
An Overview of PARP Resistance in Ovarian Cancer from a Molecular and Clinical Perspective
 ,
,  ,
,
Abstract
:1. Introduction
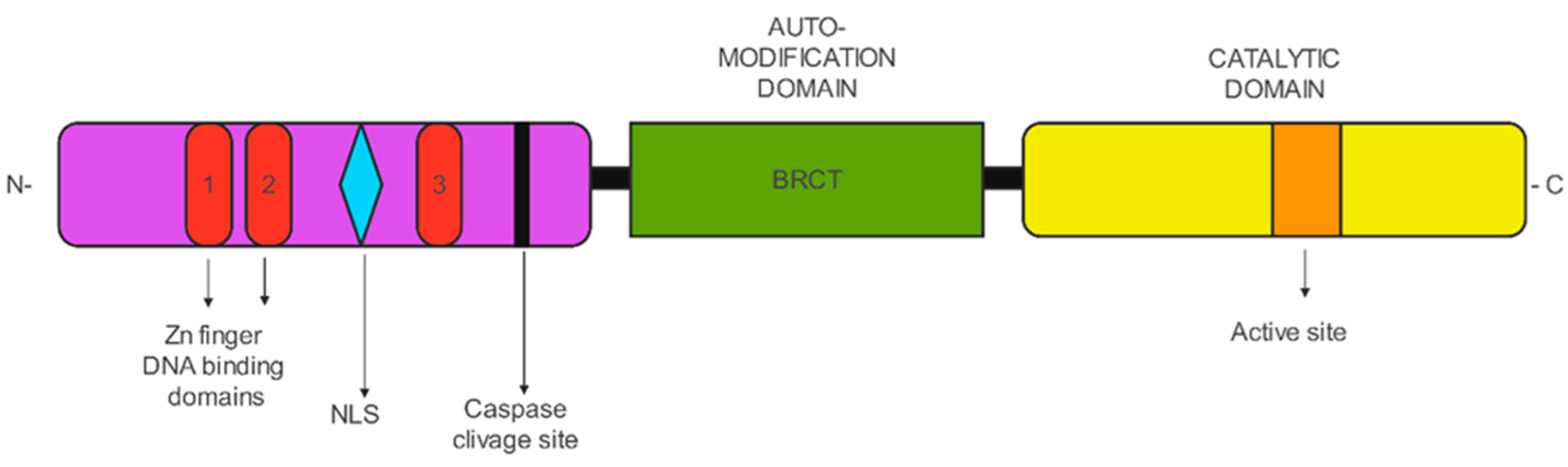
2. PARP
PARP Functions
- -
- Nucleotide excision repair (NER), a highly conserved DDR process that corrects a variety of genomic lesions [15];
- -
- -
- Mismatch repair (MMR) pathway that leads to the removal of DNA polymerase errors that appear during replication [18];
- -
- Homologous recombination (HR) that controls S and G2 phases [19]. The end effector in HR, RAD51 recombinase, is recruited by BRCA1 and BRCA2.
- -
- Non-homologous end joining recombination (NHEJ) is a different route for repairing DNA damage during the G1 phase of the cell cycle [19].
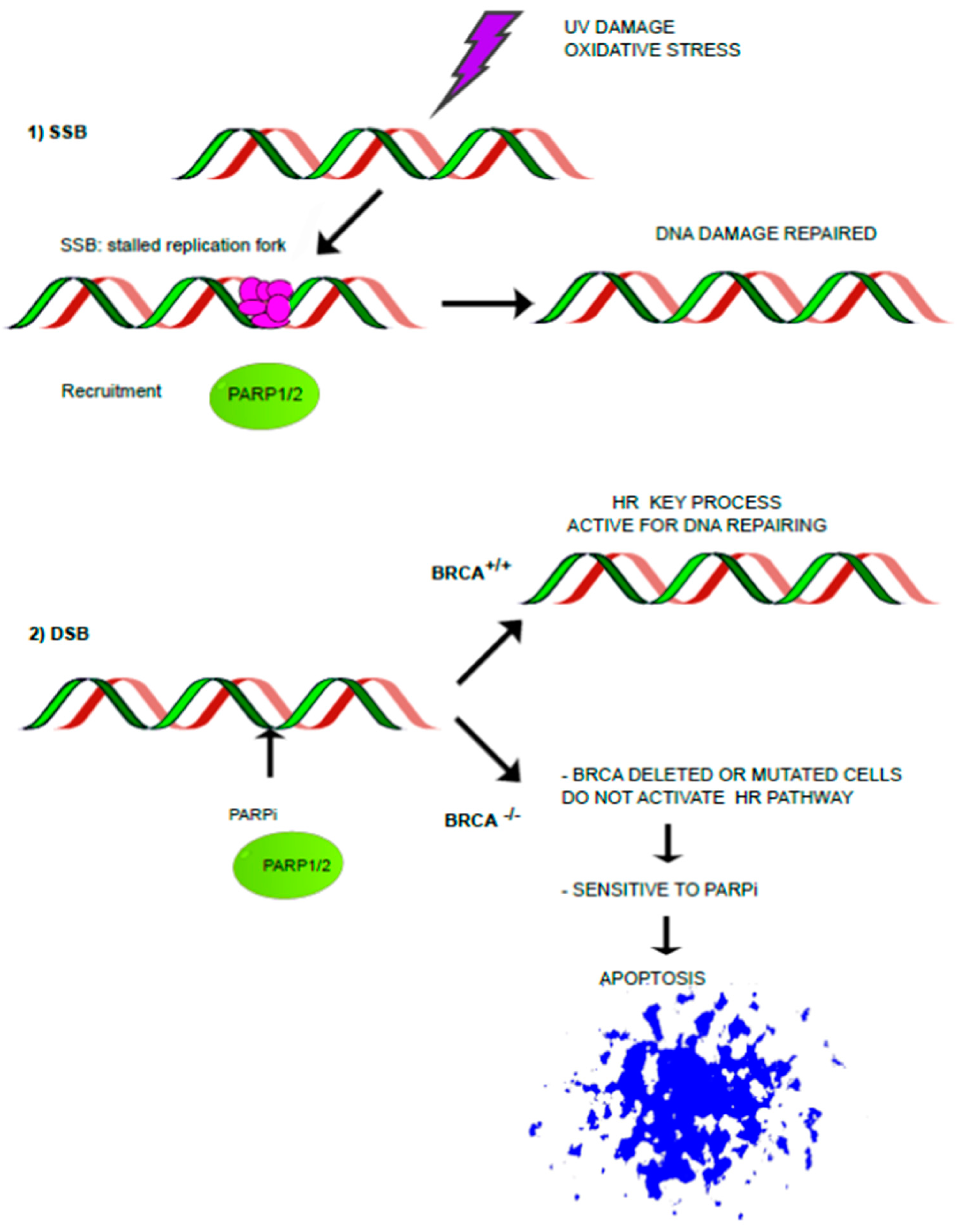
3. HRD and Ovarian Cancer
3.1. PARP-Is Resistance
3.2. Restoration of HRR
3.3. Mitigation of Replication Stress
3.4. Drug Efflux Pumps
4. Molecular Tools to Assess HRD
5. Strategies to Bypass Resistance
5.1. PARP Inhibitors and Chemotherapy
5.2. PARP Inhibitors and Immune Checkpoint Inhibitors

{kind=link}
{kind=link}
| Trial Name | Phase | PARPi | Number of Patients | Population | Treatment Arms | Primary Endpoint | ORR (%) | PFS Months | OS Months | Most Common G3-4 AEs (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| MEDIOLA (NCT02734004) [82,83,85] | I/II | olaparib | 32 + 31 | BRCA wild type PSR ovarian cancer | O + D (doublet cohort); O + D + B (triplet cohort) | ORR; safety | 31.3 vs. 77.4 | 5.5 vs. 14.7 | NA | anemia (17.6), increased lipase (11.8), neutropenia (8.8), lymphopenia (8.8) |
| GINECO BOLD (NCT04015739) [78] | II | olaparib | 74 | PSR and PRR ovarian cancer | O + D + B | DCR | 70 in PRR; 40 in PSR | 4.1 in PRR; 4.9 in PSR | 18.8 in PRR; 18.5 in PSR | NA |
| TOPACIO/KEYNOTE-162 (NCT02657889) [79] | I/II | niraparib | 62 | recurrent ovarian carcinoma, irrespective of BRCA mutation status | N + P | ORR | 18 | NA | NA | anemia (21); thrombocytopenia (9) |
| OPEB-01 (NCT04361370) [84] | II | olaparib | 22 | PSR BRCA Wild Type Ovarian Cancer | O + P + B | PFS rate of 6 months | 68.2 | NA | NA | NA |
| OPAL (NCT03574779) [80] | II | niraparib | 41 | PRR ovarian cancer | N + D + B | ORR | 17.9 | 7.6 | NA | hypertension (22), fatigue (17.1), and anemia (17.1) |
| MOONSTONE/GOG-3032 (NCT03955471) [81] | II | niraparib | 41 | PRR ovarian cancer | N + D | ORR | 12 | 2.1 | NA | NA |
| Trial Name | Phase | PARPi | Number of Patients | Population | Treatment Arms | Primary Endpoint |
|---|---|---|---|---|---|---|
| ATHENA-COMBO (NCT03522246) [87] | III | rucaparib | 1000 | Newly diagnosed advanced ovarian cancer after first line CT | R + Nivo/placebo | PFS |
| DUO-O (NCT03737643) [86] | III | olaparib | 1374 | Newly diagnosed advanced ovarian cancer after first line CT | B + O/placebo + D/placebo | PFS in non-tBRCA HRD positive patients; PFS in all non-tBRCA patients |
| FIRST (NCT03602859) [88] | III | niraparib | 1405 | Newly diagnosed advanced ovarian cancer after first line CT | N/placebo + Dosta/placebo | PFS for PD-L1 positive participants; PFS for all participants |
| KEYLYNK-001 (NCT03740165) [89] | III | olaparib | 1367 | Newly diagnosed advanced BRCA wild type ovarian cancer | CT + P/placebo followed by P/placebo + O/placebo | PFS for PD-L1 positive participants; PFS for all participants |
| ANITA (NCT03598270) [90] | III | niraparib | 414 | PSR ovarian cancer | Platinum-base CT + A/placebo followed by N + A/placebo | PFS |
| NItCHE-MITO33 (NCT04679064) [91] | III | niraparib | 427 | Recurrent ovarian cancer not candidate for platinum-base CT | CT vs. N + Dosta | OS |
5.3. PARP Inhibitors + Anti-Angiogenic Agents
5.4. PARP Inhibitors + BET Inhibitors
5.5. PARP Inhibitors + Radiation Therapy
6. ATR Inhibitors
7. CHK1 Inhibitors
8. WEE1 Inhibitors
9. Indirect Inhibition of HR
10. RAS/RAF/MEK
11. POLQ = DNA Polymerase θ
12. Rechallenge
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Summey, R.; Uyar, D. Ovarian cancer resistance to PARPi and platinum-containing chemotherapy. Cancer Drug Resist. 2022, 5, 637–646. [Google Scholar] [CrossRef]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Tan, D.S.P. The role of homologous recombination deficiency testing in ovarian cancer and its clinical implications: Do we need it? ESMO Open 2021, 6, 100144. [Google Scholar] [CrossRef] [PubMed]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar] [CrossRef]
- Langelier, M.F.; Zandarashvili, L.; Aguiar, P.M.; Black, B.E.; Pascal, J.M. NAD(+) analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nat. Commun. 2018, 9, 844. [Google Scholar] [CrossRef] [Green Version]
- Farres, J.; Martin-Caballero, J.; Martinez, C.; Lozano, J.J.; Llacuna, L.; Ampurdanes, C.; Ruiz-Herguido, C.; Dantzer, F.; Schreiber, V.; Villunger, A.; et al. Parp-2 is required to maintain hematopoiesis following sublethal gamma-irradiation in mice. Blood 2013, 122, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.S.; Challa, S.; Jones, A.; Kraus, W.L. PARPs and ADP-ribosylation in RNA biology: From RNA expression and processing to protein translation and proteostasis. Genes. Dev. 2020, 34, 302–320. [Google Scholar] [CrossRef] [PubMed]
- Yelamos, J.; Farres, J.; Llacuna, L.; Ampurdanes, C.; Martin-Caballero, J. PARP-1 and PARP-2: New players in tumour development. Am. J. Cancer Res. 2011, 1, 328–346. [Google Scholar]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; de Azambuja, E.; Azim, H.A., Jr.; Piccart, M. An update on PARP inhibitors—moving to the adjuvant setting. Nat. Rev. Clin. Oncol. 2015, 12, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Spivak, G. Nucleotide excision repair in humans. DNA Repair 2015, 36, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Dianov, G.L.; Hubscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 41, 3483–3490. [Google Scholar] [CrossRef]
- Ukraintsev, A.A.; Belousova, E.A.; Kutuzov, M.M.; Lavrik, O.I. Study of Interaction of the PARP Family DNA-Dependent Proteins with Nucleosomes Containing DNA Intermediates of the Initial Stages of BER Process. Biochemistry 2022, 87, 331–345. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu. Rev. Genet. 2015, 49, 291–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Dziadkowiec, K.N.; Gasiorowska, E.; Nowak-Markwitz, E.; Jankowska, A. PARP inhibitors: Review of mechanisms of action and BRCA1/2 mutation targeting. Prz. Menopauzalny 2016, 15, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Malanga, M.; Althaus, F.R. The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem. Cell Biol. 2005, 83, 354–364. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Huang, J. DNA double-strand break repair pathway choice: The fork in the road. Genome Instab. Dis. 2020, 1, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Trego, K.S.; Groesser, T.; Davalos, A.R.; Parplys, A.C.; Zhao, W.; Nelson, M.R.; Hlaing, A.; Shih, B.; Rydberg, B.; Pluth, J.M.; et al. Non-catalytic Roles for XPG with BRCA1 and BRCA2 in Homologous Recombination and Genome Stability. Mol. Cell 2016, 61, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, J.; Jung, K.; Luger, K. Inhibitors of PARP: Number crunching and structure gazing. Proc. Natl. Acad. Sci. USA 2022, 119, e2121979119. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschetta, M.; George, A.; Kaye, S.B.; Banerjee, S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann. Oncol. 2016, 27, 1449–1455. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef] [Green Version]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Sahnane, N.; Carnevali, I.; Formenti, G.; Casarin, J.; Facchi, S.; Bombelli, R.; Di Lauro, E.; Memoli, D.; Salvati, A.; Rizzo, F.; et al. BRCA Methylation Testing Identifies a Subset of Ovarian Carcinomas without Germline Variants that Can Benefit from PARP Inhibitor. Int. J. Mol. Sci. 2020, 21, 9708. [Google Scholar] [CrossRef]
- Cunningham, J.M.; Cicek, M.S.; Larson, N.B.; Davila, J.; Wang, C.; Larson, M.C.; Song, H.; Dicks, E.M.; Harrington, P.; Wick, M.; et al. Clinical characteristics of ovarian cancer classified by BRCA1, BRCA2, and RAD51C status. Sci. Rep. 2014, 4, 4026. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, D.J.; Konner, J.A.; Bell-McGuinn, K.M.; Bhatia, J.; Sabbatini, P.; Aghajanian, C.A.; Offit, K.; Barakat, R.R.; Spriggs, D.R.; Kauff, N.D. Survival in epithelial ovarian cancer: A multivariate analysis incorporating BRCA mutation status and platinum sensitivity. Ann. Oncol. 2011, 22, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.M.; Brady, M.F.; Harrell, M.I.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Burger, R.A.; Tewari, K.S.; et al. Mutations in Homologous Recombination Genes and Outcomes in Ovarian Carcinoma Patients in GOG 218: An NRG Oncology/Gynecologic Oncology Group Study. Clin. Cancer Res. 2018, 24, 777–783. [Google Scholar] [CrossRef] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Barber, L.J.; Sandhu, S.; Chen, L.; Campbell, J.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Rodrigues, D.N.; Reis Filho, J.S.; Moreno, V.; et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 2013, 229, 422–429. [Google Scholar] [CrossRef]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Tobalina, L.; Armenia, J.; Irving, E.; O’Connor, M.J.; Forment, J.V. A meta-analysis of reversion mutations in BRCA genes identifies signatures of DNA end-joining repair mechanisms driving therapy resistance. Ann. Oncol. 2021, 32, 103–112. [Google Scholar] [CrossRef]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Ter Brugge, P.; Kristel, P.; van der Burg, E.; Boon, U.; de Maaker, M.; Lips, E.; Mulder, L.; de Ruiter, J.; Moutinho, C.; Gevensleben, H.; et al. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw148. [Google Scholar] [CrossRef]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swisher, E.M.; Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Coleman, R.L.; Aghajanian, C.; Konecny, G.E.; O’Malley, D.M.; et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat. Commun. 2021, 12, 2487. [Google Scholar] [CrossRef]
- Cao, L.; Xu, X.; Bunting, S.F.; Liu, J.; Wang, R.H.; Cao, L.L.; Wu, J.J.; Peng, T.N.; Chen, J.; Nussenzweig, A.; et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell 2009, 35, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef] [PubMed]
- He, Y.J.; Meghani, K.; Caron, M.C.; Yang, C.; Ronato, D.A.; Bian, J.; Sharma, A.; Moore, J.; Niraj, J.; Detappe, A.; et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 2018, 563, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.R.; Cuella-Martin, R.; Barazas, M.; Liu, R.; Oliveira, C.; Oliver, A.W.; Bilham, K.; Holt, A.B.; Blackford, A.N.; Heierhorst, J.; et al. The ASCIZ-DYNLL1 axis promotes 53BP1-dependent non-homologous end joining and PARP inhibitor sensitivity. Nat. Commun. 2018, 9, 5406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paes Dias, M.; Tripathi, V.; van der Heijden, I.; Cong, K.; Manolika, E.M.; Bhin, J.; Gogola, E.; Galanos, P.; Annunziato, S.; Lieftink, C.; et al. Loss of nuclear DNA ligase III reverts PARP inhibitor resistance in BRCA1/53BP1 double-deficient cells by exposing ssDNA gaps. Mol. Cell 2021, 81, 4692–4708.e9. [Google Scholar] [CrossRef] [PubMed]
- Thakar, T.; Moldovan, G.L. The emerging determinants of replication fork stability. Nucleic Acids Res. 2021, 49, 7224–7238. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolinjivadi, A.M.; Sannino, V.; De Antoni, A.; Zadorozhny, K.; Kilkenny, M.; Techer, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881.e7. [Google Scholar] [CrossRef] [Green Version]
- Dungrawala, H.; Bhat, K.P.; Le Meur, R.; Chazin, W.J.; Ding, X.; Sharan, S.K.; Wessel, S.R.; Sathe, A.A.; Zhao, R.; Cortez, D. RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol. Cell 2017, 67, 374–386.e5. [Google Scholar] [CrossRef]
- Freimund, A.E.; Beach, J.A.; Christie, E.L.; Bowtell, D.D.L. Mechanisms of Drug Resistance in High-Grade Serous Ovarian Cancer. Hematol. Oncol. Clin. N. Am. 2018, 32, 983–996. [Google Scholar] [CrossRef]
- Lawlor, D.; Martin, P.; Busschots, S.; Thery, J.; O’Leary, J.J.; Hennessy, B.T.; Stordal, B. PARP Inhibitors as P-glyoprotein Substrates. J. Pharm. Sci. 2014, 103, 1913–1920. [Google Scholar] [CrossRef]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat. Commun. 2019, 10, 1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaya, H.; Nakai, H.; Takamatsu, S.; Mandai, M.; Matsumura, N. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Sci. Rep. 2020, 10, 2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquard, A.M.; Eklund, A.C.; Joshi, T.; Krzystanek, M.; Favero, F.; Wang, Z.C.; Richardson, A.L.; Silver, D.P.; Szallasi, Z.; Birkbak, N.J. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark. Res. 2015, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujade-Lauraine, E.; Christinat, Y.; D’incalci, M.; Schouten, P.; Buisson, A.; Heukamp, L.; Gorp, T.V.; Kramer, C.; Mckee, T.; Marchini, S.; et al. Homologous recombination deficiency testing in advanced ovarian cancer: Description of the ENGOT HRD European initiative. Int. J. Gynecol. Cancer 2021, 31 (Suppl. S3), A208. [Google Scholar]
- Christinat, Y.; Ho, L.; Clément, S.; Genestie, C.; Sehouli, J.; Martín, A.G.; Denison, U.; Fujiwara, K.; Vergote, I.; Tognon, G.; et al. 2022-RA-567-ESGO The Geneva HRD test: Clinical validation on 469 samples from the PAOLA-1 trial. Ovarian Cancer 2022, 32 (Suppl. S2), A238.2–A239. [Google Scholar] [CrossRef]
- Willing, E.-M.; Vollbrecht, C.; Voessing, C.; Weist, P.; Schallenberg, S.; Jori, B.; Tiemann, M.; Bataillon, G.; Harter, P.; Pignata, S.; et al. 2022-RA-873-ESGO Validation study of the ‘NOGGO-GIS ASSAY’ based on ovarian cancer samples from the first-line PAOLA-1/ENGOT-ov25 phase-III trial. Int. J. Gynecol. Cancer 2022, 32 (Suppl. S2), A370. [Google Scholar]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.; Sonke, G.S.; Colombo, N.; Spacek, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Gray, H.J.; Bell-McGuinn, K.; Fleming, G.F.; Cristea, M.; Xiong, H.; Sullivan, D.; Luo, Y.; McKee, M.D.; Munasinghe, W.; Martin, L.P. Phase I combination study of the PARP inhibitor veliparib plus carboplatin and gemcitabine in patients with advanced ovarian cancer and other solid malignancies. Gynecol. Oncol. 2018, 148, 507–514. [Google Scholar] [CrossRef]
- Hjortkjaer, M.; Kanstrup, H.; Jakobsen, A.; Steffensen, K.D. Veliparib and topotecan for patients with platinum-resistant or partially platinum-sensitive relapse of epithelial ovarian cancer with BRCA negative or unknown BRCA status. Cancer Treat. Res. Commun. 2018, 14, 7–12. [Google Scholar] [CrossRef]
- Kummar, S.; Oza, A.M.; Fleming, G.F.; Sullivan, D.M.; Gandara, D.R.; Naughton, M.J.; Villalona-Calero, M.A.; Morgan, R.J., Jr.; Szabo, P.M.; Youn, A.; et al. Randomized Trial of Oral Cyclophosphamide and Veliparib in High-Grade Serous Ovarian, Primary Peritoneal, or Fallopian Tube Cancers, or BRCA-Mutant Ovarian Cancer. Clin. Cancer Res. 2015, 21, 1574–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freyer, G.; Floquet, A.; Tredan, O.; Langlois-Jacques, C.; Selle, F.; Abdeddaim, C.; Leary, A.; Dubot, C.; Fabbro, M.; Gladieff, L.; et al. 733P Bevacizumab (Bev), olaparib (Ola) and durvalumab (Durva) in patients with recurrent advanced ovarian cancer (AOC): The GINECO BOLD study. Ann. Oncol. 2021, 32, S734–S735. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Gaillard, S.; Hendrickson, A.W.; Moroney, J.; Yeku, O.; Diver, E.; Gunderson, C.; Arend, R.; Ratner, E.; Samnotra, V.; et al. An open-label phase II study of dostarlimab (TSR-042), bevacizumab (bev), and niraparib combination in patients (pts) with platinum-resistant ovarian cancer (PROC): Cohort A of the OPAL trial. Gynecol. Oncol. 2021, 162, S17–S18. [Google Scholar] [CrossRef]
- Randall, L.M.; O’Malley, D.M.; Monk, B.J.; Coleman, R.L.; Gaillard, S.; Adams, S.F.; Duska, L.R.; Cappuccini, F.; Dalton, H.; Holloway, R.W.; et al. MOONSTONE/GOG-3032: Interim analysis of a phase 2 study of niraparib + dostarlimab in patients (pts) with platinum-resistant ovarian cancer (PROC). J. Clin. Oncol. 2022, 40 (Suppl. S16), 5573. [Google Scholar] [CrossRef]
- Drew, Y.; Kaufman, B.; Banerjee, S.; Lortholary, A.; Hong, S.H.; Park, Y.H.; Zimmermann, S.; Roxburgh, P.; Ferguson, M.; Alvarez, R.H.; et al. 1190PD—Phase II study of olaparib + durvalumab (MEDIOLA): Updated results in germline BRCA-mutated platinum-sensitive relapsed (PSR) ovarian cancer (OC). Ann. Oncol. 2019, 30, v485–v486. [Google Scholar] [CrossRef]
- Drew, Y.; Penson, R.T.; O’Malley, D.M.; Kim, J.W.; Zimmermann, S.; Roxburgh, P.; Sohn, J.; Stemmer, S.M.; Bastian, S.; Ferguson, M.; et al. 814MO Phase II study of olaparib (O) plus durvalumab (D) and bevacizumab (B) (MEDIOLA): Initial results in patients (pts) with non-germline BRCA-mutated (non-gBRCAm) platinum sensitive relapsed (PSR) ovarian cancer (OC). Ann. Oncol. 2020, 31, S615–S616. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lim, M.C.; Kim, B.G.; Ngoi, N.Y.; Choi, C.H.; Park, S.Y.; Tan, D.S.; Go, Y.; Lee, J.Y. A single-arm phase II study of olaparib maintenance with pembrolizumab and bevacizumab in BRCA non-mutated patients with platinum-sensitive recurrent ovarian cancer (OPEB-01). J. Gynecol. Oncol. 2021, 32, e31. [Google Scholar] [CrossRef]
- Banerjee, S.; Imbimbo, M.; Roxburgh, P.; Kim, J.W.; Kim, M.H.; Plummer, R.; Stemmer, S.; You, B.; Ferguson, M.; Penson, R.T.; et al. 529MO Phase II study of olaparib plus durvalumab with or without bevacizumab (MEDIOLA): Final analysis of overall survival in patients with non-germline BRCA-mutated platinum-sensitive relapsed ovarian cancer. Ann. Oncol. 2022, 33, S788–S789. [Google Scholar] [CrossRef]
- Harter, P.; Trillsch, F.; Okamoto, A.; Reuss, A.; Kim, J.-W.; Rubio-Pérez, M.J.; Vardar, M.A.; Scambia, G.; Tredan, O.; Nyvang, G.-B.; et al. Durvalumab with paclitaxel/carboplatin (PC) and bevacizumab (bev), followed by maintenance durvalumab, bev, and olaparib in patients (pts) with newly diagnosed advanced ovarian cancer (AOC) without a tumor BRCA1/2 mutation (non-tBRCAm): Results from the randomized, placebo (pbo)-controlled phase III DUO-O trial. J. Clin. Oncol. 2023, 41 (Suppl. S17), LBA5506. [Google Scholar]
- Monk, B.J.; Coleman, R.L.; Fujiwara, K.; Wilson, M.K.; Oza, A.M.; Oaknin, A.; O’Malley, D.M.; Lorusso, D.; Westin, S.N.; Safra, T.; et al. ATHENA (GOG-3020/ENGOT-ov45): A randomized, phase III trial to evaluate rucaparib as monotherapy (ATHENA-MONO) and rucaparib in combination with nivolumab (ATHENA-COMBO) as maintenance treatment following frontline platinum-based chemotherapy in ovarian cancer. Int. J. Gynecol. Cancer 2021, 31, 1589–1594. [Google Scholar] [PubMed]
- Hardy-Bessard, A.-C.; Moore, K.N.; Mirza, M.R.; Asselain, B.; Redondo, A.; Pfisterer, J.; Pignata, S.; Provencher, D.M.; Cibula, D.; Reyners, A.K.L.; et al. ENGOT-OV44/FIRST study: A randomized, double-blind, adaptive, phase III study of platinum-based therapy with dostarlimab (TSR-042) + niraparib versus standard-of-care (SOC) platinum-based therapy as first-line treatment of stage 3/4 non-mucinous epithelial ovarian cancer (OC). J. Clin. Oncol. 2019, 37 (Suppl. S15), TPS5600. [Google Scholar]
- Vergote, I.; Sehouli, J.; Salutari, V.; Zola, P.; Madry, R.; Wenham, R.M.; Korach, J.; Pautier, P.; Cibula, D.; Lheureux, S.; et al. ENGOT-OV43/KEYLYNK-001: A phase III, randomized, double-blind, active- and placebo-controlled study of pembrolizumab plus chemotherapy with olaparib maintenance for first-line treatment of BRCA-nonmutated advanced epithelial ovarian cancer. J. Clin. Oncol. 2019, 37 (Suppl. S15), TPS5603. [Google Scholar] [CrossRef]
- Gonzalez Martin, A.; Sanchez Lorenzo, L.; Colombo, N.; dePont Christensen, R.; Heitz, F.; Meirovitz, M.; Selle, F.; van Gorp, T.; Alvarez, N.; Sanchez, J.; et al. A phase III, randomized, double blinded trial of platinum based chemotherapy with or without atezolizumab followed by niraparib maintenance with or without atezolizumab in patients with recurrent ovarian, tubal, or peritoneal cancer and platinum treatment free interval of more than 6 months: ENGOT-Ov41/GEICO 69-O/ANITA Trial. Int. J. Gynecol. Cancer 2021, 31, 617–622. [Google Scholar]
- Musacchio, L.; Salutari, V.; Pignata, S.; Braicu, E.; Cibula, D.; Colombo, N.; Frenel, J.S.; Zagouri, F.; Carbone, V.; Ghizzoni, V.; et al. Randomized phase III trial on niraparib-TSR-042 (dostarlimab) versus physician’s choice chemotherapy in recurrent ovarian, fallopian tube, or primary peritoneal cancer patients not candidate for platinum retreatment: NItCHE trial (MITO 33). Int. J. Gynecol. Cancer 2021, 31, 1369–1373. [Google Scholar] [CrossRef] [PubMed]
- Cannistra, S.A.; Matulonis, U.A.; Penson, R.T.; Hambleton, J.; Dupont, J.; Mackey, H.; Douglas, J.; Burger, R.A.; Armstrong, D.; Wenham, R.; et al. Phase II study of bevacizumab in patients with platinum-resistant ovarian cancer or peritoneal serous cancer. J. Clin. Oncol. 2007, 25, 5180–5186. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Berlin, S.; Ivy, P.; Tyburski, K.; Krasner, C.; Zarwan, C.; Berkenblit, A.; Campos, S.; Horowitz, N.; Cannistra, S.A.; et al. Cediranib, an oral inhibitor of vascular endothelial growth factor receptor kinases, is an active drug in recurrent epithelial ovarian, fallopian tube, and peritoneal cancer. J. Clin. Oncol. 2009, 27, 5601–5606. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C.; Blank, S.V.; Goff, B.A.; Judson, P.L.; Teneriello, M.G.; Husain, A.; Sovak, M.A.; Yi, J.; Nycum, L.R. OCEANS: A randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J. Clin. Oncol. 2012, 30, 2039–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poveda, A.M.; Selle, F.; Hilpert, F.; Reuss, A.; Savarese, A.; Vergote, I.; Witteveen, P.; Bamias, A.; Scotto, N.; Mitchell, L.; et al. Bevacizumab Combined with Weekly Paclitaxel, Pegylated Liposomal Doxorubicin, or Topotecan in Platinum-Resistant Recurrent Ovarian Cancer: Analysis by Chemotherapy Cohort of the Randomized Phase III AURELIA Trial. J. Clin. Oncol. 2015, 33, 3836–3838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Perol, D.; Gonzalez-Martin, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Maenpaa, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Leary, A.; Pignata, S.; Cropet, C.; Gonzalez-Martin, A.; Marth, C.; Nagao, S.; Vergote, I.; Colombo, N.; Maenpaa, J.; et al. Olaparib plus bevacizumab first-line maintenance in ovarian cancer: Final overall survival results from the PAOLA-1/ENGOT-ov25 trial. Ann. Oncol. 2023, 34, 681–692. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.F.; Brady, M.F.; Matulonis, U.A.; Miller, A.; Kohn, E.C.; Swisher, E.M.; Cella, D.; Tew, W.P.; Cloven, N.G.; Muller, C.Y.; et al. Olaparib with or without Cediranib versus Platinum-Based Chemotherapy in Recurrent Platinum-Sensitive Ovarian Cancer (NRG-GY004): A Randomized, Open-Label, Phase III Trial. J. Clin. Oncol. 2022, 40, 2138–2147. [Google Scholar] [CrossRef]
- Lee, J.M.; Moore, R.G.; Ghamande, S.; Park, M.S.; Diaz, J.P.; Chapman, J.; Kendrick, J.; Slomovitz, B.M.; Tewari, K.S.; Lowe, E.S.; et al. Cediranib in Combination with Olaparib in Patients without a Germline BRCA1/2 Mutation and with Recurrent Platinum-Resistant Ovarian Cancer: Phase IIb CONCERTO Trial. Clin. Cancer Res. 2022, 28, 4186–4193. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Tomao, F.; Benedetti Panici, P.; Nicoletto, M.O.; Tognon, G.; Bologna, A.; Lissoni, A.A.; DeCensi, A.; Lapresa, M.; Mancari, R.; et al. Randomized phase II trial of weekly paclitaxel vs. cediranib-olaparib (continuous or intermittent schedule) in platinum-resistant high-grade epithelial ovarian cancer. Gynecol. Oncol. 2022, 164, 505–513. [Google Scholar] [CrossRef]
- Nicum, S.; Holmes, J.; McGregor, N.; Dunn, R.; Collins, L.; Kaye, S.; McNeish, I.; Glasspool, R.M.; Hall, M.; Roux, R.; et al. 722O Randomised phase II trial of olaparib compared to weekly paclitaxel or olaparib plus cediranib in patients with platinum-resistant ovarian cancer (OCTOVA). Ann. Oncol. 2021, 32, S725–S726. [Google Scholar] [CrossRef]
- Lee, J.Y.; Yi, J.Y.; Kim, H.S.; Lim, J.; Kim, S.; Nam, B.H.; Kim, H.S.; Kim, J.W.; Choi, C.H.; Kim, B.G.; et al. An umbrella study of biomarker-driven targeted therapy in patients with platinum-resistant recurrent ovarian cancer: A Korean Gynecologic Oncology Group study (KGOG 3045), AMBITION. JPN J. Clin. Oncol. 2019, 49, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Oaknin, A.; Garg, S.; Bruce, J.P.; Madariaga, A.; Dhani, N.C.; Bowering, V.; White, J.; Accardi, S.; Tan, Q.; et al. EVOLVE: A Multicenter Open-Label Single-Arm Clinical and Translational Phase II Trial of Cediranib Plus Olaparib for Ovarian Cancer after PARP Inhibition Progression. Clin. Cancer Res. 2020, 26, 4206–4215. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, aal1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakashev, S.; Zhu, H.; Yokoyama, Y.; Zhao, B.; Fatkhutdinov, N.; Kossenkov, A.V.; Wilson, A.J.; Simpkins, F.; Speicher, D.; Khabele, D.; et al. BET Bromodomain Inhibition Synergizes with PARP Inhibitor in Epithelial Ovarian Cancer. Cell Rep. 2017, 21, 3398–3405. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, A.; Johnston, P.; Woodcock, M.; Joiner, M.; Marples, B. PARP-1, PARP-2, and the cellular response to low doses of ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 410–419. [Google Scholar] [CrossRef]
- Noel, G.; Godon, C.; Fernet, M.; Giocanti, N.; Megnin-Chanet, F.; Favaudon, V. Radiosensitization by the poly(ADP-ribose) polymerase inhibitor 4-amino-1,8-naphthalimide is specific of the S phase of the cell cycle and involves arrest of DNA synthesis. Mol. Cancer Ther. 2006, 5, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [Green Version]
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.D.; Wethington, S.L.; Pagan, C.; Latif, N.; Tanyi, J.; Martin, L.P.; Morgan, M.; Burger, R.A.; Haggerty, A.; Zarrin, H.; et al. Combination ATR and PARP Inhibitor (CAPRI): A phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol. Oncol. 2021, 163, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Wilson, Z.; Odedra, R.; Wallez, Y.; Wijnhoven, P.W.G.; Hughes, A.M.; Gerrard, J.; Jones, G.N.; Bargh-Dawson, H.; Brown, E.; Young, L.A.; et al. ATR Inhibitor AZD6738 (Ceralasertib) Exerts Antitumor Activity as a Monotherapy and in Combination with Chemotherapy and the PARP Inhibitor Olaparib. Cancer Res. 2022, 82, 1140–1152. [Google Scholar] [CrossRef] [PubMed]
- Chiappa, M.; Guffanti, F.; Anselmi, M.; Lupi, M.; Panini, N.; Wiesmuller, L.; Damia, G. Combinations of ATR, Chk1 and Wee1 Inhibitors with Olaparib Are Active in Olaparib Resistant Brca1 Proficient and Deficient Murine Ovarian Cells. Cancers 2022, 14, 1807. [Google Scholar] [CrossRef]
- Smith, H.L.; Prendergast, L.; Curtin, N.J. Exploring the Synergy between PARP and CHK1 Inhibition in Matched BRCA2 Mutant and Corrected Cells. Cancers 2020, 12, 878. [Google Scholar] [CrossRef] [Green Version]
- Do, K.T.; Kochupurakkal, B.; Kelland, S.; de Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors. Clin. Cancer Res. 2021, 27, 4710–4716. [Google Scholar] [CrossRef]
- Miller, W.H.; Shields, A.F.; Provencher, D.; Gilbert, L.; Shapiro, G.; Oza, A.M.; Spratlin, J.; Lheureux, S.; Bhat, G.; Salvador, S.; et al. 537P A phase I/II study of oral chk1 inhibitor LY2880070 in combination with low-dose gemcitabine in patients with advanced or metastatic ovarian cancer. Ann. Oncol. 2022, 33, S793–S794. [Google Scholar] [CrossRef]
- Westin, S.N.; Coleman, R.L.; Fellman, B.M.; Yuan, Y.; Sood, A.K.; Soliman, P.T.; Wright, A.A.; Horowitz, N.S.; Campos, S.M.; Konstantinopoulos, P.A.; et al. EFFORT: EFFicacy Of adavosertib in parp ResisTance: A randomized two-arm non-comparative phase II study of adavosertib with or without olaparib in women with PARP-resistant ovarian cancer. J. Clin. Oncol. 2021, 39 (Suppl. S15), 5505. [Google Scholar] [CrossRef]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; Weberpals, J.I.; Wahner Hendrickson, A.E.; et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef]
- Moore, K.N.; Chambers, S.K.; Hamilton, E.P.; Chen, L.M.; Oza, A.M.; Ghamande, S.A.; Konecny, G.E.; Plaxe, S.C.; Spitz, D.L.; Geenen, J.J.J.; et al. Adavosertib with Chemotherapy in Patients with Primary Platinum-Resistant Ovarian, Fallopian Tube, or Peritoneal Cancer: An Open-Label, Four-Arm, Phase II Study. Clin. Cancer Res. 2022, 28, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; Lim, J.S.J.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase I Trial of the PARP Inhibitor Olaparib and AKT Inhibitor Capivasertib in Patients with BRCA1/2- and Non-BRCA1/2-Mutant Cancers. Cancer Discov. 2020, 10, 1528–1543. [Google Scholar] [CrossRef]
- Westin, S.N.; Labrie, M.; Litton, J.K.; Blucher, A.; Fang, Y.; Vellano, C.P.; Marszalek, J.R.; Feng, N.; Ma, X.; Creason, A.; et al. Phase Ib Dose Expansion and Translational Analyses of Olaparib in Combination with Capivasertib in Recurrent Endometrial, Triple-Negative Breast, and Ovarian Cancer. Clin. Cancer Res. 2021, 27, 6354–6365. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Braicu, I.; Berger, R.; Mahner, S.; Sehouli, J.; Pujade-Lauraine, E.; Cassier, P.A.; Moll, U.M.; Ulmer, H.; Leunen, K.; et al. Part I of GANNET53: A European Multicenter Phase I/II Trial of the Hsp90 Inhibitor Ganetespib Combined with Weekly Paclitaxel in Women with High-Grade, Platinum-Resistant Epithelial Ovarian Cancer-A Study of the GANNET53 Consortium. Front. Oncol. 2019, 9, 832. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yucel, H.; Davis, R.E.; Farkkila, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A first-in-class Polymerase Theta Inhibitor selectively targets Homologous-Recombination-Deficient Tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Selle, F.; Scambia, G.; Asselain, B.; Marmé, F.; Lindemann, K.; Colombo, N.; Madry, R.; Glasspool, R.M.; Dubot, C.; et al. LBA33—Maintenance olaparib rechallenge in patients (pts) with ovarian carcinoma (OC) previously treated with a PARP inhibitor (PARPi): Phase IIIb OReO/ENGOT Ov-38 trial. Ann. Oncol. 2021, 32 (Suppl. S5), S1283–S1346. [Google Scholar] [CrossRef]
- Moubarak, M.; Harter, P.; Ataseven, B.; Traut, A.; Welz, J.; Baert, T.; Heitz, F. Re-treatment with PARPi in patients with recurrent epithelial ovarian cancer: A single institutional experience. Gynecol. Oncol. Rep. 2022, 40, 100939. [Google Scholar] [CrossRef]


| The Nucleotide and the Protein Sequence Changes | Allele Frequencies | |
|---|---|---|
| Reversion mutation #1 | c.1046A > G (p.E349G) | 7.7% |
| Reversion mutation #2 | c.1047G > T (p.E349D) | 0.45% |
| Reversion mutation #3 | c.1039_1077del39 (p.L347_P359del) | 0.16% |
| Reversion mutation #4 | c.1035_1055del21 (p.D345_K351del) | 0.13% |
| ClinicalTrials.gov Identifier | Phase | PARPi | Number of Patients | Population | Treatment Arms | Primary Endpoint | PFS Months | OS Months | Most Common G3-4 AEs (%) |
|---|---|---|---|---|---|---|---|---|---|
| NCT01081951 [71] | II | olaparib | 162 | Platinum-sensitive, recurrent HGSOC, with or without BRCA 1/2 mutations | Olaparib plus CT → olaparib maintenance vs. CT | PFS | 12.2 vs. 9.6 (HR 0.51 [95% CI 0.34–0.77]; p = 0.0012) | 33.8 vs. 37.6 months (HR 1.17 [95% CI 0.79–1.73]; p = 0.44) | Neutropenia (43 vs. 35); anaemia (9 vs. 7) |
| NCT0247058 [72] | III | veliparib | 1140 | Untreated HGSOC | CT + placebo →placebo vs. CT + veliparib →placebo vs. CT + veliparib → veliparib | PFS in veliparib-throught group vs. control group | 23.5 vs. 17.3 in ITT (HR 0.68; 95% CI, 0.56 to 0.83; p < 0.001) | NA | Neutropenia (58 vs. 49); anemia (38 vs. 26); thrombocytopenia (28 vs. 8) |
| NCT01690598 [74] | I/II | veliparib | 27 | relapsed advanced platinum resistant or partially platinum sensitive HGSOC with negative or unknown BRCA1/2 status | Veliparib plus topotecan | ORR | 2.8 (95% CI [2.6–3.6]) | 7.1 months (95% CI [4.8–10.8]) | Infection (22.2); neutropenia (11.1); fatigue (7.4); nausea (7.4) |
| NCT01306032 [75] | II | veliparib | 75 | Pretreated BRCA-mutant ovarian cancer | Veliparib plus cyclophsphamide vs. cyclophsphamide | ORR | 2.3 vs. 2.1 (p = 0.68) | NA | Lymphopenia (17.3 vs. 4); anemia (2.7 vs. 0); neutropenia (2.7 vs. 2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordani, N.; Bianchi, T.; Ammoni, L.C.; Cortinovis, D.L.; Cazzaniga, M.E.; Lissoni, A.A.; Landoni, F.; Canova, S. An Overview of PARP Resistance in Ovarian Cancer from a Molecular and Clinical Perspective. Int. J. Mol. Sci. 2023, 24, 11890. https://doi.org/10.3390/ijms241511890
Cordani N, Bianchi T, Ammoni LC, Cortinovis DL, Cazzaniga ME, Lissoni AA, Landoni F, Canova S. An Overview of PARP Resistance in Ovarian Cancer from a Molecular and Clinical Perspective. International Journal of Molecular Sciences. 2023; 24(15):11890. https://doi.org/10.3390/ijms241511890
Chicago/Turabian StyleCordani, Nicoletta, Tommaso Bianchi, Luca Carlofrancesco Ammoni, Diego Luigi Cortinovis, Marina Elena Cazzaniga, Andrea Alberto Lissoni, Fabio Landoni, and Stefania Canova. 2023. "An Overview of PARP Resistance in Ovarian Cancer from a Molecular and Clinical Perspective" International Journal of Molecular Sciences 24, no. 15: 11890. https://doi.org/10.3390/ijms241511890
APA StyleCordani, N., Bianchi, T., Ammoni, L. C., Cortinovis, D. L., Cazzaniga, M. E., Lissoni, A. A., Landoni, F., & Canova, S. (2023). An Overview of PARP Resistance in Ovarian Cancer from a Molecular and Clinical Perspective. International Journal of Molecular Sciences, 24(15), 11890. https://doi.org/10.3390/ijms241511890