
Dynamics of the Second Extracellular Loop Control Transducer Coupling of Peptide-Activated GPCRs
 and
and
Abstract
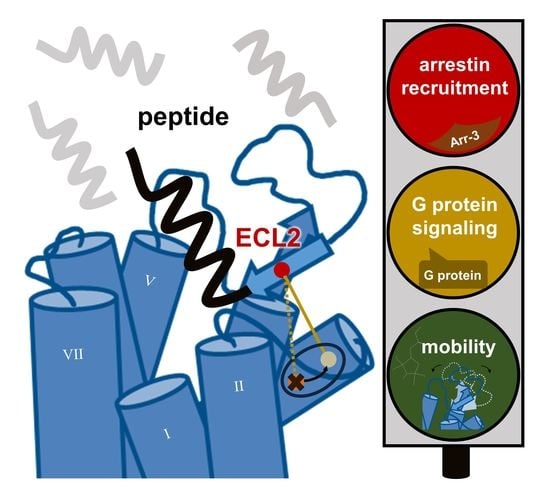
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
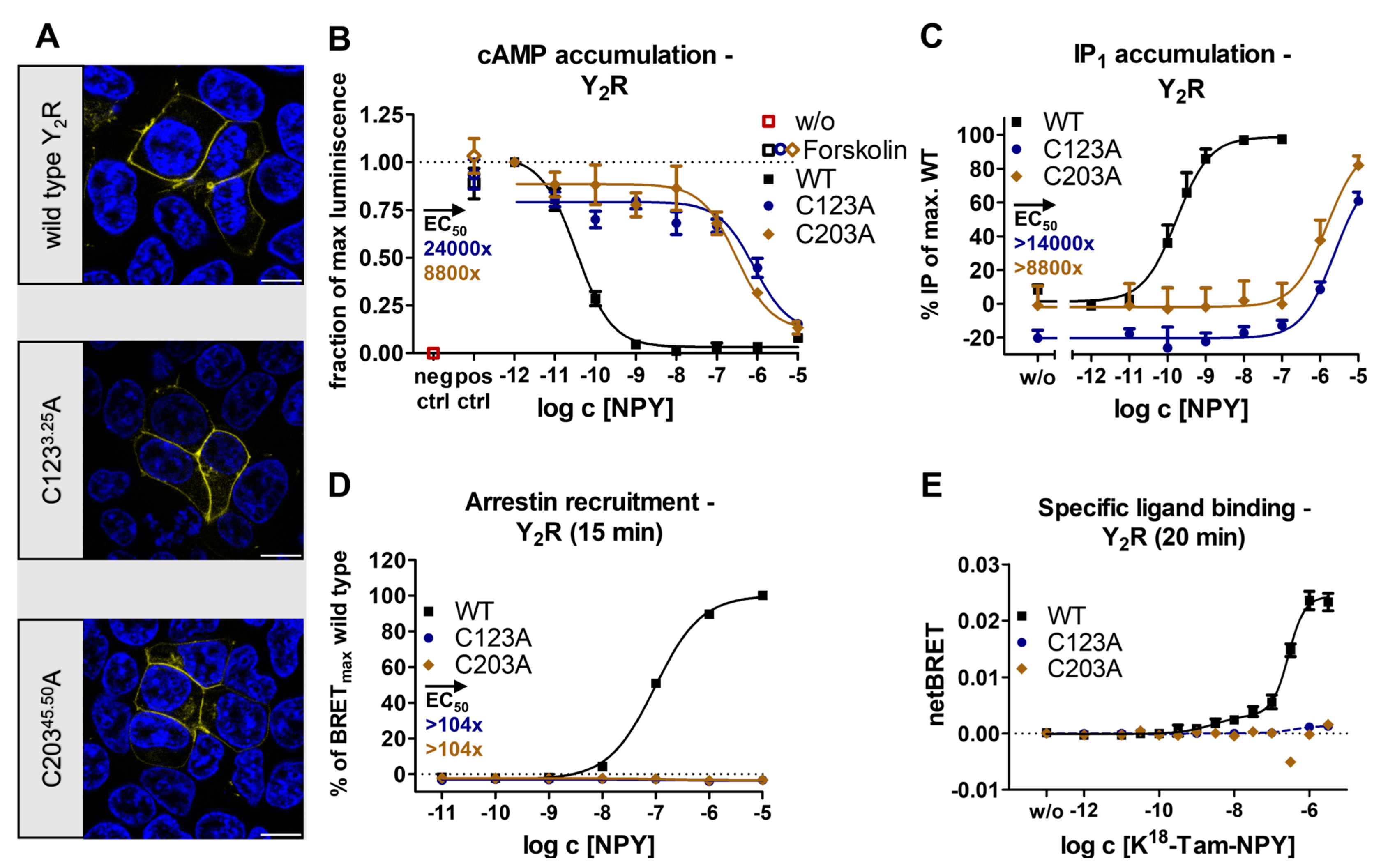
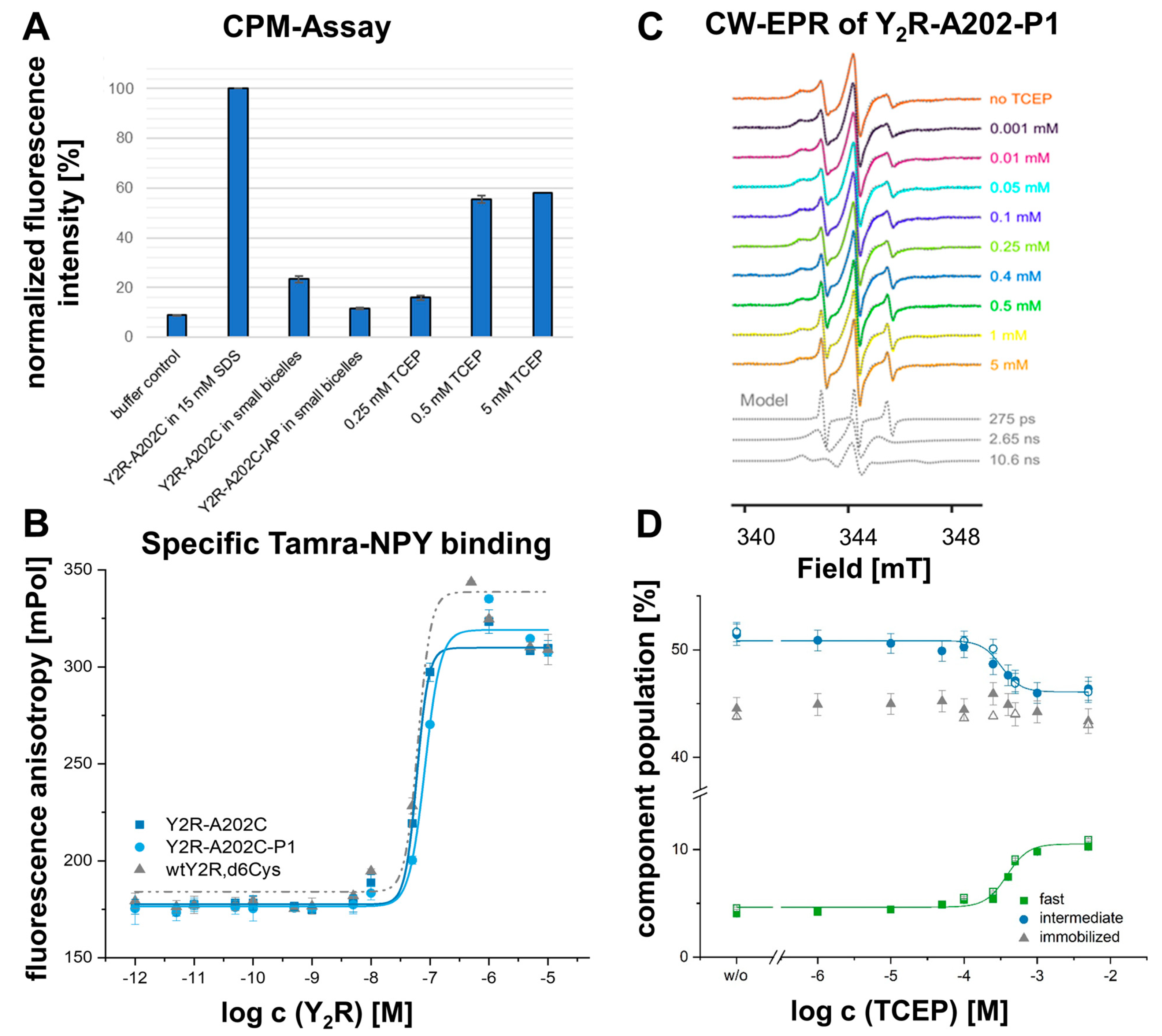
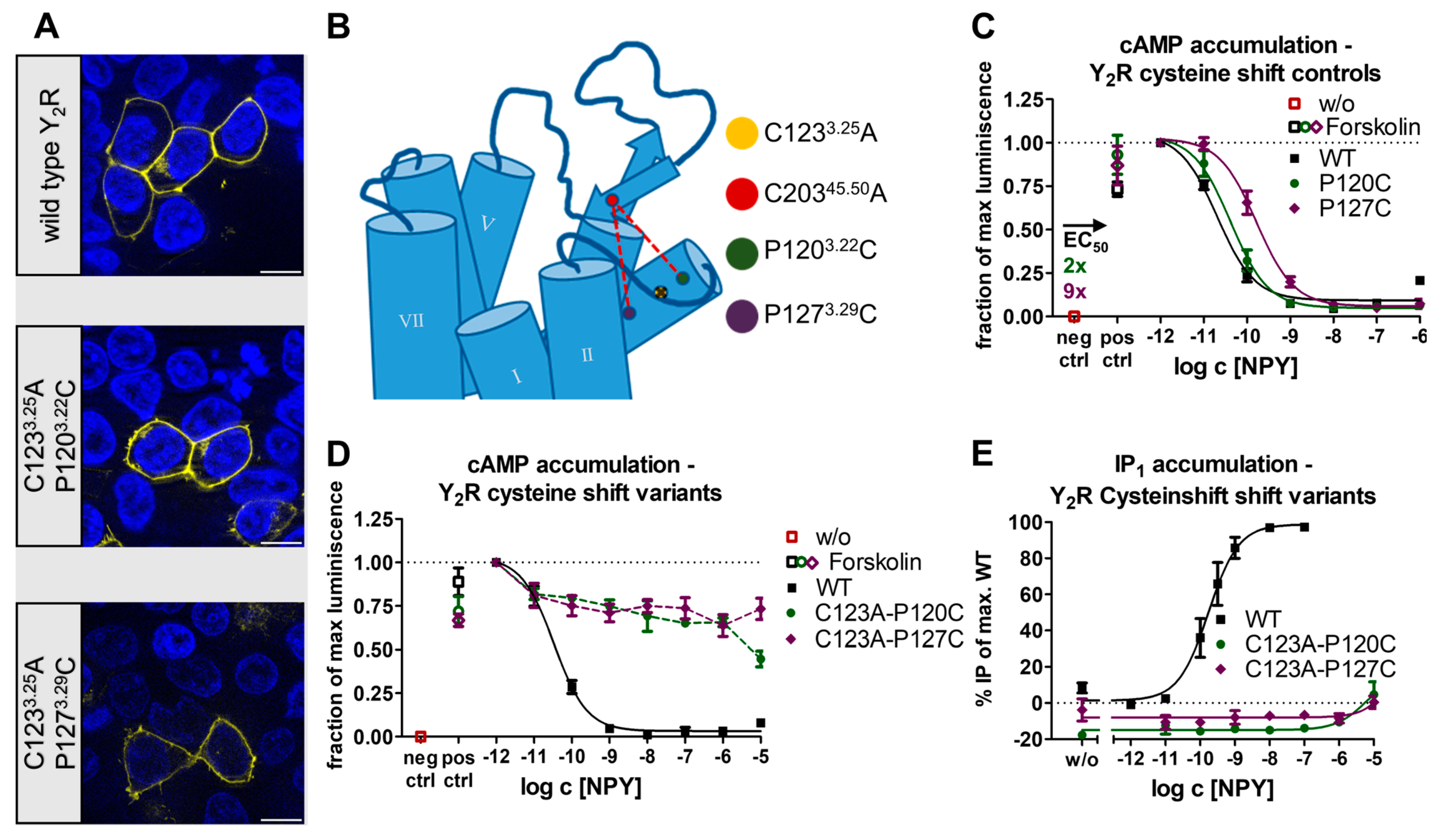
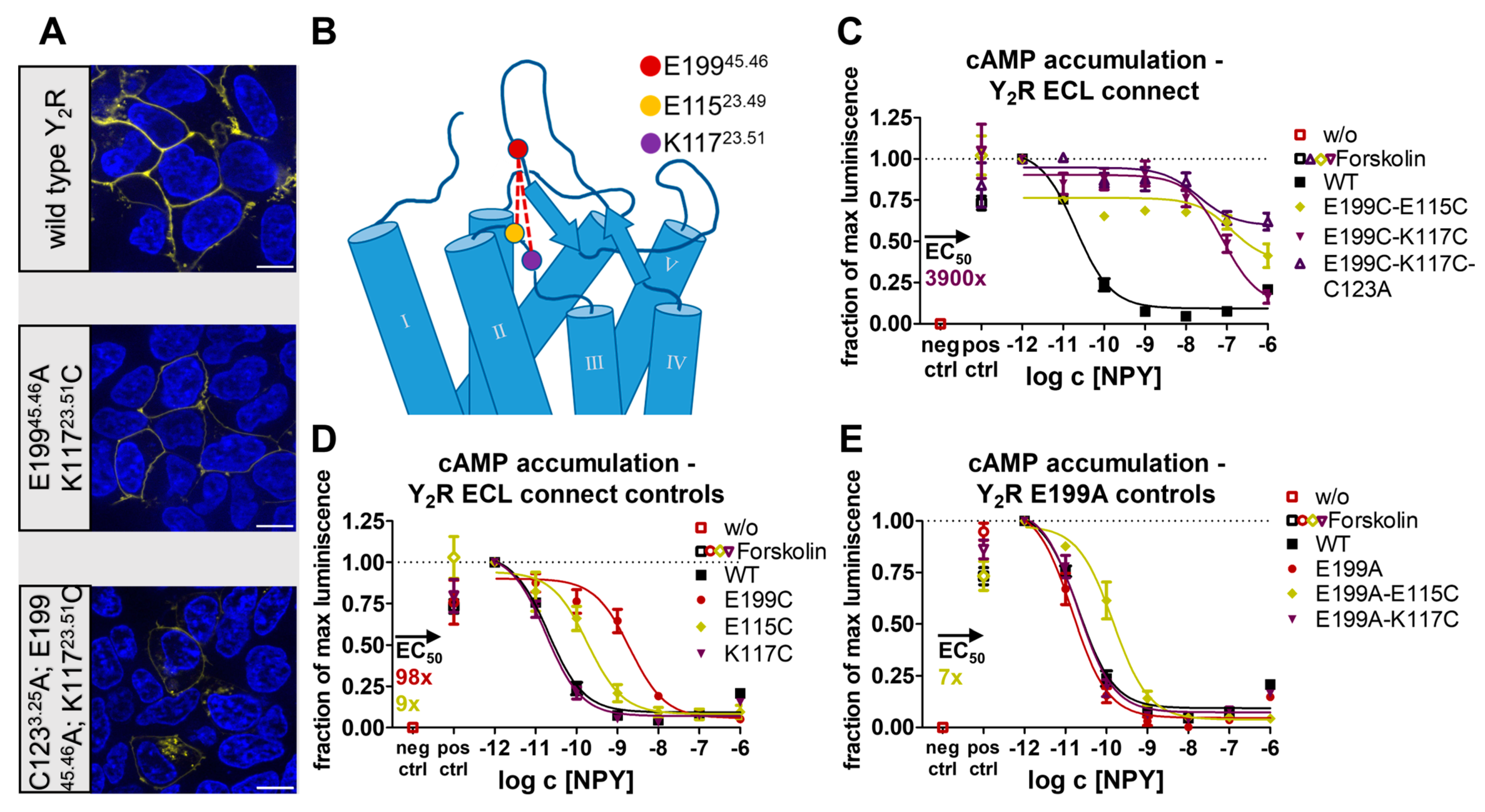
2.1. The Conserved Disulfide Controls ECL2 Dynamics and Function of the Y2R
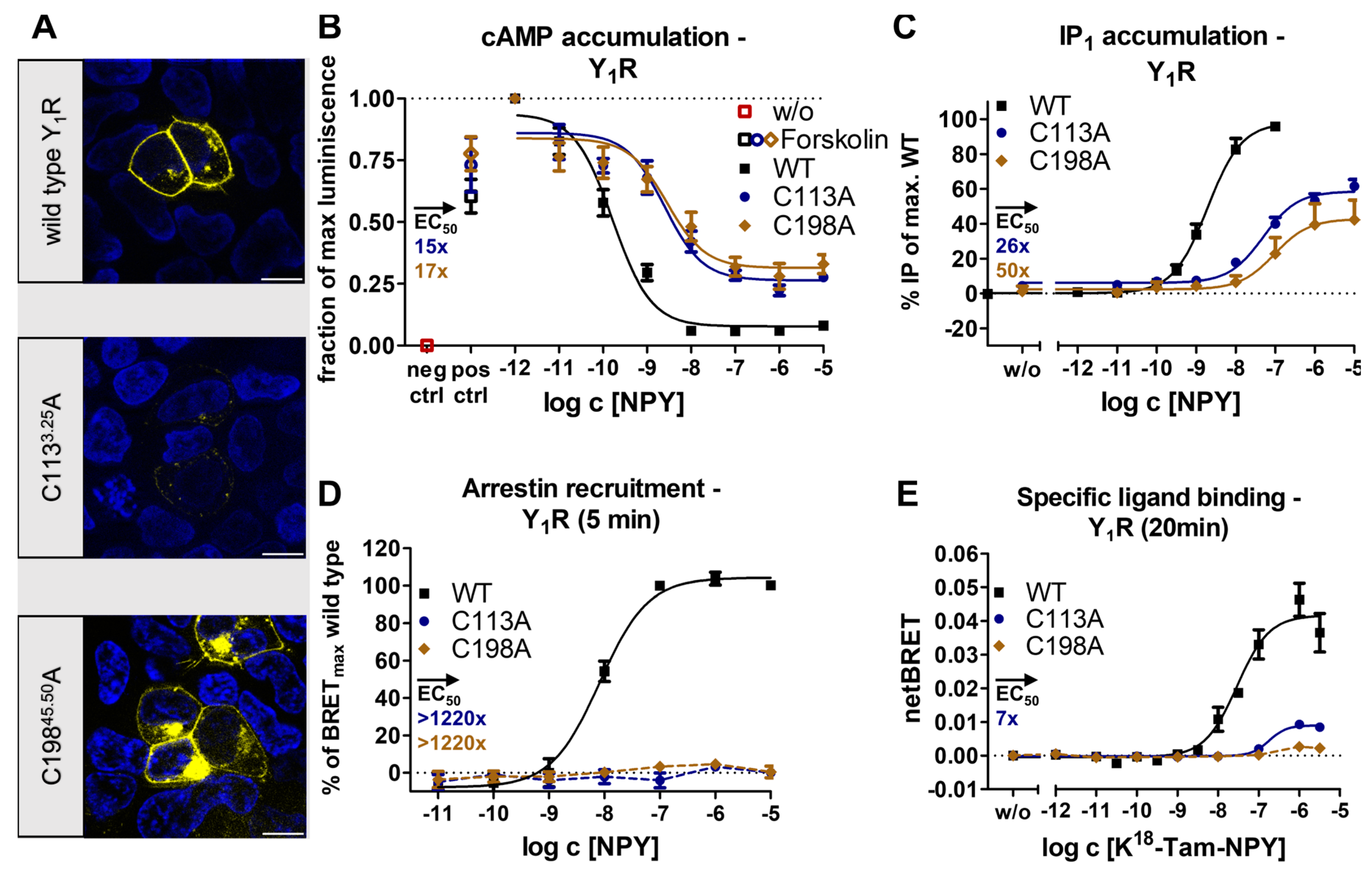
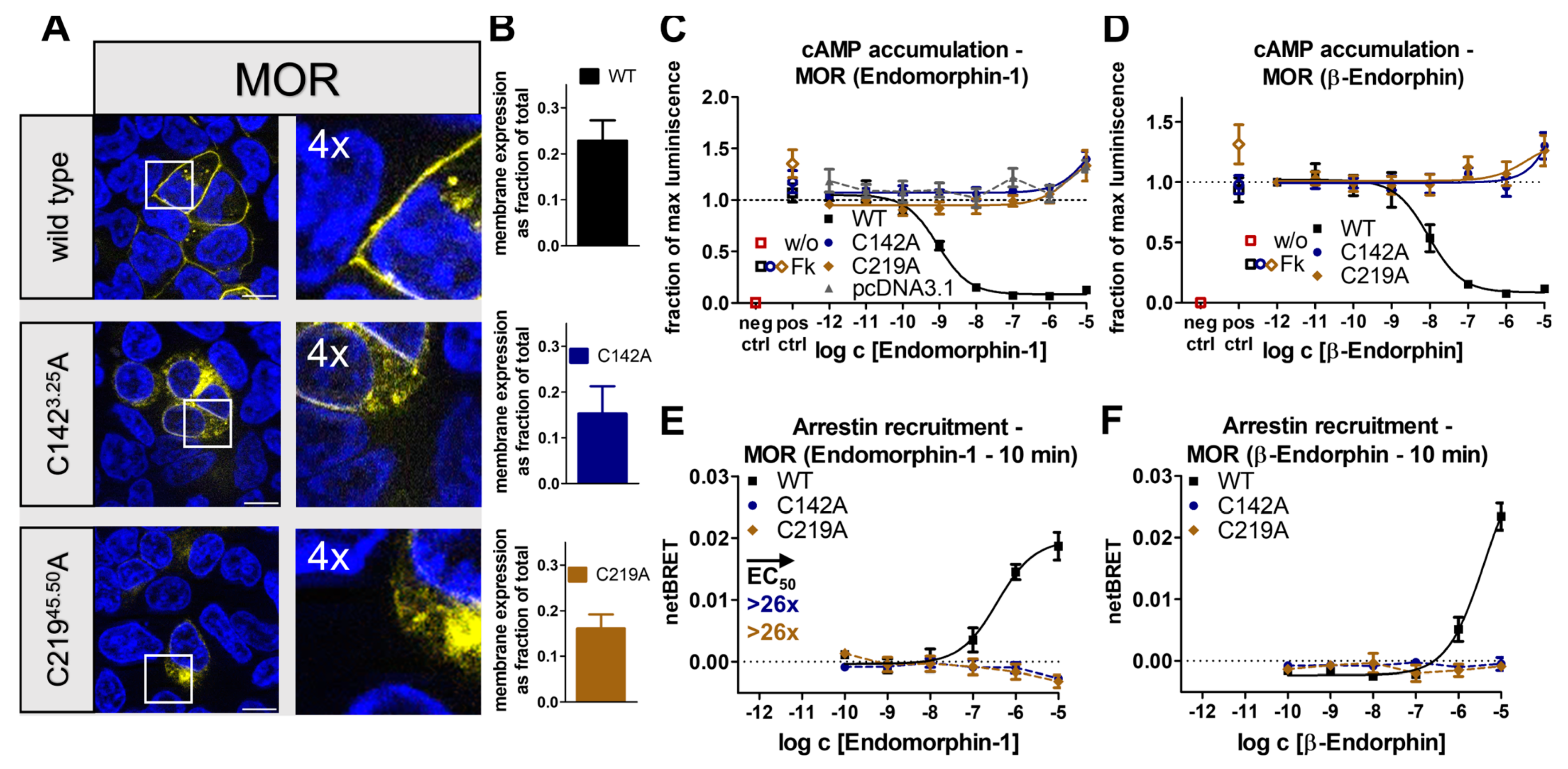
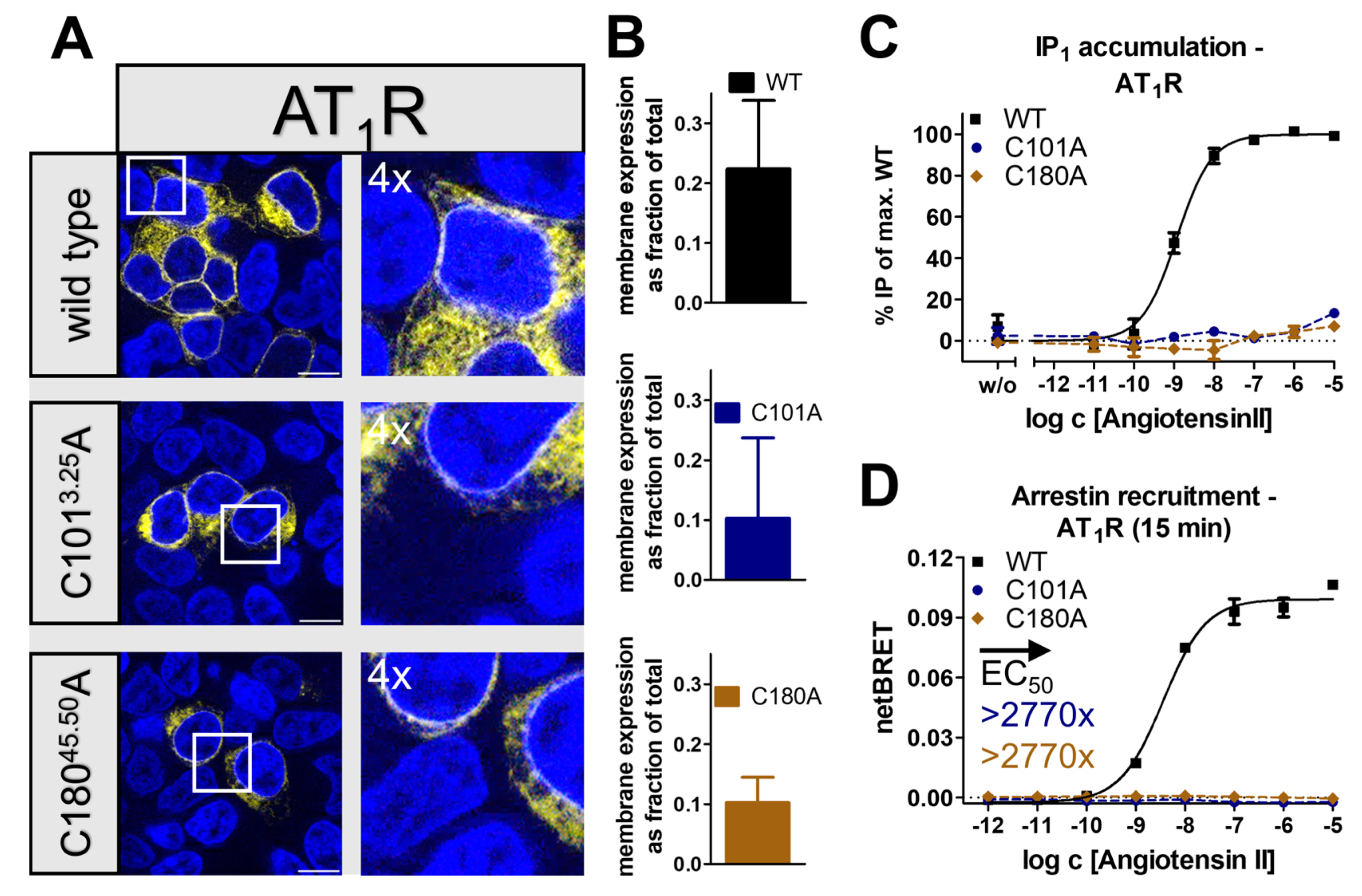
2.2. Receptor- and Pathway-Specific Role of the Conserved Disulfide in Different Peptide-Activated Rhodopsin-like GPCRs
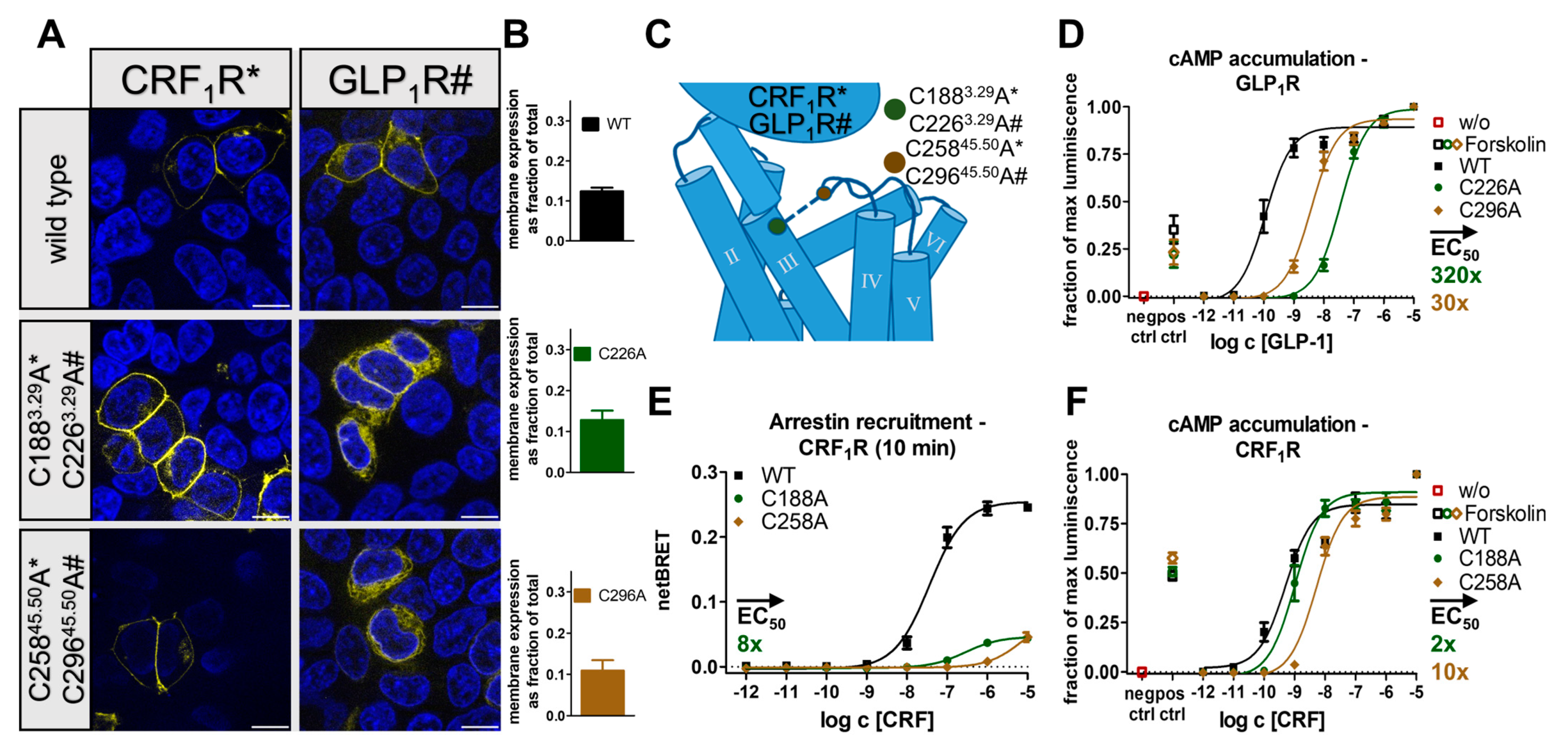
2.3. The Conserved Disulfide Bond Is Not Required for G-Protein Activation of Secretin-like GPCRs
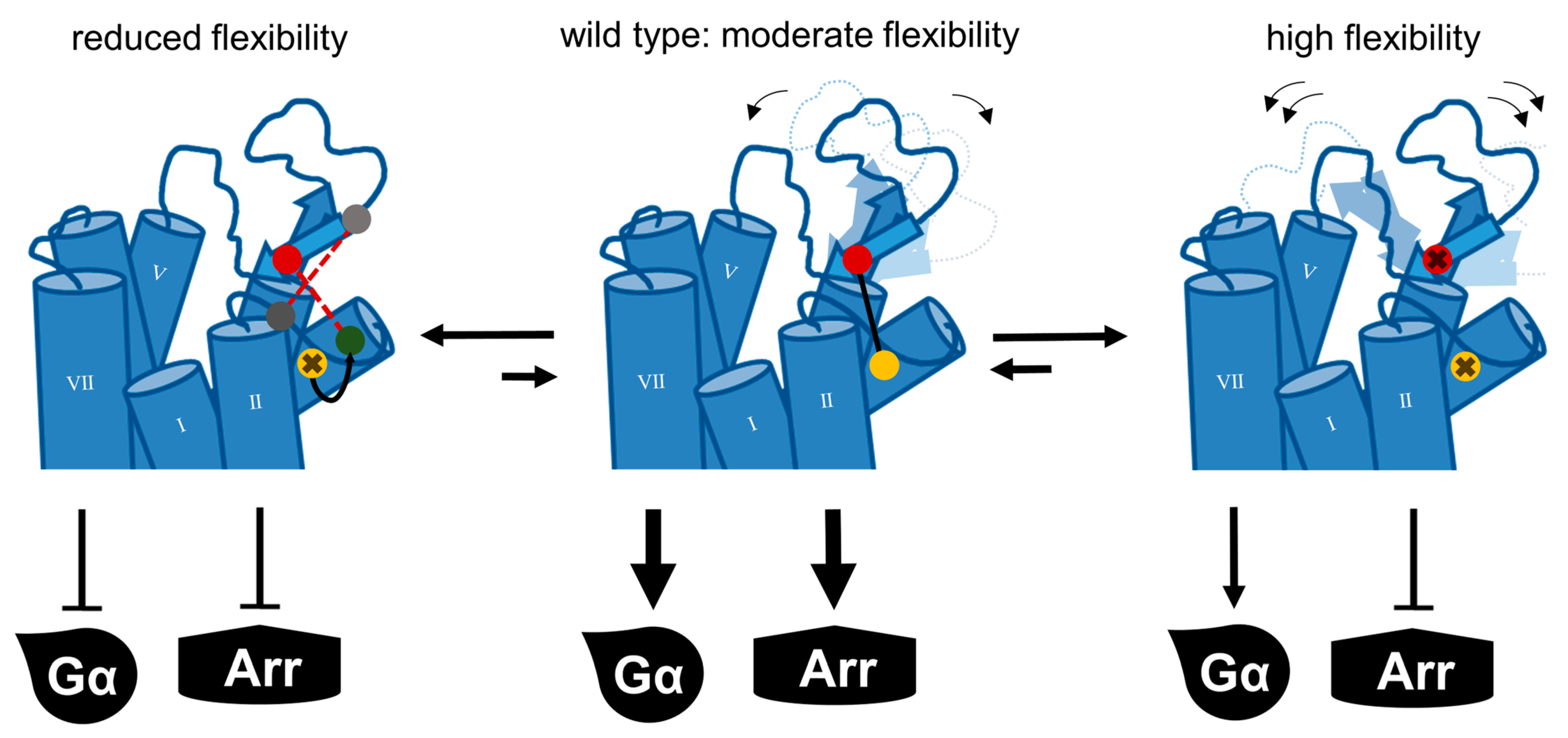
3. Discussion
4. Conclusions
5. Methods
5.1. Generation of Plasmids
5.2. Peptide Synthesis
5.3. Cell Culture
5.4. Fluorescence Microscopy
5.5. cAMP Reporter Gene Assay
5.6. Inositol Phosphate Accumulation Assay
5.7. Arrestin Recruitment Assay
5.8. NanoBRET Ligand Binding Assay
5.9. Cell Surface ELISA
5.10. In Vitro Sample Preparation of Y2R-A20245.49C, Δ6Cys in Small DMPC/DHPC Bicelles for CW-EPR
5.11. CPM Assay
5.12. Fluorescence Polarization Assay
5.13. Continuous-Wave (CW)-EPR Measurements
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, A.; Coin, I. Capturing Peptide-GPCR Interactions and Their Dynamics. Molecules 2020, 25, 4724. [Google Scholar] [CrossRef] [PubMed]
- Elgeti, M.; Hubbell, W.L. DEER Analysis of GPCR Conformational Heterogeneity. Biomolecules 2021, 11, 778. [Google Scholar] [CrossRef]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wüthrich, K. GPCR drug discovery: Integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 2019, 18, 59–82. [Google Scholar] [CrossRef]
- Deganutti, G.; Liang, Y.-L.; Zhang, X.; Khoshouei, M.; Clydesdale, L.; Belousoff, M.J.; Venugopal, H.; Truong, T.T.; Glukhova, A.; Keller, A.N.; et al. Dynamics of GLP-1R peptide agonist engagement are correlated with kinetics of G protein activation. Nat. Commun. 2022, 13, 92. [Google Scholar] [CrossRef]
- Liang, Y.-L.; Belousoff, M.J.; Fletcher, M.M.; Zhang, X.; Khoshouei, M.; Deganutti, G.; Koole, C.; Furness, S.G.B.; Miller, L.J.; Hay, D.L.; et al. Structure and Dynamics of Adrenomedullin Receptors AM1 and AM2 Reveal Key Mechanisms in the Control of Receptor Phenotype by Receptor Activity-Modifying Proteins. ACS Pharmacol. Transl. Sci. 2020, 3, 263–284. [Google Scholar] [CrossRef]
- Schihada, H.; Kowalski-Jahn, M.; Turku, A.; Schulte, G. Deconvolution of WNT-induced Frizzled conformational dynamics with fluorescent biosensors. Biosens. Bioelectron. 2021, 177, 112948. [Google Scholar] [CrossRef]
- Schulte, G.; Wright, S.C. Frizzleds as GPCRs - More Conventional Than We Thought! Trends Pharmacol. Sci. 2018, 39, 828–842. [Google Scholar] [CrossRef]
- Wingler, L.M.; Lefkowitz, R.J. Conformational Basis of G Protein-Coupled Receptor Signaling Versatility. Trends Cell Biol. 2020, 30, 736–747. [Google Scholar] [CrossRef]
- Brian, K.K.; Xavier, D. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol. Sci. 2007, 28, 397–406. [Google Scholar] [CrossRef]
- Hauser, A.S.; Kooistra, A.J.; Munk, C.; Heydenreich, F.M.; Veprintsev, D.B.; Bouvier, M.; Babu, M.M.; Gloriam, D.E. GPCR activation mechanisms across classes and macro/microscales. Nat. Struct. Mol. Biol. 2021, 28, 879–888. [Google Scholar] [CrossRef]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; Shakhnovich, E.I.; et al. Common activation mechanism of class A GPCRs. eLife 2019, 8, e50279. [Google Scholar] [CrossRef]
- Rader, A.J.; Anderson, G.; Isin, B.; Khorana, H.G.; Bahar, I.; Klein-Seetharaman, J. Identification of core amino acids stabilizing rhodopsin. Proc. Natl. Acad. Sci. USA 2004, 101, 7246–7251. [Google Scholar] [CrossRef]
- Dohlman, H.G.; Caron, M.G.; DeBlasi, A.; Frielle, T.; Lefkowitz, R.J. Role of extracellular disulfide-bonded cysteines in the ligand binding function of the beta 2-adrenergic receptor. Biochemistry 1990, 29, 2335–2342. [Google Scholar] [CrossRef]
- Noda, K.; Saad, Y.; Graham, R.M.; Karnik, S.S. The high affinity state of the beta 2-adrenergic receptor requires unique interaction between conserved and non-conserved extracellular loop cysteines. J. Biol. Chem. 1994, 269, 6743–6752. [Google Scholar] [CrossRef]
- Elling, C.E.; Raffetseder, U.; Nielsen, S.M.; Schwartz, T.W. Disulfide bridge engineering in the tachykinin NK1 receptor. Biochemistry 2000, 39, 667–675. [Google Scholar] [CrossRef]
- Cook, J.V.; Eidne, K.A. An intramolecular disulfide bond between conserved extracellular cysteines in the gonadotropin-releasing hormone receptor is essential for binding and activation. Endocrinology 1997, 138, 2800–2806. [Google Scholar] [CrossRef]
- Zeng, F.Y.; Soldner, A.; Schöneberg, T.; Wess, J. Conserved extracellular cysteine pair in the M3 muscarinic acetylcholine receptor is essential for proper receptor cell surface localization but not for G protein coupling. J. Neurochem. 1999, 72, 2404–2414. [Google Scholar] [CrossRef]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Han, S.; Keller, M.; Kaiser, A.; Bender, B.J.; Bosse, M.; Burkert, K.; Kögler, L.M.; Wifling, D.; Bernhardt, G.; et al. Structural basis of ligand binding modes at the neuropeptide Y Y1 receptor. Nature 2018, 556, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Hartig, C.; Chen, Q.; Zhao, W.; Kaiser, A.; Zhang, X.; Zhang, H.; Qu, H.; Yi, C.; Ma, L. Structural basis for ligand recognition of the neuropeptide Y Y2 receptor. Nat. Commun. 2021, 12, 737. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Tan, Q.; Han, S.; Diemar, A.; Löbner, K.; Wang, H.; Schüß, C.; Behr, V.; Mörl, K.; Wang, M.; et al. Receptor-specific recognition of NPY peptides revealed by structures of NPY receptors. Sci. Adv. 2022, 8, eabm1232. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhuang, Y.; DiBerto, J.F.; Zhou, X.E.; Schmitz, G.P.; Yuan, Q.; Jain, M.K.; Liu, W.; Melcher, K.; Jiang, Y.; et al. Structures of the entire human opioid receptor family. Cell 2023, 186, 413–427.e17. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Wingler, L.M.; Skiba, M.A.; McMahon, C.; Staus, D.P.; Kleinhenz, A.L.W.; Suomivuori, C.-M.; Latorraca, N.R.; Dror, R.O.; Lefkowitz, R.J.; Kruse, A.C. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 2020, 367, 888–892. [Google Scholar] [CrossRef]
- Shi, L.; Javitch, J.A. The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc. Natl. Acad. Sci. USA 2004, 101, 440–445. [Google Scholar] [CrossRef]
- Unal, H.; Karnik, S.S. Domain coupling in GPCRs: The engine for induced conformational changes. Trends Pharmacol. Sci. 2012, 33, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Woolley, M.J.; Conner, A.C. Understanding the common themes and diverse roles of the second extracellular loop (ECL2) of the GPCR super-family. Mol. Cell. Endocrinol. 2017, 449, 3–11. [Google Scholar] [CrossRef]
- Yi, M.; Li, H.; Wu, Z.; Yan, J.; Liu, Q.; Ou, C.; Chen, M. A Promising Therapeutic Target for Metabolic Diseases: Neuropeptide Y Receptors in Humans. Cell. Physiol. Biochem. 2018, 45, 88–107. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.-Y.; Chen, W.-C.; Shi, Y.-C.; Wang, C.-M.; Lin, S.; He, H.-F. Regulation of neuropeptide Y in body microenvironments and its potential application in therapies: A review. Cell Biosci. 2021, 11, 151. [Google Scholar] [CrossRef]
- Kostenis, E.; Waelbroeck, M.; Milligan, G. Techniques: Promiscuous Galpha proteins in basic research and drug discovery. Trends Pharmacol. Sci. 2005, 26, 595–602. [Google Scholar] [CrossRef]
- Swanson, D.M.; Wong, V.D.; Jablonowski, J.A.; Shah, C.; Rudolph, D.A.; Dvorak, C.A.; Seierstad, M.; Dvorak, L.K.; Morton, K.; Nepomuceno, D.; et al. The discovery and synthesis of JNJ 31020028, a small molecule antagonist of the Neuropeptide Y Y₂ receptor. Bioorg. Med. Chem. Lett. 2011, 21, 5552–5556. [Google Scholar] [CrossRef]
- Hubbell, W.L.; Cafiso, D.S.; Altenbach, C. Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 2000, 7, 735–739. [Google Scholar] [CrossRef]
- Torricella, F.; Pierro, A.; Mileo, E.; Belle, V.; Bonucci, A. Nitroxide spin labels and EPR spectroscopy: A powerful association for protein dynamics studies. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140653. [Google Scholar] [CrossRef]
- Witte, K.; Kaiser, A.; Schmidt, P.; Splith, V.; Thomas, L.; Berndt, S.; Huster, D.; Beck-Sickinger, A.G. Oxidative in vitro folding of a cysteine deficient variant of the G protein-coupled neuropeptide Y receptor type 2 improves stability at high concentration. Biol. Chem. 2013, 394, 1045–1056. [Google Scholar] [CrossRef]
- Laugwitz, J.M.; Haeri, H.H.; Kaiser, A.; Krug, U.; Hinderberger, D.; Beck-Sickinger, A.G.; Schmidt, P. Probing the Y2 Receptor on Transmembrane, Intra- and Extra-Cellular Sites for EPR Measurements. Molecules 2020, 25, 4143. [Google Scholar] [CrossRef]
- Krug, U.; Gloge, A.; Schmidt, P.; Becker-Baldus, J.; Bernhard, F.; Kaiser, A.; Montag, C.; Gauglitz, M.; Vishnivetskiy, S.A.; Gurevich, V.V.; et al. The Conformational Equilibrium of the Neuropeptide Y2 Receptor in Bilayer Membranes. Angew. Chem. Int. Ed Engl. 2020, 59, 23854–23861. [Google Scholar] [CrossRef]
- Schmidt, P.; Vogel, A.; Schwarze, B.; Seufert, F.; Licha, K.; Wycisk, V.; Kilian, W.; Hildebrand, P.W.; Mitschang, L. Towards Probing Conformational States of Y2 Receptor Using Hyperpolarized 129Xe NMR. Molecules 2023, 28, 1424. [Google Scholar] [CrossRef] [PubMed]
- Wanka, L.; Babilon, S.; Kaiser, A.; Mörl, K.; Beck-Sickinger, A.G. Different mode of arrestin-3 binding at the human Y1 and Y2 receptor. Cell. Signal. 2018, 50, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Johnson, P.S.; Zöllner, C.; Wang, W.; Wang, Z.; Montes, A.E.; Seidleck, B.K.; Blaschak, C.J.; Surratt, C.K. Mutation of human mu opioid receptor extracellular "disulfide cysteine" residues alters ligand binding but does not prevent receptor targeting to the cell plasma membrane. Brain Res. Mol. Brain Res. 1999, 72, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Sumita, C.; Annette, R.; Alan, R.; Gintzler. Biochemical demonstration of mu-opioid receptor association with Gsα: Enhancement following morphine exposure. Brain Res. Mol. Brain Res. 2005, 135, 217–224. [Google Scholar] [CrossRef]
- Shen, K.F.; Crain, S.M. Cholera toxin-A subunit blocks opioid excitatory effects on sensory neuron action potentials indicating mediation by Gs-linked opioid receptors. Brain Res. 1990, 525, 225–231. [Google Scholar] [CrossRef]
- Avidor-Reiss, T.; Bayewitch, M.; Levy, R.; Matus-Leibovitch, N.; Nevo, I.; Vogel, Z. Adenylylcyclase supersensitization in mu-opioid receptor-transfected Chinese hamster ovary cells following chronic opioid treatment. J. Biol. Chem. 1995, 270, 29732–29738. [Google Scholar] [CrossRef] [Green Version]
- Nicoli, A.; Dunkel, A.; Giorgino, T.; Graaf, C.d.; Di Pizio, A. Classification Model for the Second Extracellular Loop of Class A GPCRs. J. Chem. Inf. Model. 2022, 62, 511–522. [Google Scholar] [CrossRef]
- Wheatley, M.; Wootten, D.; Conner, M.T.; Simms, J.; Kendrick, R.; Logan, R.T.; Poyner, D.R.; Barwell, J. Lifting the lid on GPCRs: The role of extracellular loops. Br. J. Pharmacol. 2012, 165, 1688–1703. [Google Scholar] [CrossRef] [Green Version]
- Wootten, D.; Simms, J.; Miller, L.J.; Christopoulos, A.; Sexton, P.M. Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc. Natl. Acad. Sci. USA 2013, 110, 5211–5216. [Google Scholar] [CrossRef]
- Mann, R.J.; Al-Sabah, S.; Maturana, R.L.d.; Sinfield, J.K.; Donnelly, D. Functional coupling of Cys-226 and Cys-296 in the glucagon-like peptide-1 (GLP-1) receptor indicates a disulfide bond that is close to the activation pocket. Peptides 2010, 31, 2289–2293. [Google Scholar] [CrossRef]
- Oakley, R.H.; Laporte, S.A.; Holt, J.A.; Caron, M.G.; Barak, L.S. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 2000, 275, 17201–17210. [Google Scholar] [CrossRef] [Green Version]
- Aydin, Y.; Coin, I. Biochemical insights into structure and function of arrestins. FEBS J. 2021, 288, 2529–2549. [Google Scholar] [CrossRef]
- Wanka, L.; Behr, V.; Beck-Sickinger, A.G. Arrestin-dependent internalization of rhodopsin-like G protein-coupled receptors. Biol. Chem. 2022, 403, 133–149. [Google Scholar] [CrossRef]
- Wacker, D.; Wang, S.; McCorvy, J.D.; Betz, R.M.; Venkatakrishnan, A.J.; Levit, A.; Lansu, K.; Schools, Z.L.; Che, T.; Nichols, D.E.; et al. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 2017, 168, 377–389.e12. [Google Scholar] [CrossRef] [Green Version]
- Rath, P.; Bovee-Geurts, P.H.; DeGrip, W.J.; Rothschild, K.J. Photoactivation of rhodopsin involves alterations in cysteine side chains: Detection of an S-H band in the Meta I--Meta II FTIR difference spectrum. Biophys. J. 1994, 66, 2085–2091. [Google Scholar] [CrossRef] [Green Version]
- Parthier, C.; Reedtz-Runge, S.; Rudolph, R.; Stubbs, M.T. Passing the baton in class B GPCRs: Peptide hormone activation via helix induction? Trends Biochem. Sci. 2009, 34, 303–310. [Google Scholar] [CrossRef]
- Liang, Y.-L.; Belousoff, M.J.; Zhao, P.; Koole, C.; Fletcher, M.M.; Truong, T.T.; Julita, V.; Christopoulos, G.; Xu, H.E.; Zhang, Y.; et al. Toward a Structural Understanding of Class B GPCR Peptide Binding and Activation. Mol. Cell 2020, 77, 656–668.e5. [Google Scholar] [CrossRef]
- Wolf, S.; Grünewald, S. Sequence, structure and ligand binding evolution of rhodopsin-like G protein-coupled receptors: A crystal structure-based phylogenetic analysis. PLoS ONE 2015, 10, e0123533. [Google Scholar] [CrossRef]
- Duan, J.; Shen, D.-D.; Zhou, X.E.; Bi, P.; Liu, Q.-F.; Tan, Y.-X.; Zhuang, Y.-W.; Zhang, H.-B.; Xu, P.-Y.; Huang, S.-J.; et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 2020, 11, 4121. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, X.; Zhao, L.; Wang, Y.; Ye, C.; Zou, X.; Dai, A.; Cong, Z.; Chen, J.; Zhou, Q.; et al. Molecular insights into differentiated ligand recognition of the human parathyroid hormone receptor 2. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Koole, C.; Wootten, D.; Simms, J.; Savage, E.E.; Miller, L.J.; Christopoulos, A.; Sexton, P.M. Second extracellular loop of human glucagon-like peptide-1 receptor (GLP-1R) differentially regulates orthosteric but not allosteric agonist binding and function. J. Biol. Chem. 2012, 287, 3659–3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, L.; Zarzycka, B.; Zaidi, S.A.; Katritch, V.; Coin, I. Structural insight into the activation of a class B G-protein-coupled receptor by peptide hormones in live human cells. eLife 2017, 6, e27711. [Google Scholar] [CrossRef] [PubMed]
- Gkountelias, K.; Tselios, T.; Venihaki, M.; Deraos, G.; Lazaridis, I.; Rassouli, O.; Gravanis, A.; Liapakis, G. Alanine scanning mutagenesis of the second extracellular loop of type 1 corticotropin-releasing factor receptor revealed residues critical for peptide binding. Mol. Pharmacol. 2009, 75, 793–800. [Google Scholar] [CrossRef] [Green Version]
- Pal, K.; Melcher, K.; Xu, H.E. Structure and mechanism for recognition of peptide hormones by Class B G-protein-coupled receptors. Acta Pharmacol. Sin. 2012, 33, 300–311. [Google Scholar] [CrossRef]
- Coin, I.; Katritch, V.; Sun, T.; Xiang, Z.; Siu, F.Y.; Beyermann, M.; Stevens, R.C.; Wang, L. Genetically encoded chemical probes in cells reveal the binding path of urocortin-I to CRF class B GPCR. Cell 2013, 155, 1258–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Qiao, A.; Yang, L.; van Eps, N.; Frederiksen, K.S.; Yang, D.; Dai, A.; Cai, X.; Zhang, H.; Yi, C.; et al. Structure of the glucagon receptor in complex with a glucagon analogue. Nature 2018, 553, 106–110. [Google Scholar] [CrossRef]
- Graaf, C.d.; Song, G.; Cao, C.; Zhao, Q.; Wang, M.-W.; Wu, B.; Stevens, R.C. Extending the Structural View of Class B GPCRs. Trends Biochem. Sci. 2017, 42, 946–960. [Google Scholar] [CrossRef]
- Liu, H.; Naismith, J.H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Mäde, V.; Bellmann-Sickert, K.; Kaiser, A.; Meiler, J.; Beck-Sickinger, A.G. Position and length of fatty acids strongly affect receptor selectivity pattern of human pancreatic polypeptide analogues. ChemMedChem 2014, 9, 2463–2474. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, A.; Beck-Sickinger, A.G.; Zhao, Q.; Wu, B. IP accumulation assay. BIO-PROTOCOL 2022. [Google Scholar] [CrossRef]
- Wolf, P.; Mohr, A.; Gavins, G.; Behr, V.; Mörl, K.; Seitz, O.; Beck-Sickinger, A.G. Orthogonal Peptide-Templated Labeling Elucidates Lateral ETA R/ETB R Proximity and Reveals Altered Downstream Signaling. Chembiochem 2022, 23, e202100340. [Google Scholar] [CrossRef]
- Rudolf, K.; Eberlein, W.; Engel, W.; Wieland, H.A.; Willim, K.D.; Entzeroth, M.; Wienen, W.; Beck-Sickinger, A.G.; Doods, H.N. The first highly potent and selective non-peptide neuropeptide Y Y1 receptor antagonist: BIBP3226. Eur. J. Pharmacol. 1994, 271, R11–R13. [Google Scholar] [CrossRef]
- Shoblock, J.R.; Welty, N.; Nepomuceno, D.; Lord, B.; Aluisio, L.; Fraser, I.; Motley, S.T.; Sutton, S.W.; Morton, K.; Galici, R.; et al. In vitro and in vivo characterization of JNJ-31020028 (N-(4-{4-2-(diethylamino)-2-oxo-1-phenylethylpiperazin-1-yl}-3-fluorophenyl)-2-pyridin-3-ylbenzamide), a selective brain penetrant small molecule antagonist of the neuropeptide Y Y(2) receptor. Psychopharmacology 2010, 208, 265–277. [Google Scholar] [CrossRef]
- Schmidt, P.; Bender, B.J.; Kaiser, A.; Gulati, K.; Scheidt, H.A.; Hamm, H.E.; Meiler, J.; Beck-Sickinger, A.G.; Huster, D. Improved in Vitro Folding of the Y2 G Protein-Coupled Receptor into Bicelles. Front. Mol. Biosci. 2017, 4, 100. [Google Scholar] [CrossRef] [Green Version]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wygas, M.M.; Laugwitz, J.M.; Schmidt, P.; Elgeti, M.; Kaiser, A. Dynamics of the Second Extracellular Loop Control Transducer Coupling of Peptide-Activated GPCRs. Int. J. Mol. Sci. 2023, 24, 12197. https://doi.org/10.3390/ijms241512197
Wygas MM, Laugwitz JM, Schmidt P, Elgeti M, Kaiser A. Dynamics of the Second Extracellular Loop Control Transducer Coupling of Peptide-Activated GPCRs. International Journal of Molecular Sciences. 2023; 24(15):12197. https://doi.org/10.3390/ijms241512197
Chicago/Turabian StyleWygas, Marcel M., Jeannette M. Laugwitz, Peter Schmidt, Matthias Elgeti, and Anette Kaiser. 2023. "Dynamics of the Second Extracellular Loop Control Transducer Coupling of Peptide-Activated GPCRs" International Journal of Molecular Sciences 24, no. 15: 12197. https://doi.org/10.3390/ijms241512197
APA StyleWygas, M. M., Laugwitz, J. M., Schmidt, P., Elgeti, M., & Kaiser, A. (2023). Dynamics of the Second Extracellular Loop Control Transducer Coupling of Peptide-Activated GPCRs. International Journal of Molecular Sciences, 24(15), 12197. https://doi.org/10.3390/ijms241512197