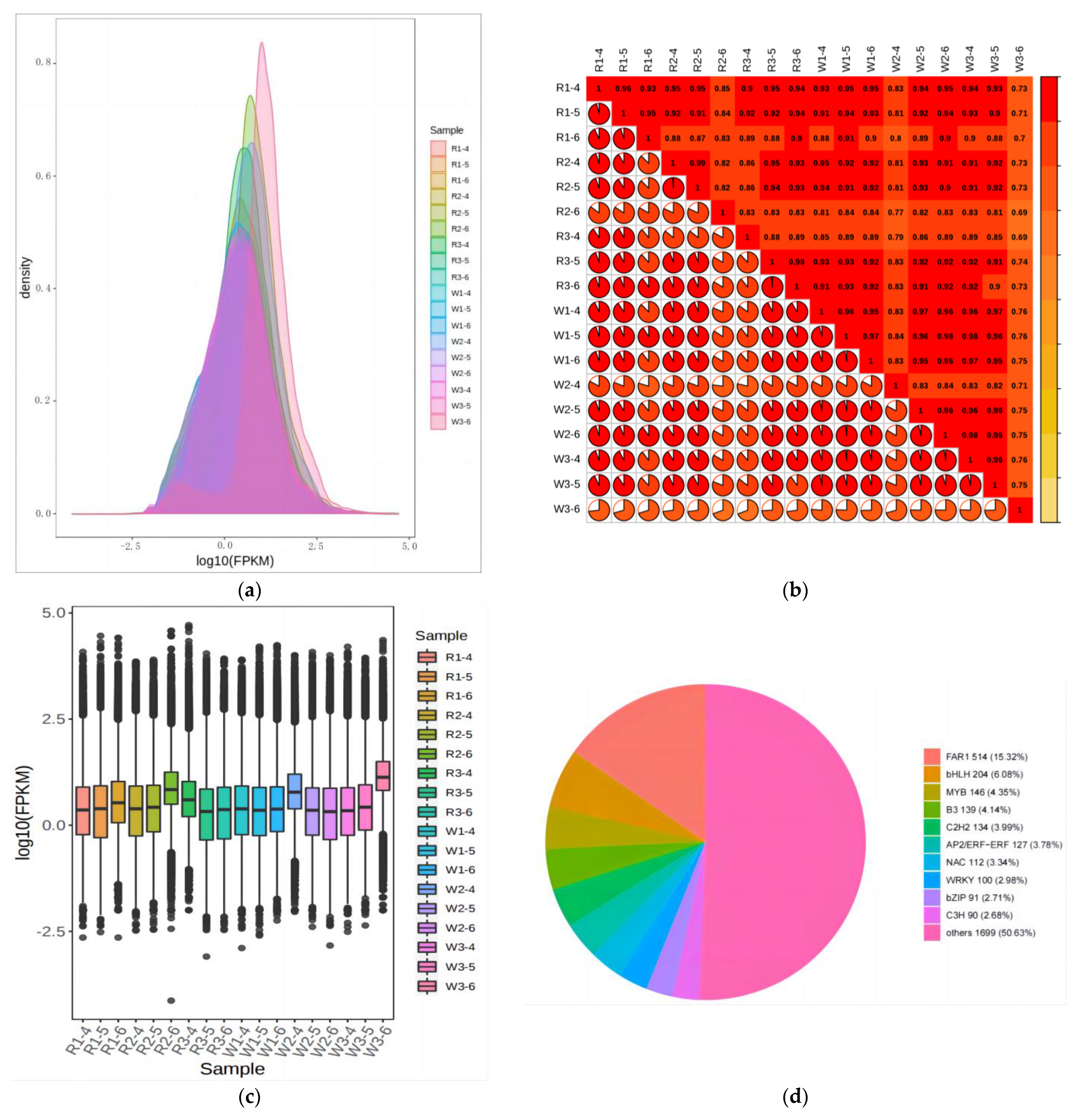
2.2. Qualitative and Quantitative Analysis of Metabolites in Quinoa Seedlings under Different Treatments
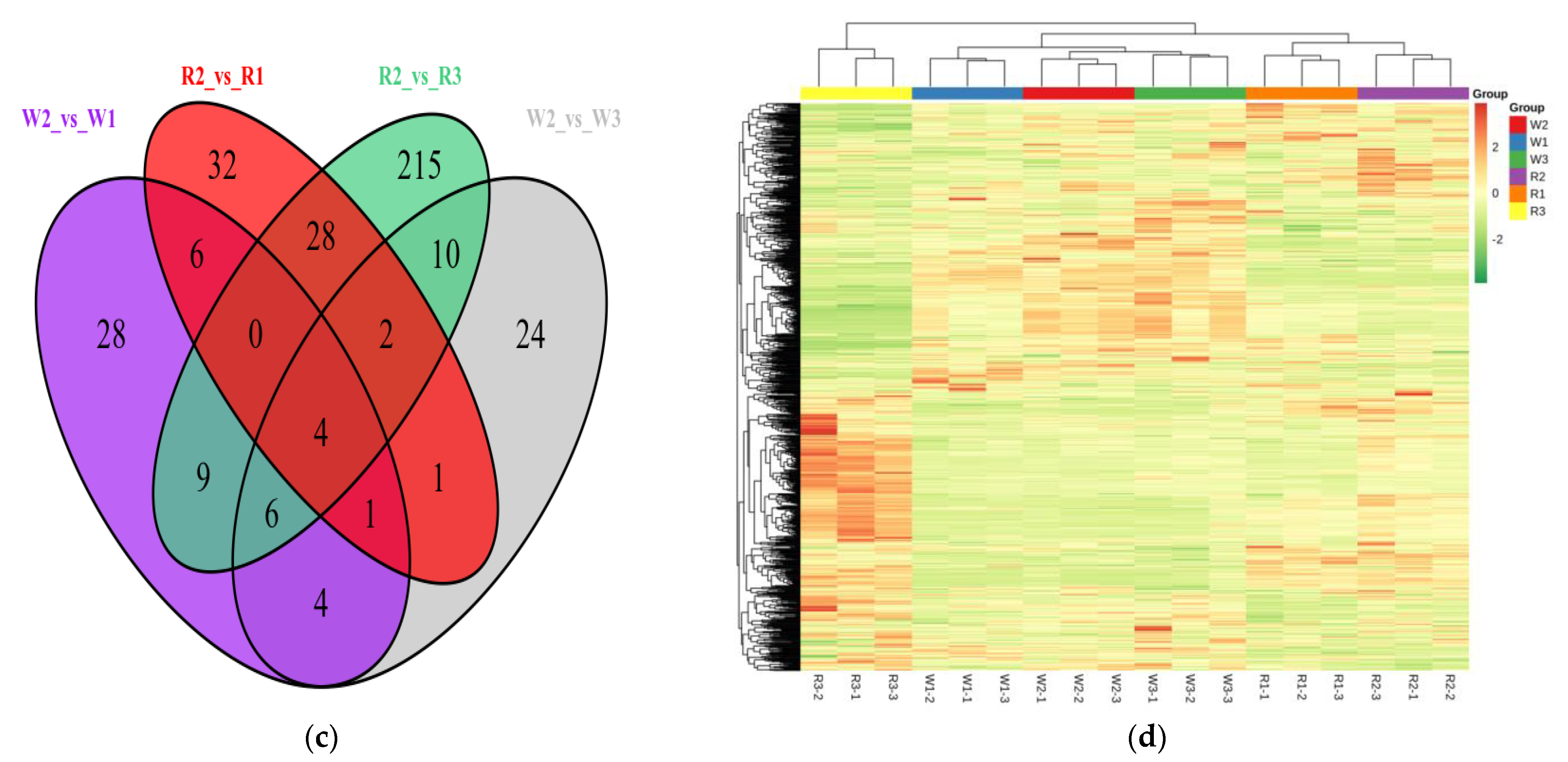
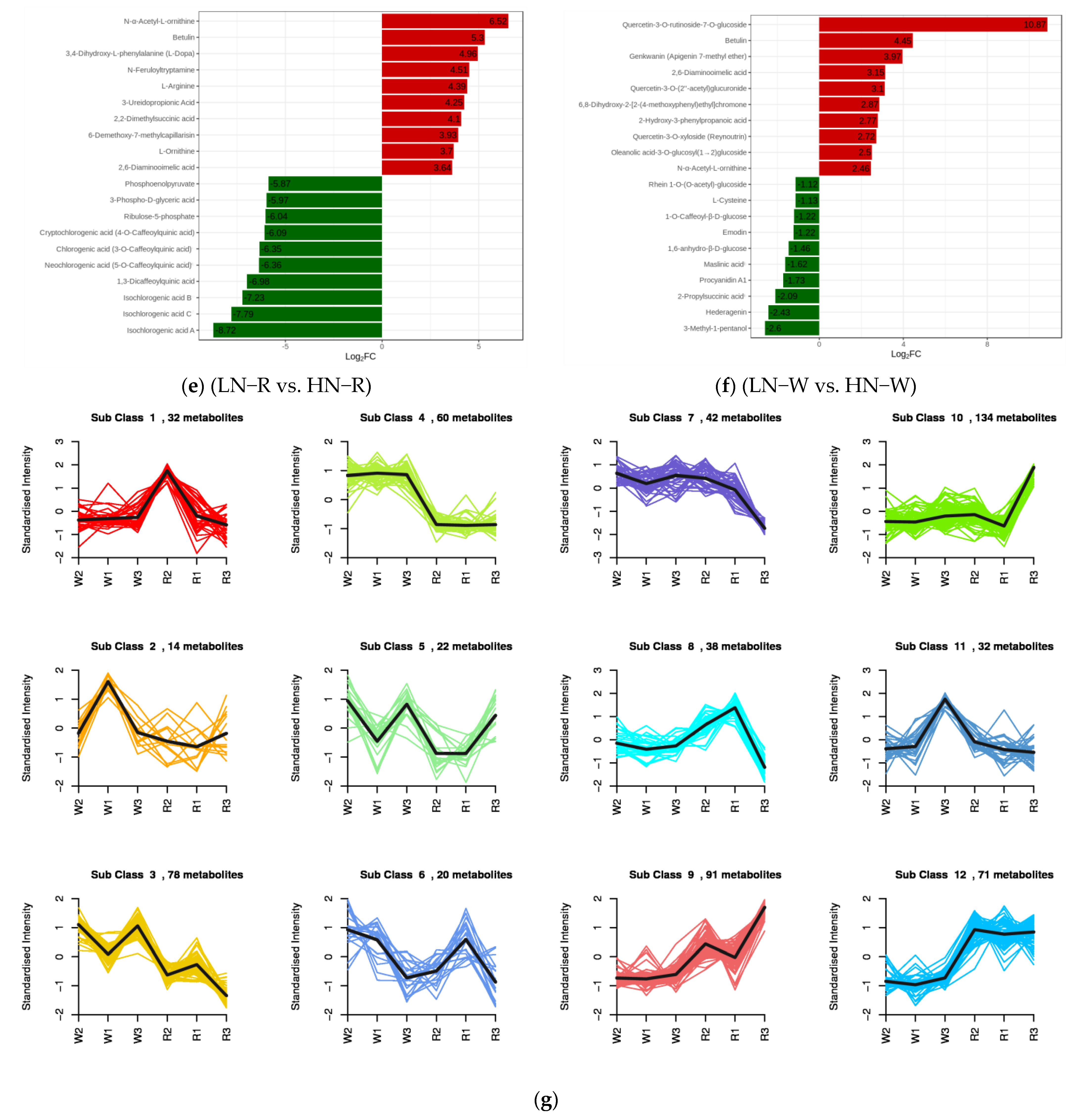
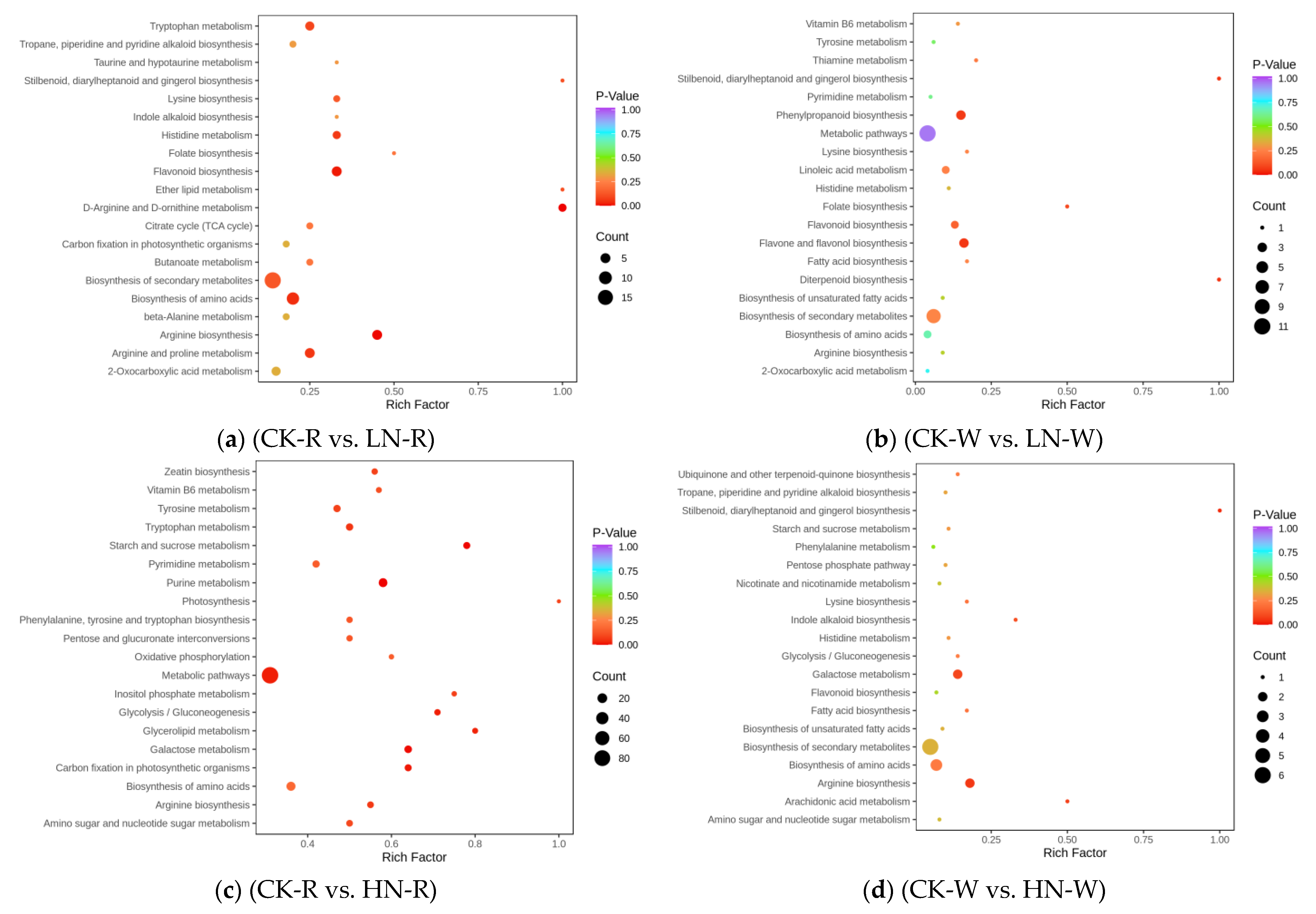
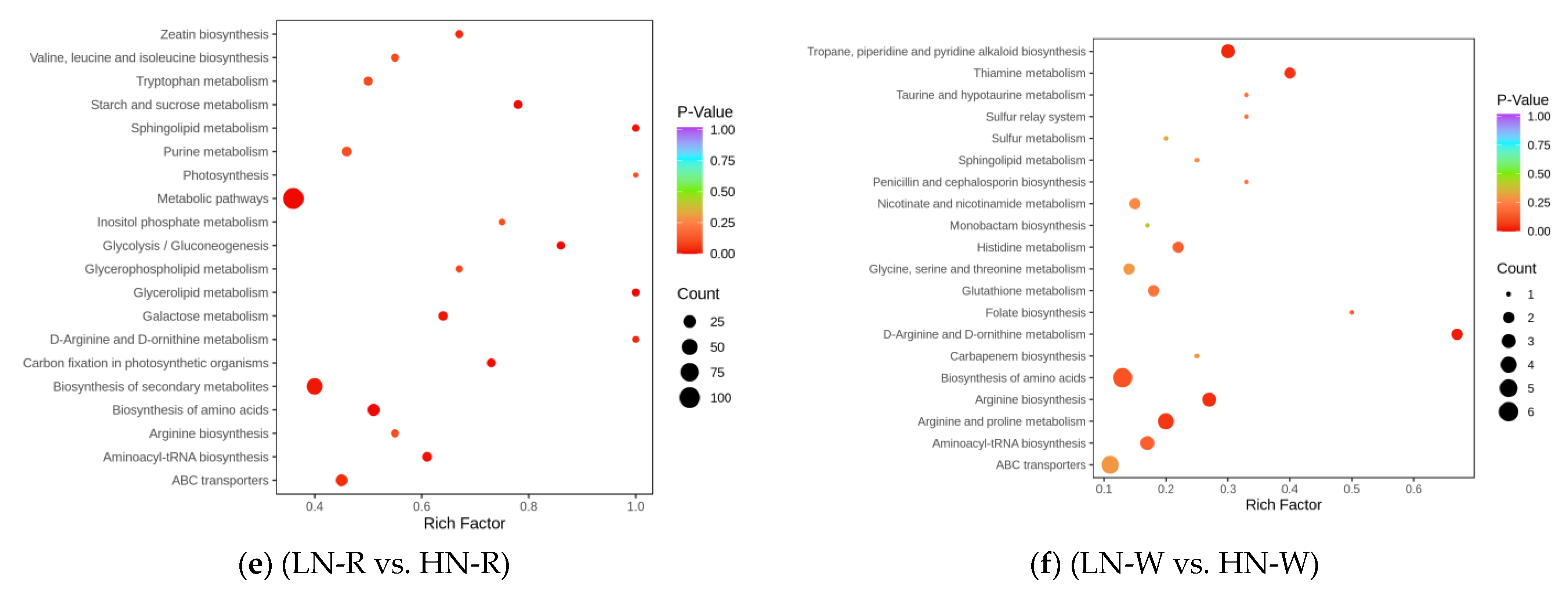
2.3. Screening of DEMs and Enrichment Analysis of the KEGG Pathways
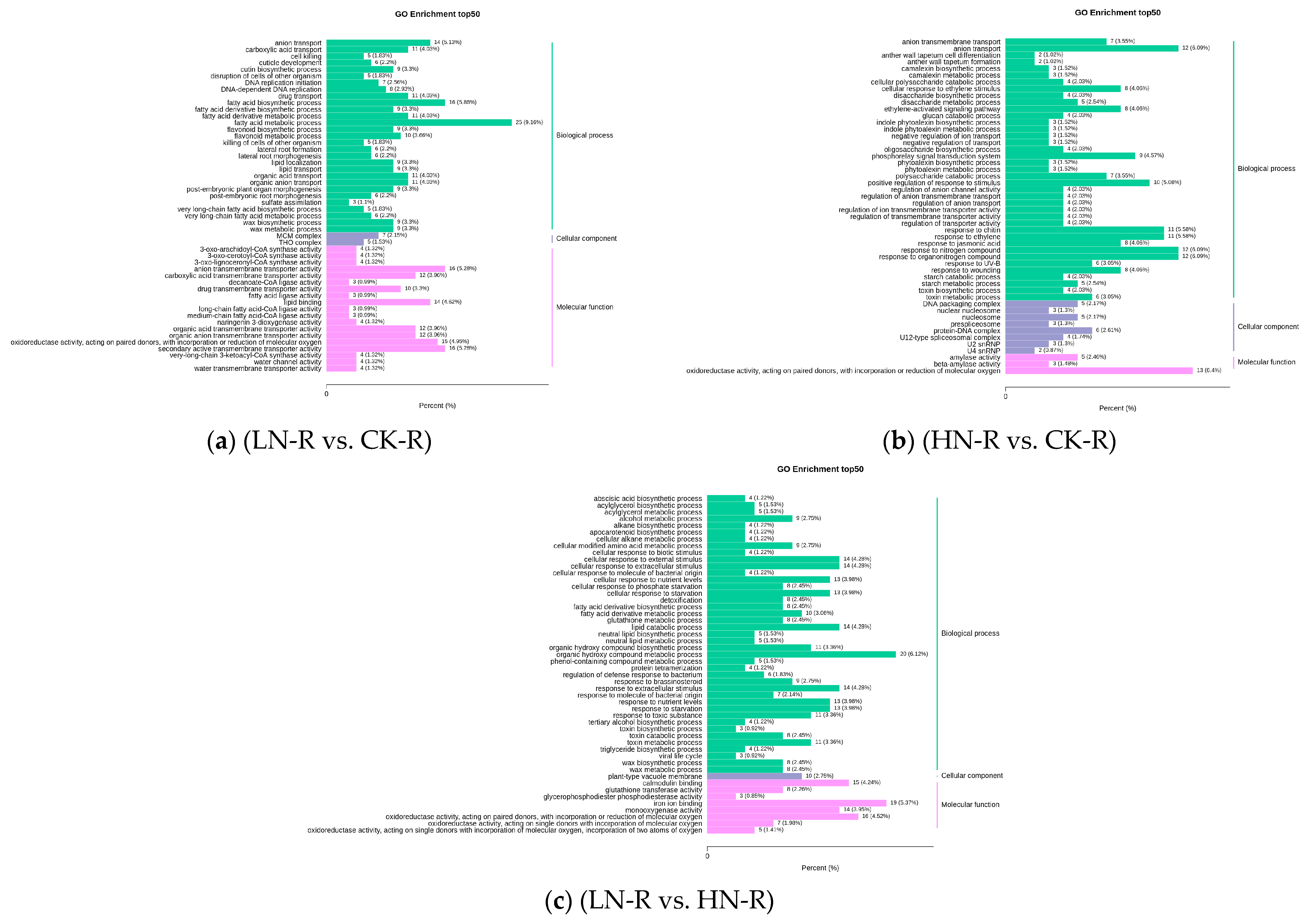
2.4. Transcriptome Analysis of Quinoa Seedlings under Differential Nitrogen Application
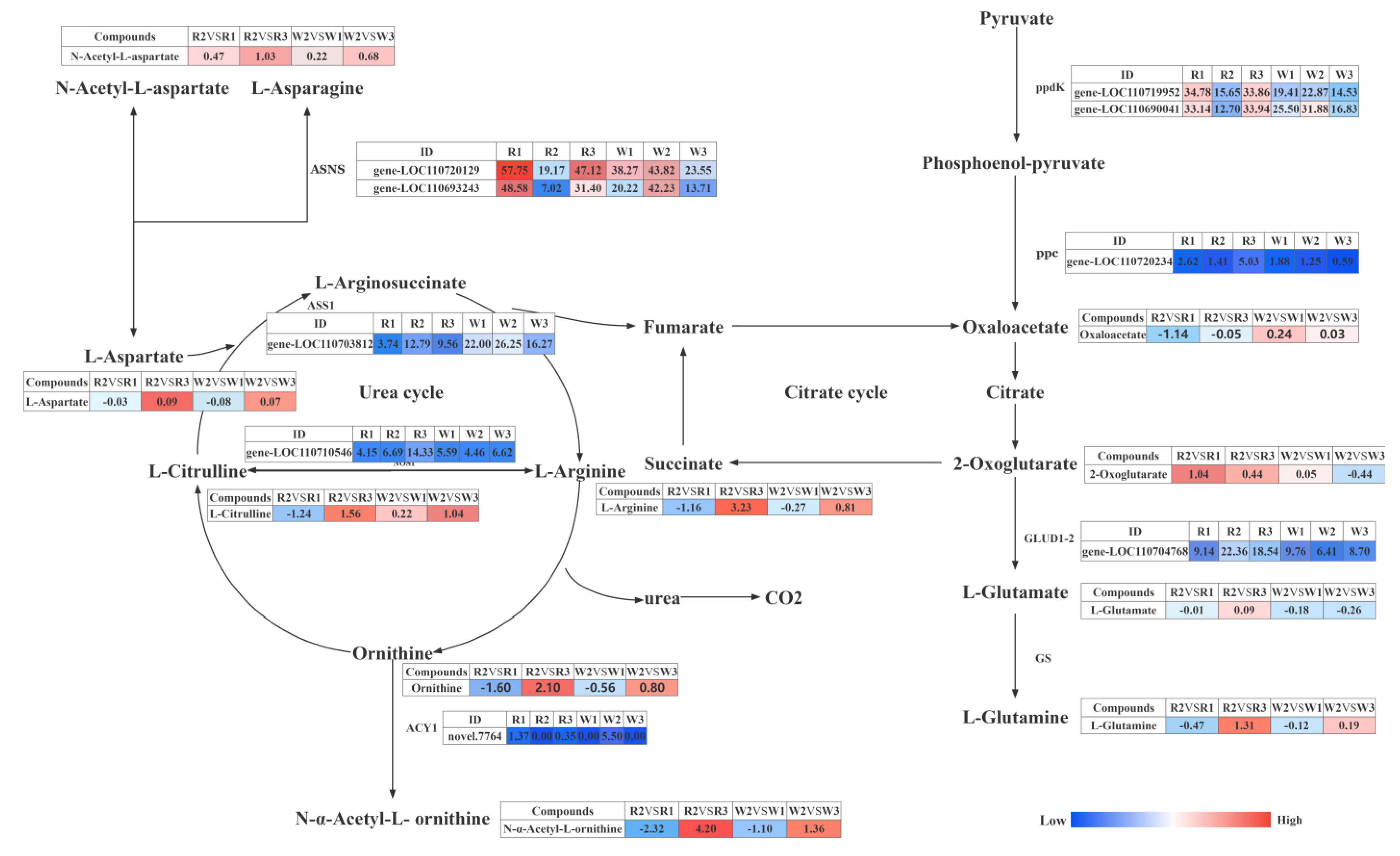
2.5. Transcriptome and Metabolome Analysis of the Nitrogen Regulation Mechanism in Quinoa Seedlings
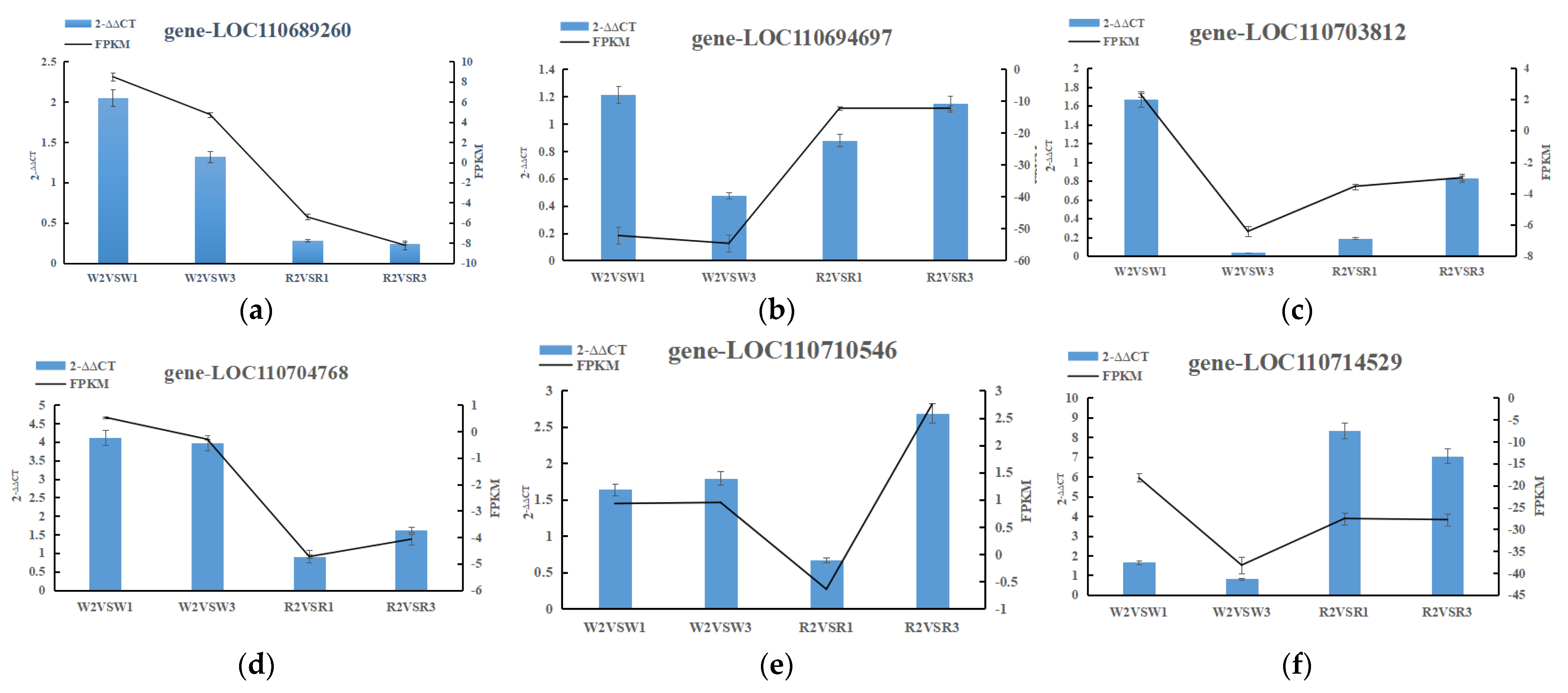
2.6. RT-qPCR
3. Discussion
4. Materials and Methods
4.1. Materials and Treatment
4.2. Morphological Data Collection
4.3. Metabolite Extraction and Qualitative and Quantitative Analysis
4.4. Transcriptome Sequencing and Data Analysis
4.5. Combined Transcriptome and Metabolome Analyses
4.6. RT-qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jarvis, D.E.; Ho, Y.S.; Lightfoot, D.J.; Schmöckel, S.M.; Li, B.; Borm, T.J.A.; Ohyanagi, H.; Mineta, K.; Michell, C.T.; Saber, N.; et al. The genome of Chenopodium quinoa. Nature 2017, 542, 307–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega-Gálvez, A.; Miranda, M.; Vergara, J.; Uribe, E.; Puente, L.; Martínez, E.A. Nutrition facts and functional potential of quinoa (Chenopodium quinoa willd.), an ancient Andean grain: A review. J. Sci. Food Agric. 2010, 90, 2541–2547. [Google Scholar] [CrossRef] [PubMed]
- Sobota, A.; Świeca, M.; Gęsiński, K.; Wirkijowska, A.; Bochnak, J. Yellow-coated quinoa (Chenopodium quinoa Willd)—physicochemical, nutritional, and antioxidant properties. J. Sci. Food Agric. 2020, 100, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- James, L.E.A. Quinoa (Chenopodium quinoa Willd.): Composition, chemistry, nutritional, and functional properties. Adv. Food Nutr. Res. 2009, 58, 1–31. [Google Scholar] [CrossRef]
- Nowak, V.; Du, J.; Charrondière, U.R. Assessment of the nutritional composition of quinoa (Chenopodium quinoa Willd). Food Chem. 2016, 193, 47–54. [Google Scholar] [CrossRef]
- Escribano, J.; Cabanes, J.; Jimenez-Atienzar, M.; Ibanez-Tremolada, M.; Rayda Gomez-Pando, L.; Garcia-Carmona, F. Characterization of betalains, saponins and antioxidant power in differently colored quinoa (Chenopodium quinoa) varieties. Food Chem. 2017, 234, 285–294. [Google Scholar] [CrossRef]
- Yao, T.; Rong, T. Phytochemicals in quinoa and amaranth grains and their antioxidant, anti-inflammatory, and potential health beneficial effects: A review. Mol. Nutr. Food Res. 2017, 61, 1600767. [Google Scholar] [CrossRef]
- Mhada, M.; Metougui, M.L.; El Hazzam, K.; El Kacimi, K.; Yasri, A. Variations of Saponins, Minerals and Total Phenolic Compounds Due to Processing and Cooking of Quinoa (Chenopodium quinoa Willd.) Seeds. Foods 2020, 9, 660. [Google Scholar] [CrossRef]
- Hussain, M.I.; Farooq, M.; Syed, Q.A.; Ishaq, A.; Al-Ghamdi, A.A.; Hatamleh, A.A. Botany, Nutritional Value, Phytochemical Composition and Biological Activities of Quinoa. Plants 2021, 10, 2258. [Google Scholar] [CrossRef]
- Sales, A.C.; Campos, C.N.S.; de Souza Junior, J.P. Silicon mitigates nutritional stress in quinoa (Chenopodium quinoa Willd.). Sci. Rep. 2021, 11, 14665. [Google Scholar] [CrossRef]
- Carciochi, R.A.; Galván-D’alessandro, L.; Vandendriessche, P.; Chollet, S. Effect of Germination and Fermentation Process on the Antioxidant Compounds of Quinoa Seeds. Plant Foods Hum. Nutr. 2016, 71, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lietz, G.; Bal, W.; Watson, A.; Morfey, B.; Seal, C. Effects of Quinoa (Chenopodium quinoa Willd.) Consumption on Markers of CVD Risk. Nutrients 2018, 10, 777. [Google Scholar] [CrossRef] [Green Version]
- Zeyneb, H.; Pei, H.; Cao, X.; Wang, Y.; Win, Y.; Gong, L. In vitro study of the effect of quinoa and quinoa polysaccharides on human gut microbiota. Food Sci. Nutr. 2021, 9, 5735–5745. [Google Scholar] [CrossRef]
- Korotkova, E.I.; Avramchik, O.A.; Kagiya, T.V.; Karbainov, Y.A.; Tcherdyntseva, N.V. Study of antioxidant properties of a water-soluble Vitamin E derivative—Tocopherol monoglucoside (TMG) by differential pulse voltammetry. Talanta 2004, 63, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Zhang, J.; Li, C. Effects of starter nitrogen fertilizer on soybean root activity, leaf photosynthesis and grain yield. PLoS ONE 2017, 12, e0174841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Li, G.; Li, W.; Liu, J.; Liu, M.; Jiang, C.; Li, Z. Nitrogen Fertilizer Deep Placement for Increased Grain Yield and Nitrogen Recovery Efficiency in Rice Grown in Subtropical China. Front. Plant Sci. 2017, 8, 1227. [Google Scholar] [CrossRef]
- Chung, R.S.; Wang, C.-H.; Wang, C.-W.; Wang, Y.-P. Influence of organic matter and inorganic fertilizer on the growth and nitrogen accumulation of corn plants. J. Plant Nutr. 2000, 23, 297–311. [Google Scholar] [CrossRef]
- Ahmed, M.; Rauf, M.; Mukhtar, Z.; Saeed, N.A. Excessive use of nitrogenous fertilizers: An unawareness causing serious threats to environment and human health. Environ. Sci. Pollut. Res. 2017, 24, 26983–26987. [Google Scholar] [CrossRef]
- Congreves, K.A.; Otchere, O.; Ferland, D.; Farzadfar, S.; Williams, S.; Arcand, M.M. Nitrogen Use Efficiency Definitions of Today and Tomorrow. Front. Plant Sci. 2021, 12, 637108. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, C.; Ma, X.; Liu, Q.; An, J.; Xu, S.; Xie, X.; Geng, J. Combining Organic Fertilizer with Controlled-Release Urea to Reduce Nitrogen Leaching and Promote Wheat Yields. Front. Plant Sci. 2021, 12, 802137. [Google Scholar] [CrossRef]
- Dong, N.-Q.; Lin, H.-X. Higher yield with less nitrogen fertilizer. Nat. Plants 2020, 6, 1078–1079. [Google Scholar] [CrossRef] [PubMed]
- Fradgley, N.S.; Bentley, A.R.; Swarbreck, S.M. Defining the physiological determinants of low nitrogen requirement in wheat. Biochem. Soc. Trans. 2021, 49, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Mosleth, E.F.; Wan, Y.; Lysenko, A.; Chope, G.A.; Penson, S.P.; Shewry, P.R.; Hawkesford, M.J. A novel approach to identify genes that determine grain protein deviation in cereals. Plant Biotechnol. J. 2015, 13, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.; Todd, C.D.; Trovato, M.; Forlani, G.; Funck, D. Physiological implications of arginine metabolism in plants. Front. Plant Sci. 2015, 6, 534. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Guo, Z.; Jiang, Y.; Qin, L.; Shi, Z.; Dong, L.; Xiong, L.; Yuan, R.; Deng, W.; Wu, H.; et al. Differential Metabolomic Responses of Kentucky Bluegrass Cultivars to Low Nitrogen Stress. Front. Plant Sci. 2022, 12, 808772. [Google Scholar] [CrossRef]
- Liang, J.; He, J. Protective role of anthocyanins in plants under low nitrogen stress. Biochem. Biophys. Res. Commun. 2018, 498, 946–953. [Google Scholar] [CrossRef]
- Fiehn, O.; Kopka, J.; Dörmann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000, 18, 1157–1161. [Google Scholar] [CrossRef]
- Qiu, Y.P.; Reed, D. Gas Chroma to Graphy in Metabolomics Study; Rijeka, M., Ed.; Intech: London, UK, 2014; pp. 83–101. [Google Scholar]
- Sun, T.; Zhang, J.; Zhang, Q.; Li, X.; Li, M.; Yang, Y.; Zhou, J.; Wei, Q.; Zhou, B. Transcriptome and metabolome analyses revealed the response mechanism of apple to different phosphorus stresses. Plant Physiol. Biochem. 2021, 167, 639–650. [Google Scholar] [CrossRef]
- Huang, T.; Zhang, X.; Wang, Q.; Guo, Y.; Xie, H.; Li, L.; Zhang, P.; Liu, J.; Qin, P. Metabolome and transcriptome profiles in quinoa seedlings in response to potassium supply. BMC Plant Biol. 2022, 22, 604. [Google Scholar] [CrossRef]
- Bascuñán-Godoy, L.; Sanhueza, C.; Pinto, K.; Cifuentes, L.; Reguera, M.; Briones, V.; Zurita-Silva, A.; Álvarez, R.; Morales, A.; Silva, H. Nitrogen physiology of contrasting genotypes of Chenopodium quinoa Willd. (Amaranthaceae) open. Sci. Rep. 2018, 8, 17524. [Google Scholar] [CrossRef] [Green Version]
- Ning, W.; Fengzhen, F.; Jinfeng, J.; Peng, W.; Xingmei, Z. Effects of irrigation and nitrogen on chlorophyll content, dry matter and nitrogen accumulation in sugar beet (Beta vulgaris L.). Sci. Rep. 2021, 11, 16651. [Google Scholar] [CrossRef]
- Filho, A.B.C.; Nascimento, C.S.; Pereira, B.D.J.; Nascimento, C.S. Nitrogen fertilisation impacts greenhouse gas emissions, carbon footprint, and agronomic responses of beet intercropped with arugula. J. Environ. Manag. 2022, 307, 114568. [Google Scholar] [CrossRef] [PubMed]
- Gadallah, F.M.; El-Sawah, N.A.; Belal, H.E.; Majrashi, A.; El-Tahan, A.M.; El-Saadony, M.T.; Elrys, A.S.; El-Saadony, F.M. Nitrogen-molybdenum-manganese co-fertilization reduces nitrate accumulation and enhances spinach (Spinacia oleracea L.) yield and its quality. Saudi J. Biol. Sci. 2021, 29, 2238–2246. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, G.S.; Eskin, N.A.M.; Tkachuk, R. Nutrients and antinutrients in quinoa seed. J. Cereal Chem. 1992, 69, 85–88. [Google Scholar] [CrossRef]
- Filho, A.M.M.; Pirozi, M.R.; Borges, J.T.D.S.; Sant’Ana, H.M.P.; Chaves, J.B.P.; Coimbra, J.S.D.R. Quinoa: Nutritional, functional, and antinutritional aspects. Crit. Rev. Food Sci. Nutr. 2017, 57, 1618–1630. [Google Scholar] [CrossRef]
- Cárdenas-Castillo, J.E.; Delatorre-Herrera, J.; Bascuñán-Godoy, L.; Rodriguez, J.P. Quinoa (Chenopodium quinoa Wild.) Seed Yield and Efficiency in Soils Deficient of Nitrogen in the Bolivian Altiplano: An Analytical Review. Plants 2021, 10, 2479. [Google Scholar] [CrossRef]
- Deng, Y.; Sun, X.; Zhang, Q.; Anwar, S.; Lu, J.; Guo, H.; Qin, L.; Zhang, L.; Wang, C. Comprehensive Evaluation and Physiological Response of Quinoa Genotypes to Low Nitrogen. Agronomy 2023, 13, 1597. [Google Scholar] [CrossRef]
- auf’m Erley, G.S.; Kaul, H.-P.; Kruse, M.; Aufhammer, W. Yield and nitrogen utilization efficiency of the pseudocereals amaranth, quinoa, and buckwheat under differing nitrogen fertilization. Eur. J. Agron. 2005, 22, 95–100. [Google Scholar] [CrossRef]
- Berti, M.; De Concepción, U.; Wilckens, R.; Hevia, F.; Serri, H.; Vidal, I.; Mendez, C. Fertilización Nitrogenada en Quinoa (Chenopodium quinoa Willd). J. Int. J. Agric. Nat. Resour. 2000, 27, 81–90. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Xu, L.; Wang, M.; Zhou, W.; Li, S.; Tan, W.; Wang, Q.; Xing, W.; Liu, D. Acclimation of sugar beet in morphological, physiological and BvAMT1.2 expression under low and high nitrogen supply. PLoS ONE 2022, 17, e0278327. [Google Scholar] [CrossRef]
- Tiwari, J.K.; Buckseth, T.; Zinta, R.; Saraswati, A.; Singh, R.K.; Rawat, S.; Dua, V.K.; Chakrabarti, S.K. Transcriptome analysis of potato shoots, roots and stolons under nitrogen stress. Sci. Rep. 2020, 10, 1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slocum, R.D. Genes, enzymes and regulation of arginine biosynthesis in plants. Plant Physiol. Biochem. 2005, 43, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Siddappa, S.; Marathe, G.K. What we know about plant arginases? Plant Physiol. Biochem. 2020, 156, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Feechan, A.; Yun, B.-W.; Shafiei, R.; Hofmann, A.; Taylor, P.; Xue, P.; Yang, F.-Q.; Xie, Z.-S.; Pallas, J.A.; et al. S-Nitrosylation of AtSABP3 Antagonizes the Expression of Plant Immunity. J. Biol. Chem. 2009, 284, 2131–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Ding, W.; Zhao, M.; Sun, B.; Zhang, L. Nitric oxide protects against oxidative stress under heat stress in the calluses from two ecotypes of reed. Plant Sci. 2006, 171, 449–458. [Google Scholar] [CrossRef]
- Barroso, J.B.; Corpas, F.J.; Carreras, A.; Sandalio, L.M.; Valderrama, R.; Palma, J.; Lupiáñez, J.A.; del Rı́o, L.A. Localization of Nitric-oxide Synthase in Plant Peroxisomes. J. Biol. Chem. 1999, 274, 36729–36733. [Google Scholar] [CrossRef] [Green Version]
- Girard-Thernier, C.; Pham, T.-N.; Demougeot, C. The Promise of Plant-Derived Substances as Inhibitors of Arginase. Mini-Reviews Med. Chem. 2015, 15, 798–808. [Google Scholar] [CrossRef]
- Sylvain, J.; Daniel, W.; Stuehr, D.J.; Lorenzo, L.; Michael, M.; Zhijian, T. Occurrence, structure, and evolution of nitric oxide synthase–like proteins in the plant kingdom. Sci. Signal. 2016, 9, re2. [Google Scholar] [CrossRef] [Green Version]
- Nie, Z.; Wang, J.; Rengel, Z.; Liu, H.; Zhao, P. Effects of nitrogen combined with zinc application on glutamate, glutamine, aspartate and asparagine accumulation in two winter wheat cultivars. Plant Physiol. Biochem. 2018, 127, 485–495. [Google Scholar] [CrossRef]
- Joshi, V.; Joshi, M.; Penalosa, A. Comparative analysis of tissue-specific transcriptomic responses to nitrogen stress in spinach (Spinacia oleracea). PLoS ONE 2020, 15, e0232011. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, Y.; Lin, Z.; Zhou, J.; Chai, H.; He, Q.; Li, Y.; Wang, J. The alleviative effect of exogenous phytohormones on the growth, physiology and gene expression of Tetraselmis cordiformis under high ammonia-nitrogen stress. Bioresour. Technol. 2019, 282, 339–347. [Google Scholar] [CrossRef]
- Lyu, X.; Sun, C.; Zhang, J.; Wang, C.; Zhao, S.; Ma, C.; Li, S.; Li, H.; Gong, Z.; Yan, C. Integrated Proteomics and Metabolomics Analysis of Nitrogen System Regulation on Soybean Plant Nodulation and Nitrogen Fixation. Int. J. Mol. Sci. 2022, 23, 2545. [Google Scholar] [CrossRef]
- Mauceri, A.; Aci, M.M.; Toppino, L.; Panda, S.; Meir, S.; Mercati, F.; Araniti, F.; Lupini, A.; Panuccio, M.R.; Rotino, G.L.; et al. Uncovering Pathways Highly Correlated to NUE through a Combined Metabolomics and Transcriptomics Approach in Eggplant. Plants 2022, 11, 700. [Google Scholar] [CrossRef] [PubMed]
- Oaks, A. Evidence for deamination by glutamate dehydrogenase in higher plants: Reply. Can. J. Bot. 2011, 73, 1116–1117. [Google Scholar] [CrossRef]
- Xian, L.; Zhang, Y.; Cao, Y.; Wan, T.; Gong, Y.; Dai, C. Glutamate dehydrogenase plays an important role in ammonium detoxification by submerged macrophytes. Sci. Total Environ. 2020, 722, 137. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Zhou, D.; Song, J.; Gao, J. Nitrogen Uptake and Distribution in Different Chinese Cabbage Genotypes under Low Nitrogen Stress. Int. J. Mol. Sci. 2022, 23, 1573. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Li, S.; Liao, P.; Cui, X.; Zheng, B.; Yang, Y.; Liu, D.; Zheng, Y. The transcriptome variations of Panaxnotoginseng roots treated with different forms of nitrogen fertilizers. BMC Genom. 2019, 20 (Suppl. 9), 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamani-Nour, S.; Lin, H.-C.; Walker, B.J.; Mettler-Altmann, T.; Khoshravesh, R.; Karki, S.; Bagunu, E.; Sage, T.L.; Quick, W.P.; Weber, A.P.M. Overexpression of the chloroplastic 2-oxoglutarate/malate transporter disturbs carbon and nitrogen homeostasis in rice. J. Exp. Bot. 2020, 72, 137–152. [Google Scholar] [CrossRef]
- Wang, J.; Song, K.; Sun, L.; Qin, Q.; Pan, J.; Xue, Y. Morphological and Transcriptome Analysis of Wheat Seedlings Response to Low Nitrogen Stress. Plants 2019, 8, 98. [Google Scholar] [CrossRef] [Green Version]
- Kogenaru, S.; Yan, Q.; Guo, Y.; Wang, N. RNA-seq and microarray complement each other in transcriptome profiling. BMC Genom. 2012, 13, 629. [Google Scholar] [CrossRef] [Green Version]
- Fraga, C.G.; Clowers, B.H.; Moore, R.J.; Zink, E.M. Signature-Discovery Approach for Sample Matching of a Nerve-Agent Precursor Using Liquid Chromatography−Mass Spectrometry, XCMS, and Chemometrics. Anal. Chem. 2010, 82, 4165–4173. [Google Scholar] [CrossRef]
- Eriksson, L.; Kettanehwold, N.; Trygg, J.; Wikström, C.; Wold, S. Multi- and Megavariate Data Analysis: Part I: Basic Principles and Applications; Umetrics Inc.: San Jose, CA, USA, 2006; Volume 8, pp. 263–336. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, R.; Song, Y.; He, J.; Sun, J.; Bai, J.; An, Z.; Dong, L.; Zhan, Q.; Abliz, Z. RRLC-MS/MS-based metabonomics combined with in-depth analysis of metabolic correlation network: Finding potential biomarkers for breast cancer. Anal 2009, 134, 2003–2011. [Google Scholar] [CrossRef]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the Human Adult Urinary Metabolome Variations with Age, Body Mass Index, and Gender by Implementing a Comprehensive Workflow for Univariate and OPLS Statistical Analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Ben, L.; Steven, L.S. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Geo, M.; Pertea, C.M.; Antonescu, T.-C.C.; Joshua, T.M.; Steven, L.S. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varet, H.; Loraine, B.-G.; Jean-Yves, C.; Marie-Agnès, D. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 2018, 34, 4313–4314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouhaddani, S.E.; Houwing-Duistermaat, J.; Salo, P.; Perola, M.; Jongbloed, G.; Uh, H.W. Evaluation of O2PLS in Omics data integration. In BMC Bioinformatics; BioMed Central: London, UK, 2016; Volume 17, pp. 117–132. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Gene-ID | EC | Metaname | Formula | Compound | PCC |
|---|---|---|---|---|---|---|
| CK vs. LN | gene-LOC110704768 | glutamate dehydrogenase | pme2380 | C5H6O5 | α-Ketoglutaric acid | −0.851 |
| CK vs. HN | gene-LOC110710546 | nitric oxide synthase | pme2527 | C5H12N2O2 | L-Ornithine | 0.84 |
| gene-LOC110710546 | nitric oxide synthase | pme0193 | C5H10N2O3 | L-Glutamine | 0.809 | |
| gene-LOC110710546 | nitric oxide synthase | mws0260 | C6H14N4O2 | L-Arginine | 0.864 | |
| gene-LOC110710546 | nitric oxide synthase | Zmyn000155 | C7H14N2O3 | N-α-Acetyl-L-ornithine | 0.800 |
| Quantity | Gene-ID | NCBI-Gene ID | Primer | 5′ to 3′ |
|---|---|---|---|---|
| 1 | gene-LOC110689260 | 110689260 | Forward Primer | CTCTGACTATGATTGAACA |
| Reverse Primer | CAATAGCAACCAAGAATG | |||
| 2 | gene-LOC110694697 | 110694697 | Forward Primer | TACTTCTCATACCCTATCA |
| Reverse Primer | CATCAACTTCTCACTGTA | |||
| 3 | gene-LOC110703812 | 110703812 | Forward Primer | CTTAATCCTGCTCTCAAT |
| Reverse Primer | ATTCCTGTCTCTGCTATA | |||
| 4 | gene-LOC110704768 | 110704768 | Forward Primer | GGTGTTATCATTCTTCCT |
| Reverse Primer | TCTTCAGTTCATTGTTCA | |||
| 5 | gene-LOC110710546 | 110710546 | Forward Primer | CTATATCTGACGCTCTTG |
| Reverse Primer | TCACATATTCACTCTCATC | |||
| 6 | gene-LOC110714529 | 110714529 | Forward Primer | GAATCCTCCATCTTACAG |
| Reverse Primer | CTTATCTTATGCTCTTCCA | |||
| Internal reference gene | TUB-6 | 831100 | Forward Primer | TGAGAACGCAGATGAGTGTATG |
| Reverse Primer | GAAACGAAGACAGCAAGTGACA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Wang, Q.; Huang, T.; Liu, J.; Zhang, P.; Li, L.; Xie, H.; Wang, H.; Liu, C.; Qin, P. Transcriptome and Metabolome Analyses Reveal Mechanisms Underlying the Response of Quinoa Seedlings to Nitrogen Fertilizers. Int. J. Mol. Sci. 2023, 24, 11580. https://doi.org/10.3390/ijms241411580
Li H, Wang Q, Huang T, Liu J, Zhang P, Li L, Xie H, Wang H, Liu C, Qin P. Transcriptome and Metabolome Analyses Reveal Mechanisms Underlying the Response of Quinoa Seedlings to Nitrogen Fertilizers. International Journal of Molecular Sciences. 2023; 24(14):11580. https://doi.org/10.3390/ijms241411580
Chicago/Turabian StyleLi, Hanxue, Qianchao Wang, Tingzhi Huang, Junna Liu, Ping Zhang, Li Li, Heng Xie, Hongxin Wang, Chenghong Liu, and Peng Qin. 2023. "Transcriptome and Metabolome Analyses Reveal Mechanisms Underlying the Response of Quinoa Seedlings to Nitrogen Fertilizers" International Journal of Molecular Sciences 24, no. 14: 11580. https://doi.org/10.3390/ijms241411580
APA StyleLi, H., Wang, Q., Huang, T., Liu, J., Zhang, P., Li, L., Xie, H., Wang, H., Liu, C., & Qin, P. (2023). Transcriptome and Metabolome Analyses Reveal Mechanisms Underlying the Response of Quinoa Seedlings to Nitrogen Fertilizers. International Journal of Molecular Sciences, 24(14), 11580. https://doi.org/10.3390/ijms241411580