
Benzenesulfonamide Analogs: Synthesis, Anti-GBM Activity and Pharmacoprofiling

 ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results
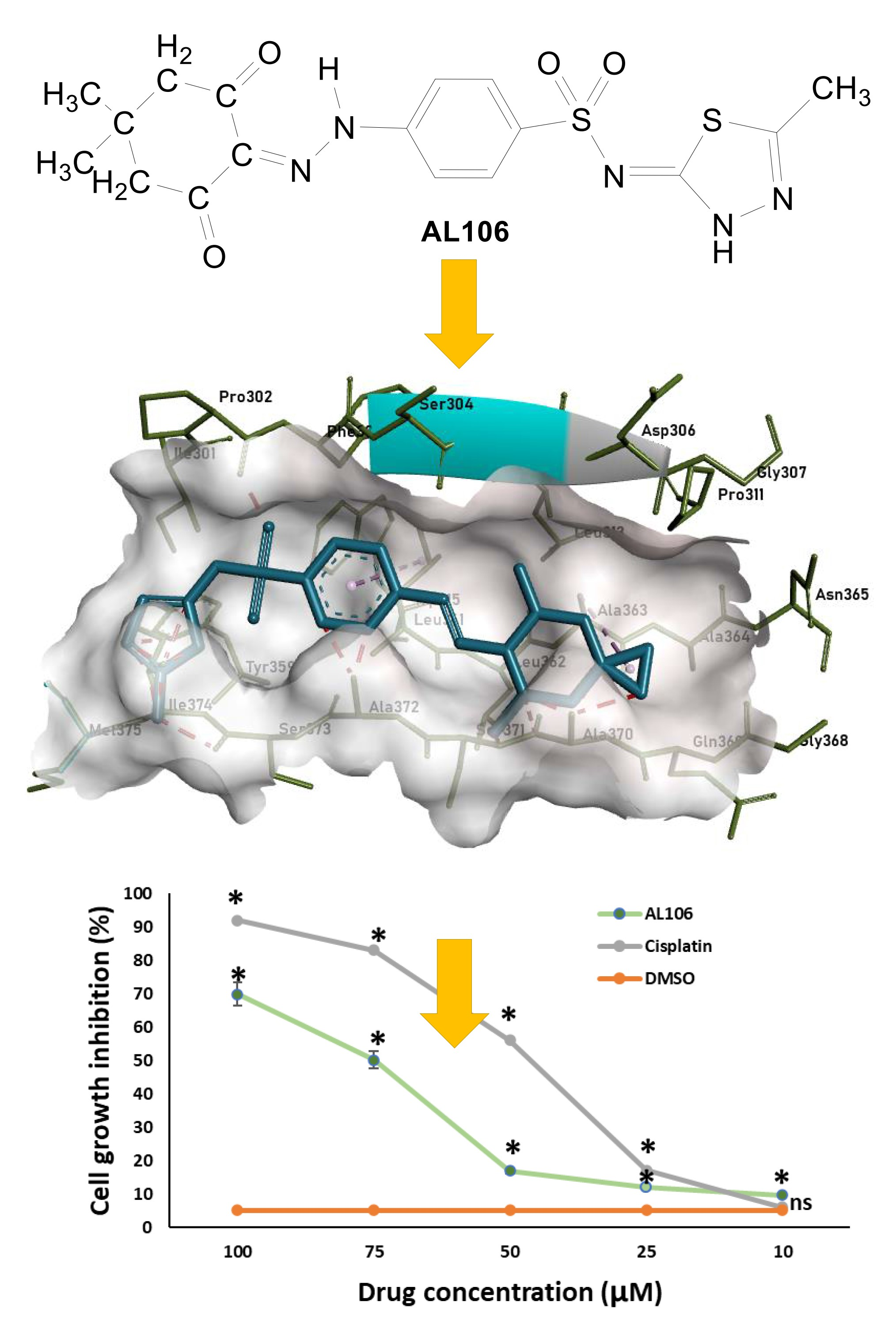
2.1. Benzenesulfonamide Derivatives’ Interaction with Receptor Tyrosine Kinase, TrkA
2.2. AL106 Effectively Reduces Cell Viability of GBM Cells
2.3. AL106 Effectively Reduces the GBM Proliferation
2.4. Benezesulfonamide Derived Compounds Exhibit Drug-like Properties
2.5. Quantitative Structure–Activity Relationship (QSAR) Analysis of Synthesized Derivatives and Their Biological Activity
3. Discussion
4. Materials and Methodology
4.1. Chemistry
4.2. Synthesis of Novel Benzenesulfonamide Derivatives
4.3. Characterization of Synthesized Benzenesulfonamide Derivatives
4.4. Computational Assessment of Benzenesulfonamide Derivatives–TrkA Interaction
4.5. Chemicals and Drug Preparation
4.6. Cell Culture
4.7. Cytotoxic Effect of Benzenesulfonamide Derivatives
4.8. Dose-Dependent Cell Viability Assay
4.9. ADMET and Drug-likeness Prediction
4.10. Quantitative Structure–Activity Relationship Models (QSAR) for AL106
4.11. Data and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C.; et al. Coactivation of Receptor Tyrosine Kinases Affects the Response of Tumor Cells to Targeted Therapies. Science 2007, 318, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef]
- Hubbard, S.R. Structural analysis of receptor tyrosine kinases. Prog. Biophys. Mol. Biol. 1999, 71, 343–358. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Descamps, S.; Toillon, R.-A.; Adriaenssens, E.; Pawlowski, V.; Cool, S.M.; Nurcombe, V.; Le Bourhis, X.; Boilly, B.; Peyrat, J.-P.; Hondermarck, H. Nerve Growth Factor Stimulates Proliferation and Survival of Human Breast Cancer Cells through Two Distinct Signaling Pathways. J. Biol. Chem. 2001, 276, 17864–17870. [Google Scholar] [CrossRef] [PubMed]
- Martins, N.M.R.; Anbu, S.; Mahmudov, K.T.; Ravishankaran, R.; da Silva, M.F.C.G.; Martins, L.M.D.R.S.; Karande, A.A.; Pombeiro, A.J.L. DNA and BSA binding and cytotoxic properties of copper(II) and iron(III) complexes with arylhydrazone of ethyl 2-cyanoacetate or formazan ligands. New J. Chem. 2017, 41, 4076–4086. [Google Scholar] [CrossRef]
- Palmucci, J.; Mahmudov, K.T.; da Silva, M.F.C.G.; Marchetti, F.; Pettinari, C.; Petrelli, D.; Vitali, L.A.; Quassinti, L.; Bramucci, M.; Lupidi, G.; et al. DNA and BSA binding, anticancer and antimicrobial properties of Co(II), Co(II/III), Cu(II) and Ag(I) complexes of arylhydrazones of barbituric acid. RSC Adv. 2015, 6, 4237–4249. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; da Silva, M.C.G.; Kopylovich, M.N.; Fernandes, A.R.; Silva, A.; Mizar, A.; Pombeiro, A.J. Di- and tri-organotin(IV) complexes of arylhydrazones of methylene active compounds and their antiproliferative activity. J. Organomet. Chem. 2014, 760, 67–73. [Google Scholar] [CrossRef]
- Verma, G.; Marella, A.; Shaquiquzzaman, M.; Akhtar, M.; Rahmat Ali, M.; Alam, M.M. A review exploring biological activities of hydrazones. J. Pharm. Bioallied Sci. 2014, 6, 69–80. [Google Scholar] [CrossRef]
- Palanivel, S.; Zhurina, A.; Doan, P.; Chandraseelan, J.G.; Khandelwal, V.K.M.; Zubkov, F.I.; Mahmudov, K.T.; Pombeiro, A.J.; Yli-Harja, O.; Kandhavelu, M. In vitro characterization of arylhydrazones of active methylene derivatives. Saudi Pharm. J. 2018, 26, 430–436. [Google Scholar] [CrossRef]
- Viswanathan, A.; Kute, D.; Musa, A.; Mani, S.K.; Sipilä, V.; Emmert-Streib, F.; Zubkov, F.; Gurbanov, A.V.; Yli-Harja, O.; Kandhavelu, M. 2-(2-(2,4-dioxopentan-3-ylidene)hydrazineyl)benzonitrile as novel inhibitor of receptor tyrosine kinase and PI3K/AKT/mTOR signaling pathway in glioblastoma. Eur. J. Med. Chem. 2019, 166, 291–303. [Google Scholar] [CrossRef]
- Xu, G.; Abad, M.C.; Connolly, P.J.; Neeper, M.P.; Struble, G.T.; Springer, B.A.; Emanuel, S.L.; Pandey, N.; Gruninger, R.H.; Adams, M.; et al. 4-Amino-6-arylamino-pyrimidine-5-carbaldehyde hydrazones as potent ErbB-2/EGFR dual kinase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4615–4619. [Google Scholar] [CrossRef]
- Abou-Seri, S.M. Synthesis and biological evaluation of novel 2,4′-bis substituted diphenylamines as anticancer agents and potential epidermal growth factor receptor tyrosine kinase inhibitors. Eur. J. Med. Chem. 2010, 45, 4113–4121. [Google Scholar] [CrossRef] [PubMed]
- Gurbanov, A.V.; Kuznetsov, M.L.; Mahmudov, K.T.; Pombeiro, A.J.L.; Resnati, G. Resonance Assisted Chalcogen Bonding as a New Synthon in the Design of Dyes. Chem. A Eur. J. 2020, 26, 14833–14837. [Google Scholar] [CrossRef]
- Gurbanov, A.V.; Kuznetsov, M.L.; Resnati, G.; Mahmudov, K.T.; Pombeiro, A.J.L. Chalcogen and Hydrogen Bonds at the Periphery of Arylhydrazone Metal Complexes. Cryst. Growth Des. 2022, 22, 3932–3940. [Google Scholar] [CrossRef]
- Ron-Harel, N.; Ghergurovich, J.M.; Notarangelo, G.; LaFleur, M.W.; Tsubosaka, Y.; Sharpe, A.H.; Rabinowitz, J.D.; Haigis, M.C. T Cell Activation Depends on Extracellular Alanine. Cell Rep. 2019, 28, 3011–3021.e4. [Google Scholar] [CrossRef]
- Hay, D.L.; Harris, P.W.R.; Kowalczyk, R.; Brimble, A.M.; Rathbone, D.L.; Barwell, J.; Conner, A.C.; Poyner, D.R. Structure–activity relationships of the N-terminus of calcitonin gene-related peptide: Key roles of alanine-5 and threonine-6 in receptor activation. Br. J. Pharmacol. 2013, 171, 415–426. [Google Scholar] [CrossRef]
- Doan, P.; Musa, A.; Murugesan, A.; Sipilä, V.; Candeias, N.R.; Emmert-Streib, F.; Ruusuvuori, P.; Granberg, K.; Yli-Harja, O.; Kandhavelu, M. Glioblastoma Multiforme Stem Cell Cycle Arrest by Alkylaminophenol through the Modulation of EGFR and CSC Signaling Pathways. Cells 2020, 9, 681. [Google Scholar] [CrossRef]
- Saravanan, K.M.; Palanivel, S.; Yli-Harja, O.; Kandhavelu, M. Identification of novel GPR17-agonists by structural bioinformatics and signaling activation. Int. J. Biol. Macromol. 2017, 106, 901–907. [Google Scholar] [CrossRef]
- Nguyen, P.; Doan, P.; Rimpilainen, T.; Mani, S.K.; Murugesan, A.; Yli-Harja, O.; Candeias, N.R.; Kandhavelu, M. Synthesis and Preclinical Validation of Novel Indole Derivatives as a GPR17 Agonist for Glioblastoma Treatment. J. Med. Chem. 2021, 64, 10908–10918. [Google Scholar] [CrossRef] [PubMed]
- Mutharasu, G.; Murugesan, A.; Mani, S.K.; Yli-Harja, O.; Kandhavelu, M. Transcriptomic analysis of glioblastoma multiforme providing new insights into GPR17 signaling communication. J. Biomol. Struct. Dyn. 2022, 40, 2586–2599. [Google Scholar] [CrossRef]
- Nguyen, P.; Doan, P.; Murugesan, A.; Ramesh, T.; Rimpilainen, T.; Candeias, N.R.; Yli-Harja, O.; Kandhavelu, M. GPR17 signaling activation by CHBC agonist induced cell death via modulation of MAPK pathway in glioblastoma. Life Sci. 2022, 291, 120307. [Google Scholar] [CrossRef] [PubMed]
- Vanderbeek, A.; Rahman, R.; Fell, G.; Ventz, S.; Chen, T.; Redd, R.; Parmigiani, G.; Cloughesy, T.F.; Wen, P.Y.; Trippa, L.; et al. The clinical trials landscape for glioblastoma: Is it adequate to develop new treatments? Neuro-Oncology 2018, 20, 1034–1043. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Sanner, M.F.; Olson, A.J.; Forli, S. The AutoDock suite at 30. Protein Sci. 2020, 30, 31–43. [Google Scholar] [CrossRef]
- Gao, Y.-D.; Huang, J.-F. An extension strategy of Discovery Studio 2.0 for non-bonded interaction energy automatic calculation at the residue level. Dongwuxue Yanjiu 2011, 32, 262–266. [Google Scholar] [CrossRef]
- Alsaid, M.S.; Al-Mishari, A.A.; Soliman, A.M.; Ragab, F.A.; Ghorab, M.M. Discovery of Benzo[g]quinazolin benzenesulfonamide derivatives as dual EGFR/HER2 inhibitors. Eur. J. Med. Chem. 2017, 141, 84–91. [Google Scholar] [CrossRef] [PubMed]
- V4. 0.100. 13345, D.S. Visualizer. Accelrys Software Inc.: San Diego, CA, USA, 2005.
- Geetha, T.; Seibenhener, M.L.; Chen, L.; Madura, K.; Wooten, M.W. p62 serves as a shuttling factor for TrkA interaction with the proteasome. Biochem. Biophys. Res. Commun. 2008, 374, 33–37. [Google Scholar] [CrossRef]
- Bessette; Giraud, S.; Loum, E.; Bessette, B.; Mathonnet, M.; Lalloué, F. P75 neurotrophin receptor is sequestered in the Golgi apparatus of the U-87 MG human glioblastoma cell line. Int. J. Oncol. 2011, 38, 391–399. [Google Scholar] [CrossRef]
- Rashid, M. Design, synthesis and ADMET prediction of bis-benzimidazole as anticancer agent. Bioorg. Chem. 2020, 96, 103576. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADMET | Descriptors | AL34 | AL56 | AL106 | AL107 | AL109 | AL110 | Sulfamethiazole |
|---|---|---|---|---|---|---|---|---|
| Physicochemical Properties | H-bond acceptors | 6 | 8 | 7 | 7 | 7 | 5 | 4 |
| H-bond donors | 2 | 4 | 2 | 2 | 3 | 3 | 2 | |
| TPSA | 141.16 | 208.23 | 167.1 | 167.1 | 179.13 | 144.99 | 134.59 | |
| Lipophilicity | iLOGP | 1.78 | −0.26 | 1.54 | 1.65 | 1.6 | 1.54 | 1.5 |
| XLOGP3 | 2.17 | 0.44 | 1.76 | 2.32 | 3.42 | 2.97 | 0.54 | |
| WLOGP | 3.99 | −0.28 | 3.07 | 2.02 | 3.15 | 3.38 | 2.13 | |
| MLOGP | 1.04 | −1.01 | 0.33 | 0.22 | 0.5 | 1.55 | −0.05 | |
| Silicos-IT Log P | 3.02 | 0.01 | 2.72 | 3.14 | 2.07 | 2.48 | 0.92 | |
| Consensus Log P | 2.4 | −0.22 | 1.88 | 1.87 | 2.15 | 2.38 | 1.01 | |
| Water Solubility | ESOL Log S | −3.7 | −2.63 | −3.52 | −4.13 | −4.65 | −4.33 | −2.14 |
| ESOL Class | Soluble | Soluble | Soluble | Moderately soluble | Moderately soluble | Moderately soluble | Soluble | |
| Ali Log S | −4.77 | −4.38 | −4.89 | −5.47 | −6.86 | −5.68 | −2.94 | |
| Ali Class | Moderately soluble | Moderately soluble | Moderately soluble | Moderately soluble | Poorly soluble | Moderately soluble | Soluble | |
| Silicos-IT LogSw | −5.78 | −4.68 | −6 | −7.38 | −6.67 | −6.75 | −3.44 | |
| Silicos-IT class | Moderately soluble | Moderately soluble | Moderately soluble | Poorly soluble | Poorly soluble | Poorly soluble | Soluble | |
| Pharmacokinetics | GI absorption | Low | Low | Low | Low | Low | Low | High |
| BBB permeant | No | No | No | No | No | No | No | |
| Pgp substrate | No | No | No | No | No | Yes | No | |
| CYP1A2 inhibitor | No | No | No | No | No | Yes | No | |
| CYP2C19 inhibitor | No | No | No | Yes | No | Yes | No | |
| CYP2C9 inhibitor | Yes | No | No | No | Yes | Yes | No | |
| CYP2D6 inhibitor | No | No | No | No | No | No | No | |
| CYP3A4 inhibitor | No | No | No | Yes | Yes | Yes | No | |
| log Kp (cm/s) | −7.04 | −8.48 | −7.62 | −7.26 | −6.67 | −6.72 | −7.57 | |
| Druglikeness | Lipinski violations | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| Training Set | ||||
|---|---|---|---|---|
| Sl. No | Compound | Chemical Name | Observed | Predicted |
| 1 | AL56 | (Z)-N-(5-methyl-1,3,4-thiadiazol-2(3H)-ylidene)-4-(2-(2,4,6-trioxotetrahydropyrimidin-5(2H)-ylidene)hydrazineyl)benzenesulfonamide | 0.2920 | 0.2939 |
| 2 | AL107 | (Z)-4-(2-(1,3-dioxo-1,3-dihydro-2H-inden-2-ylidene)hydrazineyl)-N-(5-methyl-1,3,4-thiadiazol-2(3H)-ylidene)benzenesulfonamide | 0.2569 | 0.1753 |
| 3 | AL109 | (Z)-2-(2-(4-(N-((Z)-5-methyl-1,3,4-thiadiazol-2(3H)-ylidene)sulfamoyl)phenyl)hydrazineylidene)-3-oxo-N-phenylbutanamide | 1.0000 | 1.0000 |
| 4 | AL110 | 4-(((E)-indolin-2-ylmethylene)amino)-N-((Z)-5-methyl-1,3,4-thiadiazol-2(3H)-ylidene)benzenesulfonamide | 0.5220 | 0.4761 |
| 5 | R8 | 3-(2-(4-bromophenyl)hydrazineylidene) pentane-2,4-dione | 0.4786 | 0.3883 |
| 6 | R9 | 3-(2- (4-bromophenyl)hydrazineylidene) pentane-2,4-dione | 0.4683 | 0.3806 |
| 7 | R10 | (2-(2-(2,4- dioxopentan-3-ylidene)hydrazineyl) phenyl)arsonic acid | 0.8102 | 0.7668 |
| 8 | R31 | 5-chloro-3-(2-(1-ethoxy-1,3-dioxobutan-2-ylidene)hydrazineyl)- 2-hydroxybenzenesulfonic acid | 0.7412 | 0.7758 |
| 9 | R40 | ethyl 2-(2-(4-chlorophenyl)hydrazineylidene)-3- oxobutanoate | 0.2023 | 0.0586 |
| 10 | R46 | 5-(2-(1,3-dioxo-1-phenylbutan-2- ylidene)hydrazineyl)-4-hydroxybenzene-1,3-disulfonic acid | 0.5571 | 0.5651 |
| 11 | R156 | 4-(2-(2,4,6-trioxotetrahydropyrimidin- 5(2H)-ylidene)hydrazineyl)benzoic acid | 0.5725 | 0.5923 |
| 12 | R221 | N,N-dimethyl-2-(2-(4-nitrophenyl)hydrazineylidene)-3- oxobutanamide | 0.2119 | 0.1312 |
| 13 | R235 | 3-(2-(2-nitrophenyl) hydrazineylidene)pentane-2,4-dione | 0.1831 | 0.1977 |
| 14 | R243 | ethyl 2-(2-(4-cyanophenyl) hydrazineylidene)-3-oxobutanoate | 0.0000 | 0.0456 |
| 15 | R236 | 2-(2-(2,4-dioxopentan-3- ylidene)hydrazineyl) benzoic acid | 0.1862 | 0.2609 |
| 16 | R237 | 2-(2-(2,4-dioxopentan-3-ylidene)hydrazineyl) benzenesulfonic acid | 0.1497 | 0.0000 |
| 17 | R241 | 4-(2-(2,4-dioxopentan-3-ylidene)hydrazineyl) benzonitrile | 0.1301 | 0.1559 |
| 18 | R244 | 2-(2-(4,4-dimethyl-2,6-dioxocyclohexylidene) hydrazineyl) benzoic acid | 0.4727 | 0.2739 |
| 19 | R246 | sodium 2-(2- (1,3-dioxo-1,3-diphenylpropan-2-ylidene)hydrazineyl)benzenesulfonate | 0.5716 | 0.6203 |
| 20 | R283 | 5-(2-(2,4- dioxopentan-3-ylidene)hydrazineyl)isophthalic acid | 0.4994 | 0.4164 |
| Test Set | ||||
| Sl. No | Compound | Chemical Name | ||
| 1 | AL34 | 4-(((E)-2-hydroxybenzylidene)amino)-N-((Z)-5-methyl-1,3,4-thiadiazol-2(3H)-ylidene)benzenesulfonamide | 0.5644 | 0.5212 |
| 2 | AL106 | (Z)-4-(2-(4,4-dimethyl-2,6-dioxocyclohexylidene)hydrazineyl)-N-(5-methyl-1,3,4-thiadiazol-2(3H)-ylidene)benzenesulfonamide | 0.1750 | 0.1162 |
| 3 | R2 | 3-(2-(2,4-dioxopentan-3-ylidene)hydrazineyl)-2- hydroxy-5-nitrobenzenesulfonic acid | 0.4695 | 0.4641 |
| 4 | R212 | 3-(2-(1-(dimethylamino)-1,3-dioxobutan-2- ylidene)hydrazineyl)-2-hydroxy-5-nitrobenzenesulfonic acid | 0.3247 | 0.2548 |
| 5 | R234 | 2-(2-(2,4-dioxopentan-3-ylidene) hydrazineyl)benzonitrile | 0.5204 | 0.5103 |
| 6 | R313 | 4-(2-(1-cyano-2-methoxy-2- oxoethylidene) hydrazineyl)benzoic acid | 0.5352 | 0.4491 |
| 7 | Sul | 4-amino-N-(5-methyl-1,3,4-thiadiazol-2-yl)-benzenesulfonamide | 0.5257 | 0.4384 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murugesan, A.; Konda Mani, S.; Thiyagarajan, R.; Palanivel, S.; Gurbanov, A.V.; Zubkov, F.I.; Kandhavelu, M. Benzenesulfonamide Analogs: Synthesis, Anti-GBM Activity and Pharmacoprofiling. Int. J. Mol. Sci. 2023, 24, 12276. https://doi.org/10.3390/ijms241512276
Murugesan A, Konda Mani S, Thiyagarajan R, Palanivel S, Gurbanov AV, Zubkov FI, Kandhavelu M. Benzenesulfonamide Analogs: Synthesis, Anti-GBM Activity and Pharmacoprofiling. International Journal of Molecular Sciences. 2023; 24(15):12276. https://doi.org/10.3390/ijms241512276
Chicago/Turabian StyleMurugesan, Akshaya, Saravanan Konda Mani, Ramesh Thiyagarajan, Suresh Palanivel, Atash V. Gurbanov, Fedor I. Zubkov, and Meenakshisundaram Kandhavelu. 2023. "Benzenesulfonamide Analogs: Synthesis, Anti-GBM Activity and Pharmacoprofiling" International Journal of Molecular Sciences 24, no. 15: 12276. https://doi.org/10.3390/ijms241512276
APA StyleMurugesan, A., Konda Mani, S., Thiyagarajan, R., Palanivel, S., Gurbanov, A. V., Zubkov, F. I., & Kandhavelu, M. (2023). Benzenesulfonamide Analogs: Synthesis, Anti-GBM Activity and Pharmacoprofiling. International Journal of Molecular Sciences, 24(15), 12276. https://doi.org/10.3390/ijms241512276