
Treprostinil Reconstitutes Mitochondrial Organisation and Structure in Idiopathic Pulmonary Fibrosis Cells

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
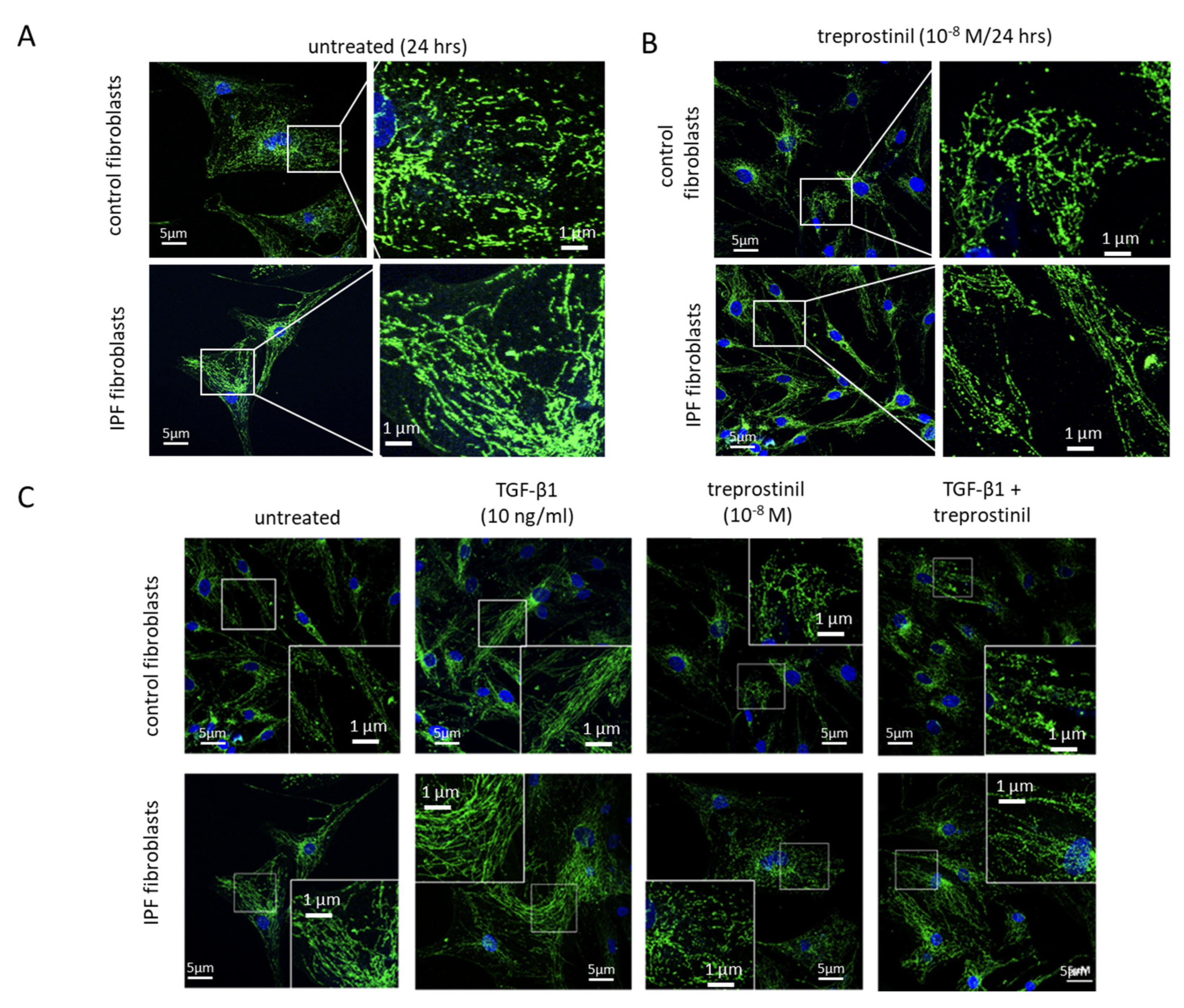
2.1. Mitochondrial Damage in IPF Cells
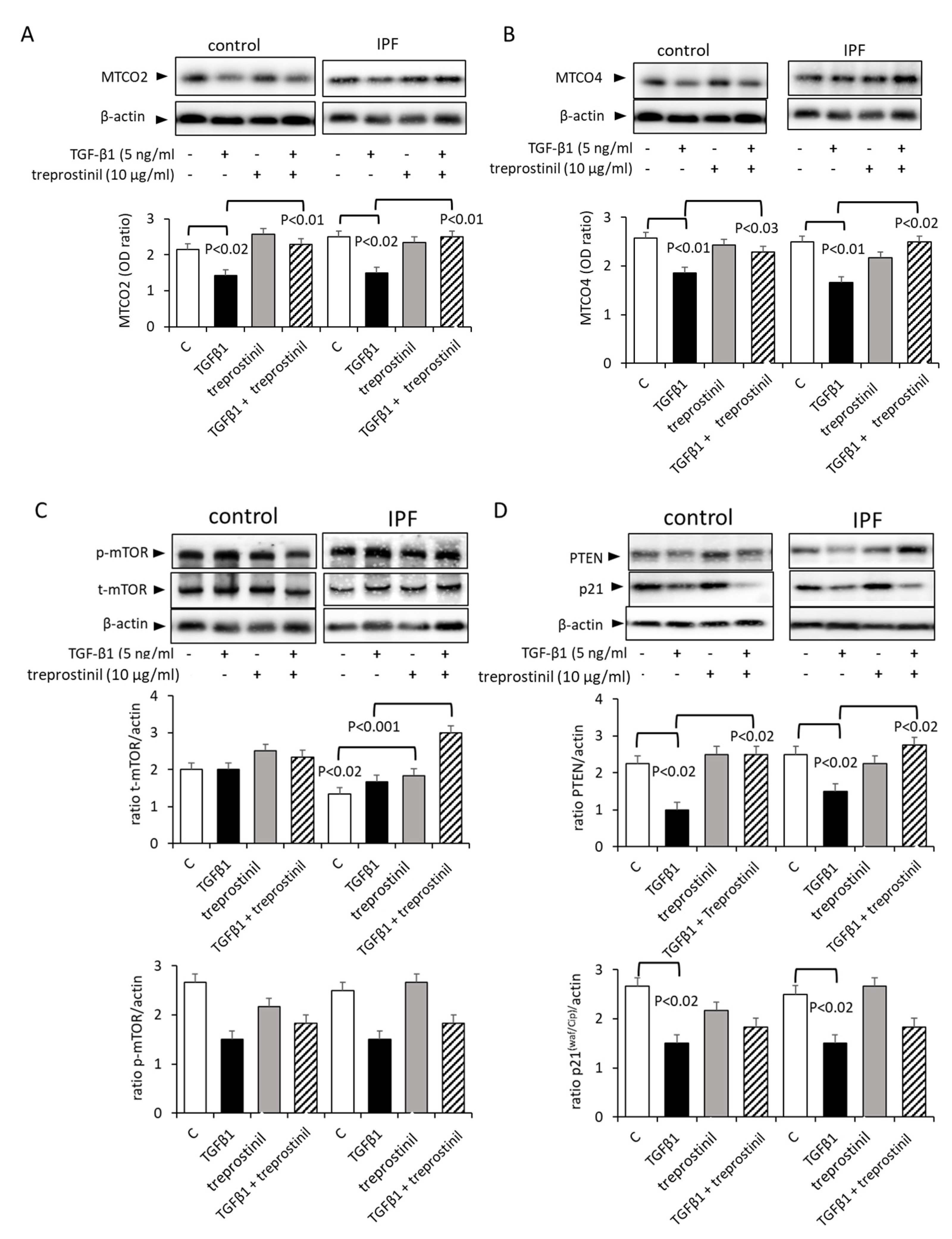
2.2. Treprostinil Modulates Mitochondrial Activity and Autophagy
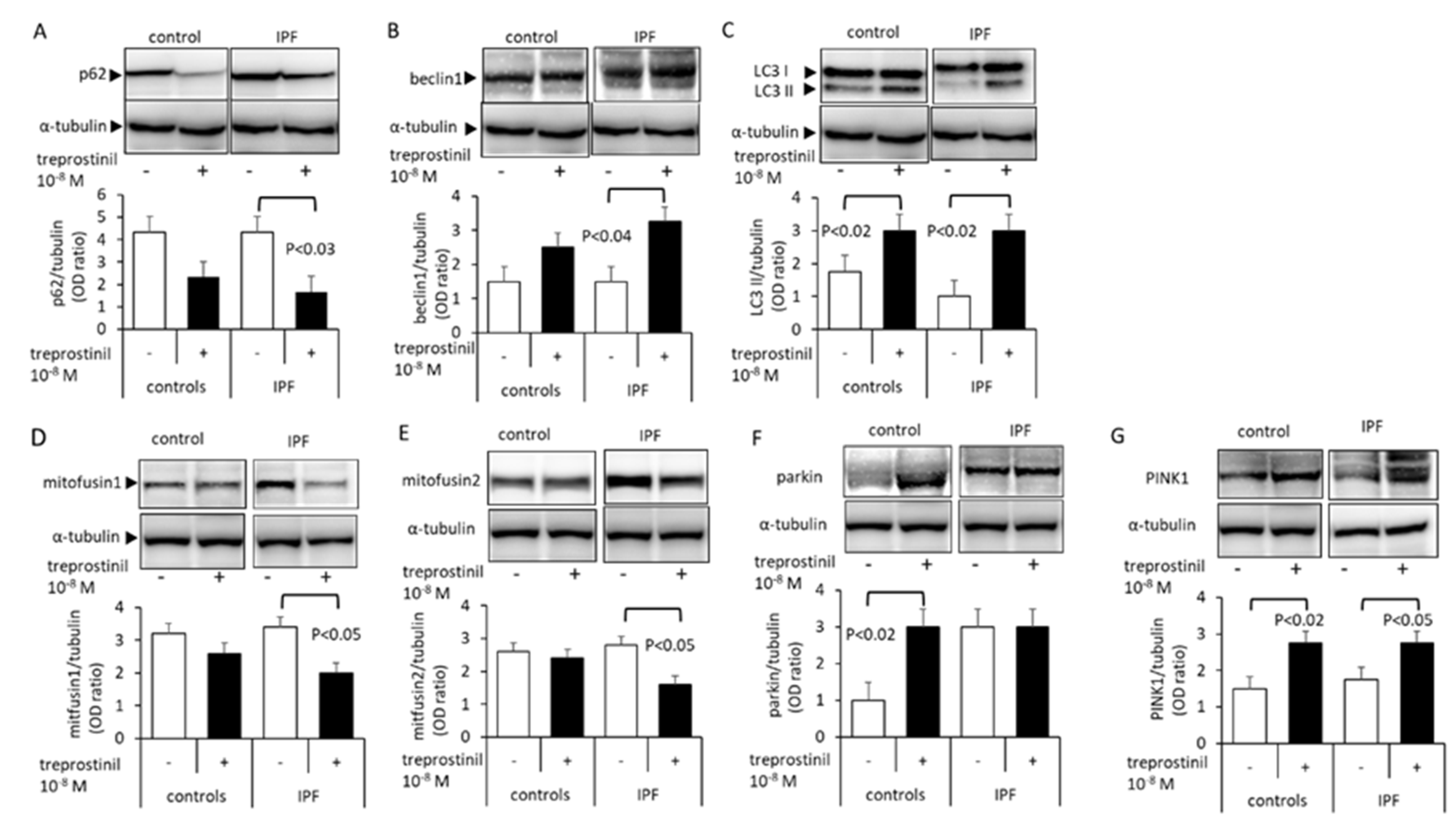
2.3. Treprostinil Affects Mitophagy- and Autophagy-Regulating Protein Expression
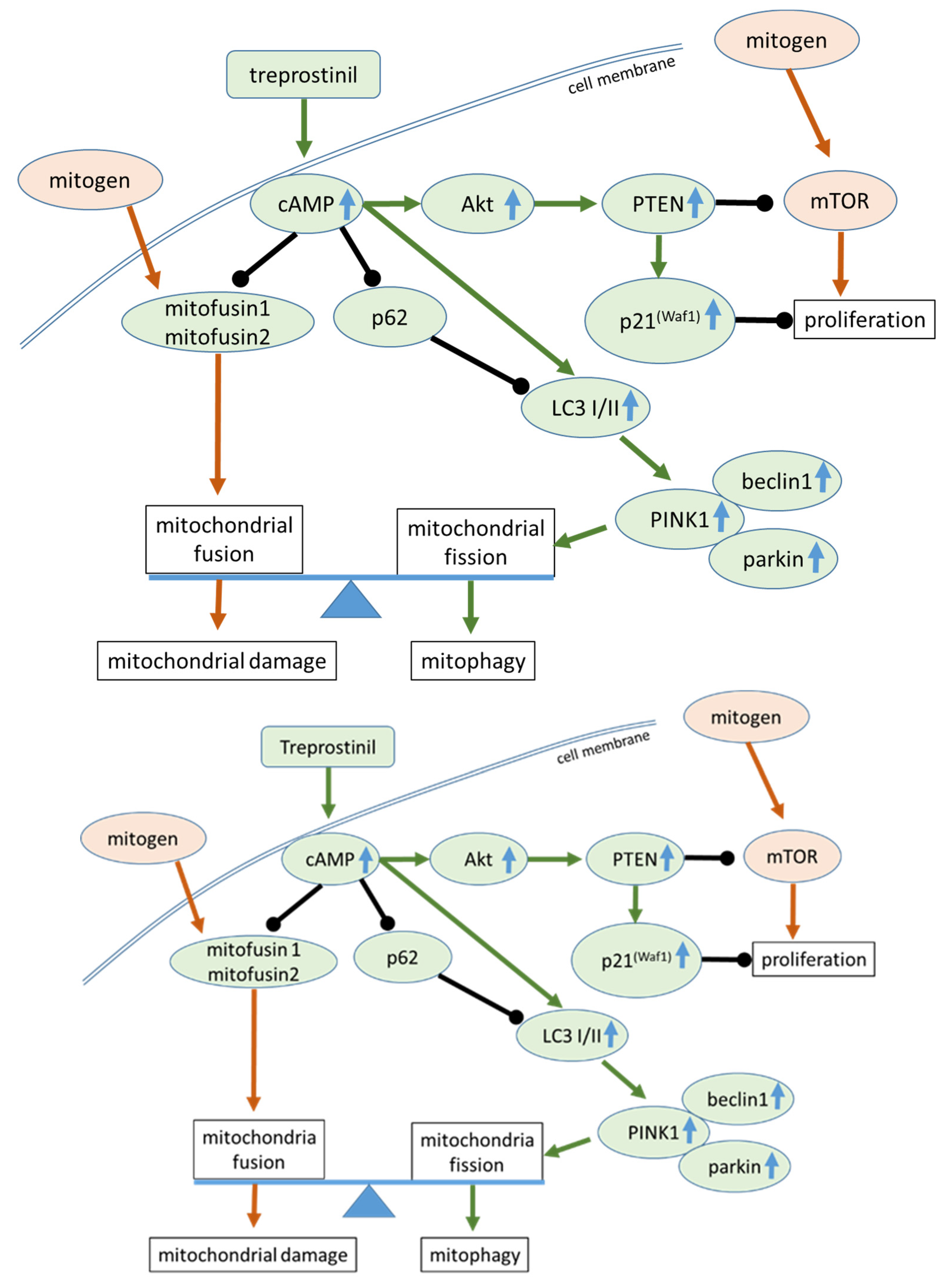
2.4. Treprostinil Induces cAMP–Akt–CREB Signalling
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Drugs
4.3. Western Blotting
4.4. Immunofluorescence Microscopy
4.5. Mitochondrial Phenotyping
4.6. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mei, Q.; Liu, Z.; Zuo, H.; Yang, Z.; Qu, J. Idiopathic Pulmonary Fibrosis: An Update on Pathogenesis. Front. Pharmacol. 2022, 12, 797292. [Google Scholar] [CrossRef]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, M.; Calyeca, J.; Rojas, M.; Mora, A.L. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020, 33, 101509. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Larson-Casey, J.L.; Andrabi, S.A.; Lee, J.H.; Meza-Perez, S.; Randall, T.D.; Carter, A.B. Mitochondrial calcium uniporter regulates PGC-1α expression to mediate metabolic reprogramming in pulmonary fibrosis. Redox Biol. 2019, 26, 101307. [Google Scholar] [CrossRef]
- Jaeger, V.K.; Lebrecht, D.; Nicholson, A.G.; Wells, A.; Bhayani, H.; Gazdhar, A.; Tamm, M.; Venhoff, N.; Geiser, T.; Walker, U.A. Mitochondrial DNA mutations and respiratory chain dysfunction in idiopathic and connective tissue disease-related lung fibrosis. Sci. Rep. 2019, 9, 5500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuliga, M.; Pechkovsky, D.; Read, J.; Waters, D.W.; Blokland, K.; Reid, A.; Hogaboam, C.; Khalil, N.; Burgess, J.; Prêle, C.M.; et al. Mitochondrial dysfunction contributes to the senescent phenotype of IPF lung fibroblasts. J. Cell. Mol. Med. 2018, 22, 5847–5861. [Google Scholar] [CrossRef]
- Lee, J.S.; La, J.; Aziz, S.; Dobrinskikh, E.; Brownell, R.; Jones, K.D.; Achtar-Zadeh, N.; Green, G.; Elicker, B.M.; A Golden, J.; et al. Molecular markers of telomere dysfunction and senescence are common findings in the usual interstitial pneumonia pattern of lung fibrosis. Histopathology 2021, 79, 67–76. [Google Scholar] [CrossRef]
- Siekacz, K.; Piotrowski, W.J.; Iwański, M.A.; Górski, P.; Białas, A.J. The Role of Interaction between Mitochondria and the Extracellular Matrix in the Development of Idiopathic Pulmonary Fibrosis. Oxidative Med. Cell. Longev. 2021, 2021, 1–12. [Google Scholar] [CrossRef]
- Qian, W.; Xia, S.; Yang, X.; Yu, J.; Guo, B.; Lin, Z.; Wei, R.; Mao, M.; Zhang, Z.; Zhao, G.; et al. Complex Involvement of the Extracellular Matrix, Immune Effect, and Lipid Metabolism in the Development of Idiopathic Pulmonary Fibrosis. Front. Mol. Biosci. 2022, 8, 800747. [Google Scholar] [CrossRef]
- Lerner, C.A.; Sundar, I.K.; Rahman, I. Mitochondrial redox system, dynamics, and dysfunction in lung inflammaging and COPD. Int. J. Biochem. Cell Biol. 2016, 81, 294–306. [Google Scholar] [CrossRef] [Green Version]
- Tsitoura, E.; Vasarmidi, E.; Bibaki, E.; Trachalaki, A.; Koutoulaki, C.; Papastratigakis, G.; Papadogiorgaki, S.; Chalepakis, G.; Tzanakis, N.; Antoniou, K.M. Accumulation of damaged mitochondria in alveolar macrophages with reduced OXPHOS related gene expression in IPF. Respir. Res. 2019, 20, 264. [Google Scholar] [CrossRef] [Green Version]
- Vasarmidi, E.; Sarantoulaki, S.; Trachalaki, A.; Margaritopoulos, G.; Bibaki, E.; Spandidos, D.A.; Tzanakis, N.; Antoniou, K. Investigation of key autophagy-and mitophagy-related proteins and gene expression in BALF cells from patients with IPF and RA-ILD. Mol. Med. Rep. 2018, 18, 3891–3897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, C.; Sun, H.; Gulati, M.; Herazo-Maya, J.D.; Chen, Y.; Osafo-Addo, A.; Brandsdorfer, C.; Winkler, J.; Blaul, C.; Faunce, J.; et al. Extracellular Mitochondrial DNA Is Generated by Fibroblasts and Predicts Death in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A.; Piplani, H.; Sin, J.; Sawaged, S.; Hamid, S.M.; Taylor, D.J.; Germano, J.D.F. At the heart of mitochondrial quality control: Many roads to the top. Cell. Mol. Life Sci. 2021, 78, 3791–3801. [Google Scholar] [CrossRef]
- Roque, W.; Cuevas-Mora, K.; Romero, F. Mitochondrial Quality Control in Age-Related Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, C.; Bitto, A.; Pulliam, D.; Nacarelli, T.; Konigsberg, M.; Van Remmen, H.; Torres, C.; Sell, C. Reduced mammalian target of rapamycin activity facilitates mitochondrial retrograde signaling and increases life span in normal human fibroblasts. Aging Cell 2013, 12, 966–977. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, J.; Nho, R. The Role of the Mammalian Target of Rapamycin (mTOR) in Pulmonary Fibrosis. Int. J. Mol. Sci. 2018, 19, 778. [Google Scholar] [CrossRef] [Green Version]
- Nathan, S.D.; Waxman, A.; Rajagopal, S.; Case, A.; Johri, S.; DuBrock, H.; De La Zerda, D.J.; Sahay, S.; King, C.; Melendres-Groves, L.; et al. Inhaled treprostinil and forced vital capacity in patients with interstitial lung disease and associated pulmonary hypertension: A post-hoc analysis of the INCREASE study. Lancet Respir. Med. 2021, 9, 1266–1274. [Google Scholar] [CrossRef]
- Chauhan, M.; Punga, T.; Punga, A.R. Muscle-specific regulation of the mTOR signaling pathway in MuSK antibody seropositive (MuSK+) experimental autoimmune Myasthenia gravis (EAMG). Neurosci. Res. 2013, 77, 102–109. [Google Scholar] [CrossRef]
- Roberts, M.J.; May, L.T.; Keen, A.C.; Liu, B.; Lam, T.; Charlton, S.J.; Rosethorne, E.M.; Halls, M.L. Inhibition of the Proliferation of Human Lung Fibroblasts by Prostacyclin Receptor Agonists is Linked to a Sustained cAMP Signal in the Nucleus. Front. Pharmacol. 2021, 12, 669227. [Google Scholar] [CrossRef]
- Lambers, C.; Boehm, P.M.; Karabacak, Y.; Samaha, E.; Benazzo, A.; Jaksch, P.; Roth, M. Combined Activation of Guanylate Cyclase and Cyclic AMP in Lung Fibroblasts as a Novel Therapeutic Concept for Lung Fibrosis. BioMed Res. Int. 2019, 2019, 1–10. [Google Scholar] [CrossRef]
- Lambers, C.; Roth, M.; Jaksch, P.; Muraközy, G.; Tamm, M.; Klepetko, W.; Ghanim, B.; Zhao, F. Treprostinil inhibits proliferation and extracellular matrix deposition by fibroblasts through cAMP activation. Sci. Rep. 2018, 8, 1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Wang, X.; Sun, Q.; Papakonstantinou, E.; Tamm, M.; Stolz, D.; Roth, M. MicroRNA-21-5p induced by IgE downregulates PTEN, thereby supporting mTOR dependent airway smooth muscle cell remodeling. Int. J. Mol. Sci. 2019, 20, 875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, J.; Huang, X.; Li, Y.; Xu, X.; Li, S.; Jiang, D.; Liu, Z.; Dai, H. Phosphatase and tensin homolog deleted on chromosome 10 contributes to phenotype transformation of fibroblasts in idiopathic pulmonary fibrosis via multiple pathways. Exp. Biol. Med. 2016, 241, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, T.; Tian, Y.; Gao, Y.; Ma, M.; Li, H.; Liu, X.; Wu, H.; Zhang, Y.; Ding, H.; Cao, M.; et al. PTEN loss regulates alveolar epithelial cell senescence in pulmonary fibrosis depending on Akt activation. Aging 2019, 11, 7492–7509. [Google Scholar] [CrossRef]
- Blumer, S.; Fang, L.; Chen, W.-C.; Khan, P.; Hostettler, K.; Tamm, M.; Roth, M.; Lambers, C. IPF-Fibroblast Erk1/2 Activity Is Independent from microRNA Cluster 17-92 but Can Be Inhibited by Treprostinil through DUSP1. Cells 2021, 10, 2836. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, L.; Liu, W.; Xiao, Z. MiR-21 regulates the apoptosis of keloid fibroblasts by caspase-8 and the mitochondria-mediated apoptotic signaling pathway via targeting FasL. Biochem. Cell Biol. 2018, 96, 548–555. [Google Scholar] [CrossRef]
- Dong, X.; Pi, Q.; Yuemaierabola, A.; Guo, W.; Tian, H. Silencing LINC00294 Restores Mitochondrial Function and Inhibits Apoptosis of Glioma Cells under Hypoxia via the miR-21-5p/CASKIN1/cAMP Axis. Oxidative Med. Cell. Longev. 2021, 2021, 1–21. [Google Scholar] [CrossRef]
- Luis-García, E.R.; Becerril, C.; Salgado-Aguayo, A.; Aparicio-Trejo, O.E.; Romero, Y.; Flores-Soto, E.; Mendoza-Milla, C.; Montaño, M.; Chagoya, V.; Pedraza-Chaverri, J.; et al. Mitochondrial Dysfunction and Alterations in Mitochondrial Permeability Transition Pore (mPTP) Contribute to Apoptosis Resistance in Idiopathic Pulmonary Fibrosis Fibroblasts. Int. J. Mol. Sci. 2021, 22, 7870. [Google Scholar] [CrossRef]
- Guo, T.; Jiang, C.-S.; Yang, S.-Z.; Zhu, Y.; He, C.; Carter, A.B.; Antony, V.B.; Peng, H.; Zhou, Y. Mitochondrial fission and bioenergetics mediate human lung fibroblast durotaxis. J. Clin. Investig. 2023, 8, e157348. [Google Scholar] [CrossRef]
- Nho, R.S.; Hergert, P. IPF Fibroblasts Are Desensitized to Type I Collagen Matrix-Induced Cell Death by Suppressing Low Autophagy via Aberrant Akt/mTOR Kinases. PLoS ONE 2014, 9, e94616. [Google Scholar] [CrossRef]
- Huang, L.S.; Berdyshev, E.V.; Tran, J.T.; Xie, L.; Chen, J.; Ebenezer, D.L.; Mathew, B.; Gorshkova, I.; Zhang, W.; Reddy, S.P.; et al. Sphingosine-1-phosphate lyase is an endogenous suppressor of pulmonary fibrosis: Role of S1P signalling and autophagy. Thorax 2015, 70, 1138–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dakhlallah, D.; Wang, Y.; Bobo, T.A.; Ellis, E.; Mo, X.; Piper, M.G.; Eubank, T.D.; Marsh, C.B. Constitutive AKT Activity Predisposes Lung Fibrosis by Regulating Macrophage, Myofibroblast and Fibrocyte Recruitment and Changes in Autophagy. Adv. Biosci. Biotechnol. 2019, 10, 346–373. [Google Scholar] [CrossRef]
- Han, Y.; Ye, L.; Du, F.; Ye, M.; Li, C.; Zhu, X.; Wang, Q.; Jiang, H.; Liu, Z.; Ma, J.; et al. Iron metabolism regulation of epithelial-mesenchymal transition in idiopathic pulmonary fibrosis. Ann. Transl. Med. 2021, 9, 1755. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.; Cherubini, E.; Scozzi, D.; Pietrangeli, V.; Tabbì, L.; Raffa, S.; Leone, L.; Visco, V.; Torrisi, M.R.; Bruno, P.; et al. Decreased expression of autophagic beclin 1 protein in idiopathic pulmonary fibrosis fibroblasts. J. Cell. Physiol. 2013, 228, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- New, M.L.; White, C.M.; McGonigle, P.; McArthur, D.G.; Dwyer-Nield, L.D.; Merrick, D.T.; Keith, R.L.; Tennis, M.A. Prostacyclin and EMT Pathway Markers for Monitoring Response to Lung Cancer Chemoprevention. Cancer Prev. Res. 2018, 11, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Zmajkovicova, K.; Menyhart, K.; Bauer, Y.; Studer, R.; Renault, B.; Schnoebelen, M.; Bolinger, M.; Nayler, O.; Gatfield, J. The Antifibrotic Activity of Prostacyclin Receptor Agonism Is Mediated through Inhibition of YAP/TAZ. Am. J. Respir. Cell Mol. Biol. 2019, 60, 578–591. [Google Scholar] [CrossRef]
- Sharma, A.; Ahmad, S.; Ahmad, T.; Ali, S.; Syed, M.A. Mitochondrial dynamics and mitophagy in lung disorders. Life Sci. 2021, 284, 119876. [Google Scholar] [CrossRef]
- Xu, G.; Wang, X.; Yu, H.; Wang, C.; Liu, Y.; Zhao, R.; Zhang, G. Beclin 1, LC3, and p62 expression in paraquat-induced pulmonary fibrosis. Hum. Exp. Toxicol. 2019, 38, 794–802. [Google Scholar] [CrossRef]
- Patel, A.S.; Morse, D.; Choi, A.M.K. Regulation and Functional Significance of Autophagy in Respiratory Cell Biology and Disease. Am. J. Respir. Cell Mol. Biol. 2013, 48, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chae, U.; Kim, H.S.; Lee, H.-S.; Lee, S.-R.; Lee, D.-S. Drp1-dependent mitochondrial fission regulates p62-mediated autophagy in LPS-induced activated microglial cells. Biosci. Biotechnol. Biochem. 2019, 83, 409–416. [Google Scholar] [CrossRef]
- Peng, J.; Xiao, X.; Li, S.; Lyu, X.; Gong, H.; Tan, S.; Dong, L.; Sanders, Y.Y.; Zhang, X. Aspirin alleviates pulmonary fibrosis through PI3K/AKT/mTOR-mediated autophagy pathway. Exp. Gerontol. 2023, 172, 112085. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Yang, L.; Dong, L.; Niu, M.; Zhang, S.; Yang, Z.; Wumaier, G.; Li, Y.; Wei, X.; Gong, Y.; et al. Cefminox, a Dual Agonist of Prostacyclin Receptor and Peroxisome Proliferator-Activated Receptor-Gamma Identified by Virtual Screening, Has Therapeutic Efficacy against Hypoxia-Induced Pulmonary Hypertension in Rats. Front. Pharmacol. 2018, 9, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, J.; Smits, J.; Schramm, R.; Langer, F.; Buhl, R.; Witt, C.; Strueber, M.; Reichenspurner, H. Lung Transplantation in Germany Since the Introduction of the Lung Allocation Score. Dtsch. Arztebl. Int. 2017, 114, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E.; Lynch, D.A.; Nicholson, A.G. An official American Thoracic Socie-ty/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Li, J.; Papakonstantinou, E.; Karakioulaki, M.; Sun, Q.; Schumann, D.; Tamm, M.; Stolz, D.; Roth, M. Secreted heat shock proteins control airway remodeling: Evidence from bronchial thermoplasty. J. Allergy Clin. Immunol. 2021, 148, 1249–1261. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Target | Source | Cat# | Dilution | Species |
|---|---|---|---|---|
| total Akt | CST 1 | 4691 | 1:2000 | rabbit |
| phosphorylated Akt | CST | 4060 | 1:2000 | rabbit |
| alpha-tubulin | TFSci 2 | 32-2700 | 1:2000 | mouse |
| beclin1 | CST | 3495 | 1:2000 | rabbit |
| total CREB | CST | 9197 | 1:1000 | rabbit |
| phosphorylated CREB | CST | 9198 | 1:1000 | rabbit |
| cytochrome C | BD 3 | 556432 | 1:1000 | rabbit |
| LC3 I/II | CST | 12741 | 1:2000 | rabbit |
| mitofusin1 | Abcam 4 | Ab221661 | 1:2000 | rabbit |
| mitofusin2 | Abcam | Ab124773 | 1:2000 | rabbit |
| total mTOR | CST | 2983 | 1:2000 | rabbit |
| phosphorylated mTOR | CST | 2974 | 1:2000 | rabbit |
| MTCO2 | Abcam | Ab79393 | 1:1000 | rabbit |
| MTCO4 | CST | 4850 | 1:2000 | rabbit |
| p21(Waf1/Cip1) | Abcam | 188224 | 1:1000 | rabbit |
| p62 | R&D 5 | MAB8028 | 1:1000 | mouse |
| parkin | Abcam | Ab77924 | 1:2000 | mouse |
| PINK1 | Abcam | Ab300623 | 1:1000 | rabbit |
| PTEN | CST | 9188 | 1:2000 | rabbit |
| anti-mouse HRP | SA 6 | A9917 | 1:2000 | goat |
| anti-mouse Alexa 488 | TFSci | A21121 | 1:500 | goat |
| anti-rabbit HRP | SA | A9169 | 1:2000 | goat |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, L.; Chen, W.-C.; Jaksch, P.; Molino, A.; Saglia, A.; Roth, M.; Lambers, C. Treprostinil Reconstitutes Mitochondrial Organisation and Structure in Idiopathic Pulmonary Fibrosis Cells. Int. J. Mol. Sci. 2023, 24, 12148. https://doi.org/10.3390/ijms241512148
Fang L, Chen W-C, Jaksch P, Molino A, Saglia A, Roth M, Lambers C. Treprostinil Reconstitutes Mitochondrial Organisation and Structure in Idiopathic Pulmonary Fibrosis Cells. International Journal of Molecular Sciences. 2023; 24(15):12148. https://doi.org/10.3390/ijms241512148
Chicago/Turabian StyleFang, Lei, Wei-Chih Chen, Peter Jaksch, Antonio Molino, Alessandro Saglia, Michael Roth, and Christopher Lambers. 2023. "Treprostinil Reconstitutes Mitochondrial Organisation and Structure in Idiopathic Pulmonary Fibrosis Cells" International Journal of Molecular Sciences 24, no. 15: 12148. https://doi.org/10.3390/ijms241512148
APA StyleFang, L., Chen, W. -C., Jaksch, P., Molino, A., Saglia, A., Roth, M., & Lambers, C. (2023). Treprostinil Reconstitutes Mitochondrial Organisation and Structure in Idiopathic Pulmonary Fibrosis Cells. International Journal of Molecular Sciences, 24(15), 12148. https://doi.org/10.3390/ijms241512148