
A Novel FLCN Variant in a Suspected Birt–Hogg–Dubè Syndrome Patient

 ,
,  ,
,  and
and
Abstract
:1. Introduction
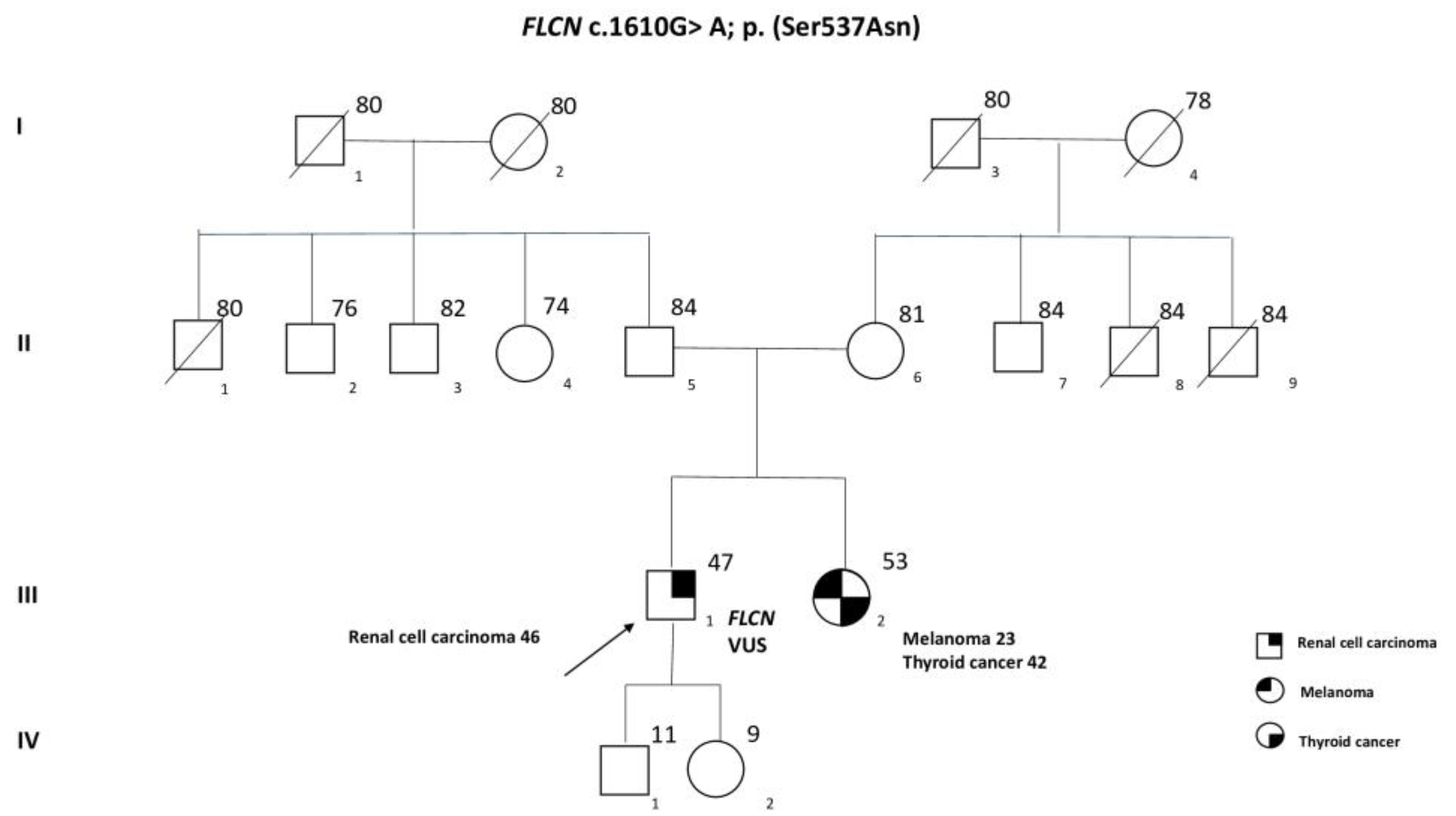
Case Presentation
2. Results
3. Discussion
4. Materials and Methods
4.1. Patient Collection
4.2. Blood Collection and DNA Extraction
4.3. Next-Generation Sequencing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stamatakis, L.; Metwalli, A.R.; Middelton, L.A.; Marston Linehan, W. Diagnosis and Management of BHD-Associated Kidney Cancer. Fam. Cancer 2013, 12, 397–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, M.; Zhang, X.; Fan, L.; Cheng, S.; Kiram, A.; Cen, S.; Chen, B.; Ye, M.; Gao, Q.; Zhu, C.; et al. A Novel FLCN Intragenic Deletion Identified by NGS in a BHDS Family and Literature Review. Front. Genet. 2021, 12, 636900. [Google Scholar] [CrossRef] [PubMed]
- Savatt, J.M.; Shimelis, H.; Moreno-De-Luca, A.; Strande, N.T.; Oetjens, M.T.; Ledbetter, D.H.; Martin, C.L.; Myers, S.M.; Finucane, B.M. Frequency of Truncating FLCN Variants and Birt-Hogg-Dubé-Associated Phenotypes in a Health Care System Population. Genet. Med. 2022, 24, 1857–1866. [Google Scholar] [CrossRef]
- Zhang, Q.; Xu, Y.; Zhang, Z.; Li, J.; Xia, Q.; Chen, Y. Folliculin Deficient Renal Cancer Cells Exhibit BRCA1 A Complex Expression Impairment and Sensitivity to PARP1 Inhibitor Olaparib. Gene 2021, 769, 145243. [Google Scholar] [CrossRef] [PubMed]
- Radzikowska, E.; Lechowicz, U.; Winek, J.; Opoka, L. Novel Folliculin Gene Mutations in Polish Patients with Birt–Hogg–Dubé Syndrome. Orphanet J. Rare Dis. 2021, 16, 302. [Google Scholar] [CrossRef]
- Zong, D.; Li, J.; Liu, X.; Guo, T.; Ouyang, R. Identification of a Novel Pathogenic Folliculin Variant in a Chinese Family With Birt–Hogg–Dubé Syndrome (Hornstein-Knickenberg Syndrome). Front. Genet. 2020, 11, 565566. [Google Scholar] [CrossRef]
- Ramírez-Calvo, M.; García-Casado, Z.; Fernández-Serra, A.; de Juan, I.; Palanca, S.; Oltra, S.; Soto, J.L.; Castillejo, A.; Barbera, V.M.; Juan-Fita, M.J.; et al. Implementation of Massive Sequencing in the Genetic Diagnosis of Hereditary Cancer Syndromes: Diagnostic Performance in the Hereditary Cancer Programme of the Valencia Community (FamCan-NGS). Hered. Cancer Clin. Pract. 2019, 17, 3. [Google Scholar] [CrossRef] [Green Version]
- Menko, F.H.; van Steensel, M.A.; Giraud, S.; Friis-Hansen, L.; Richard, S.; Ungari, S.; Nordenskjöld, M.; Hansen, T.V.O.; Solly, J.; Maher, E.R. Birt-Hogg-Dubé Syndrome: Diagnosis and Management. Lancet Oncol. 2009, 10, 1199–1206. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Clausen, L.; Stein, A.; Grønbæk-Thygesen, M.; Nygaard, L.; Søltoft, C.L.; Nielsen, S.V.; Lisby, M.; Ravid, T.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Folliculin Variants Linked to Birt-Hogg-Dubé Syndrome Are Targeted for Proteasomal Degradation. PLOS Genet. 2020, 16, e1009187. [Google Scholar] [CrossRef]
- Furuya, M.; Kobayashi, H.; Baba, M.; Ito, T.; Tanaka, R.; Nakatani, Y. Splice-Site Mutation Causing Partial Retention of Intron in the FLCN Gene in Birt-Hogg-Dubé Syndrome: A Case Report. BMC Med. Genom. 2018, 11, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daccord, C.; Good, J.-M.; Morren, M.-A.; Bonny, O.; Hohl, D.; Lazor, R. Birt–Hogg–Dubé Syndrome. Eur. Respir. Rev. 2020, 29, 200042. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Fukui, T.; Makita, N.; Shimizu, K.; Kono, J.; Masui, K.; Sato, T.; Goto, T.; Sawada, A.; Fujimoto, M.; et al. A Novel Missense Mutation in the Folliculin Gene Associated with the Renal Tumor-Only Phenotype of Birt-Hogg-Dubé Syndrome. Cancer Genet. 2022, 266–267, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Sattler, E.C.; Syunyaeva, Z.; Reithmair, M.; Dempke, W.; Steinlein, O.K. Colorectal Cancer Risk in Families with Birt-Hogg-Dubé Syndrome Increased. Eur. J. Cancer 2021, 151, 168–174. [Google Scholar] [CrossRef]
- Ather, H.; Zahid, N. Recurrent Renal Cancer in Birt–Hogg–Dubé Syndrome: A Case Report. Int. J. Surg. Case Rep. 2018, 42, 75–78. [Google Scholar] [CrossRef]
- Choi, Y.J.; Park, C.H.; Park, H.J.; Shin, J.M.; Kim, T.H.; Lee, K.-A.; Moon, D.H.; Lee, S.; Lee, S.E.; Byun, M.K. Characteristic Chest Computed Tomography Findings for Birt–Hogg–Dube Syndrome Indicating Requirement for Genetic Evaluation. Diagnostics 2023, 13, 198. [Google Scholar] [CrossRef]
- Al-Shinnag, M.; Marfan, H.; Susman, R.; Wakeling, J.; Gustafson, S.; Wood, S.; Mallett, A.J. Birt-Hogg-Dubé Syndrome and Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome: An Effective Multidisciplinary Approach to Hereditary Renal Cancer Predisposing Syndromes. Front. Oncol. 2021, 11, 738822. [Google Scholar] [CrossRef]
- Zhou, W.; Liu, K.; Xu, K.-F.; Liu, Y.; Tian, X. Clinical and Genetic Comparison of Birt–Hogg–Dubé Syndrome (Hornstein–Knickenberg Syndrome) in Chinese: A Systemic Review of Reported Cases. Int. J. Gen. Med. 2022, 15, 5111–5121. [Google Scholar] [CrossRef]
- Liu, K.; Xu, W.; Tian, X.; Xiao, M.; Zhao, X.; Zhang, Q.; Qu, T.; Song, J.; Liu, Y.; Xu, K.-F.; et al. Genotypic Characteristics of Chinese Patients with BHD Syndrome and Functional Analysis of FLCN Variants. Orphanet J. Rare Dis. 2019, 14, 223. [Google Scholar] [CrossRef] [Green Version]
- Federici, G.; Soddu, S. Variants of Uncertain Significance in the Era of High-Throughput Genome Sequencing: A Lesson from Breast and Ovary Cancers. J. Exp. Clin. Cancer Res. 2020, 39, 46. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, L.S.; Linehan, W.M. FLCN: The Causative Gene for Birt-Hogg-Dubé Syndrome. Gene 2018, 640, 28–42. [Google Scholar] [CrossRef] [PubMed]
- van Riel, L.; Jansen, P.R.; Boerrigter, B.G.; van Moorselaar, R.J.A.; van Haelst, M.M.; Wolthuis, R.M.F.; van de Beek, I.; Houweling, A.C. Correspondence on “Frequency of Truncating FLCN Variants and Birt-Hogg-Dubé-Associated Phenotypes in a Health Care System Population” by Savatt et Al. Genet. Med. 2023, 25, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| AIP | AP2S1 | ARMC5 | ATRX | BAP1 | BRK1 | CASR | CDC73 | CDKN1A | CDKN1B |
|---|---|---|---|---|---|---|---|---|---|
| CDKN2B | CDKN2C | DNMT3A | EGLN1 | EGLN2 | EPAS1 | ESR2 | FGFR1 | FH | FLCN |
| GCM2 | GNA11 | H3F3A | KIF1B | KMT2D | MAX | MDH2 | MEN1 | MERTK | MET |
| NF1 | PBRM1 | PDE11A | PDE8B | PRKACA | PRKAR1A | PTEN | RET | SDHA | SDHAF1 |
| SDHAF2 | SDHAF3 | SDHAF4 | SDHB | SDHC | SDHD | SLC25A11 | TMEM127 | Tp53 | TSC1 |
| TSC2 | VHL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bandini, E.; Zampiga, V.; Cangini, I.; Ravegnani, M.; Arcangeli, V.; Rossi, T.; Mammi, I.; Schiavi, F.; Zovato, S.; Falcini, F.; et al. A Novel FLCN Variant in a Suspected Birt–Hogg–Dubè Syndrome Patient. Int. J. Mol. Sci. 2023, 24, 12418. https://doi.org/10.3390/ijms241512418
Bandini E, Zampiga V, Cangini I, Ravegnani M, Arcangeli V, Rossi T, Mammi I, Schiavi F, Zovato S, Falcini F, et al. A Novel FLCN Variant in a Suspected Birt–Hogg–Dubè Syndrome Patient. International Journal of Molecular Sciences. 2023; 24(15):12418. https://doi.org/10.3390/ijms241512418
Chicago/Turabian StyleBandini, Erika, Valentina Zampiga, Ilaria Cangini, Mila Ravegnani, Valentina Arcangeli, Tania Rossi, Isabella Mammi, Francesca Schiavi, Stefania Zovato, Fabio Falcini, and et al. 2023. "A Novel FLCN Variant in a Suspected Birt–Hogg–Dubè Syndrome Patient" International Journal of Molecular Sciences 24, no. 15: 12418. https://doi.org/10.3390/ijms241512418
APA StyleBandini, E., Zampiga, V., Cangini, I., Ravegnani, M., Arcangeli, V., Rossi, T., Mammi, I., Schiavi, F., Zovato, S., Falcini, F., Calistri, D., & Danesi, R. (2023). A Novel FLCN Variant in a Suspected Birt–Hogg–Dubè Syndrome Patient. International Journal of Molecular Sciences, 24(15), 12418. https://doi.org/10.3390/ijms241512418