

Tumor Growth Ameliorates Cardiac Dysfunction and Suppresses Fibrosis in a Mouse Model for Duchenne Muscular Dystrophy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
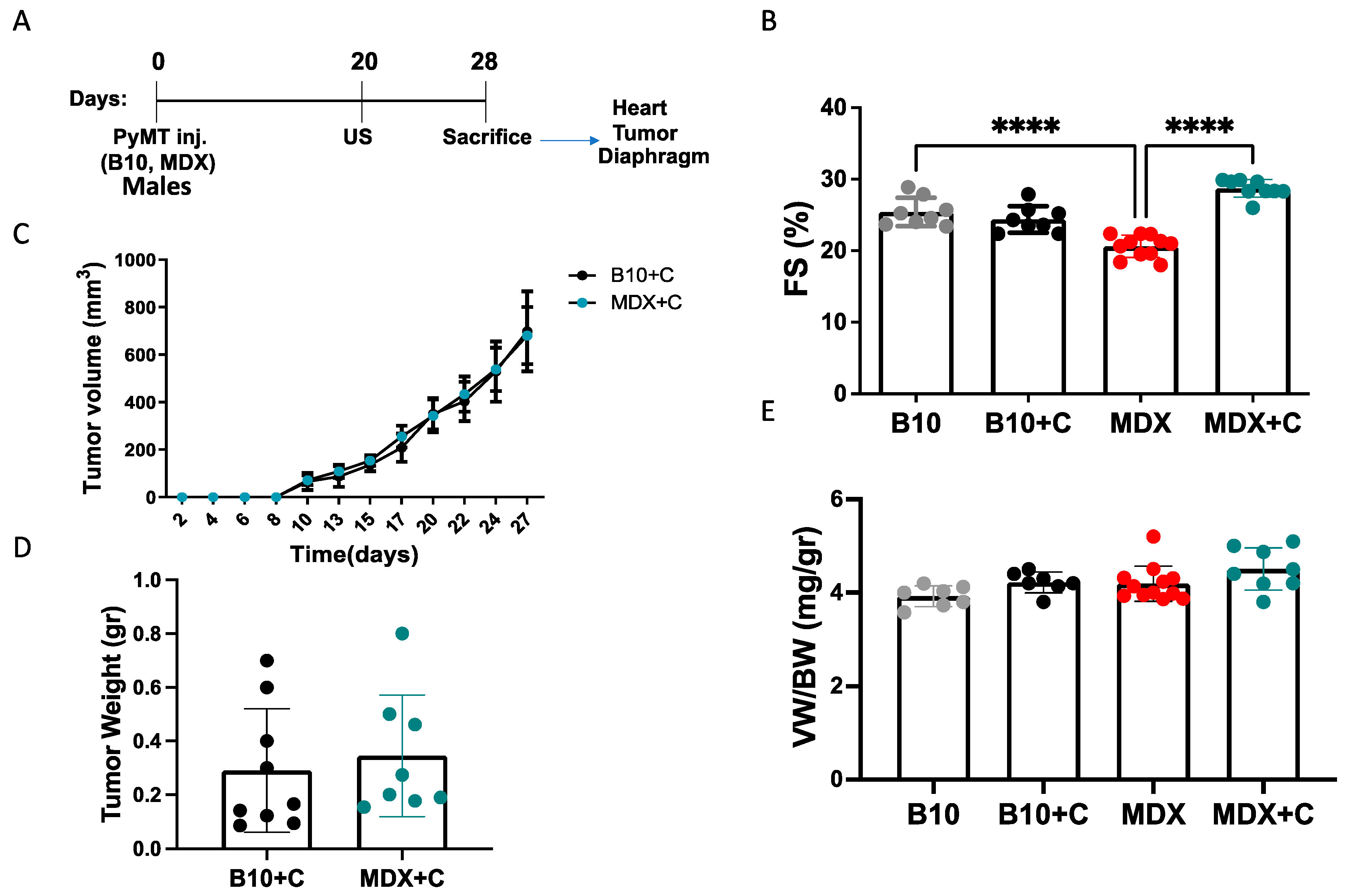
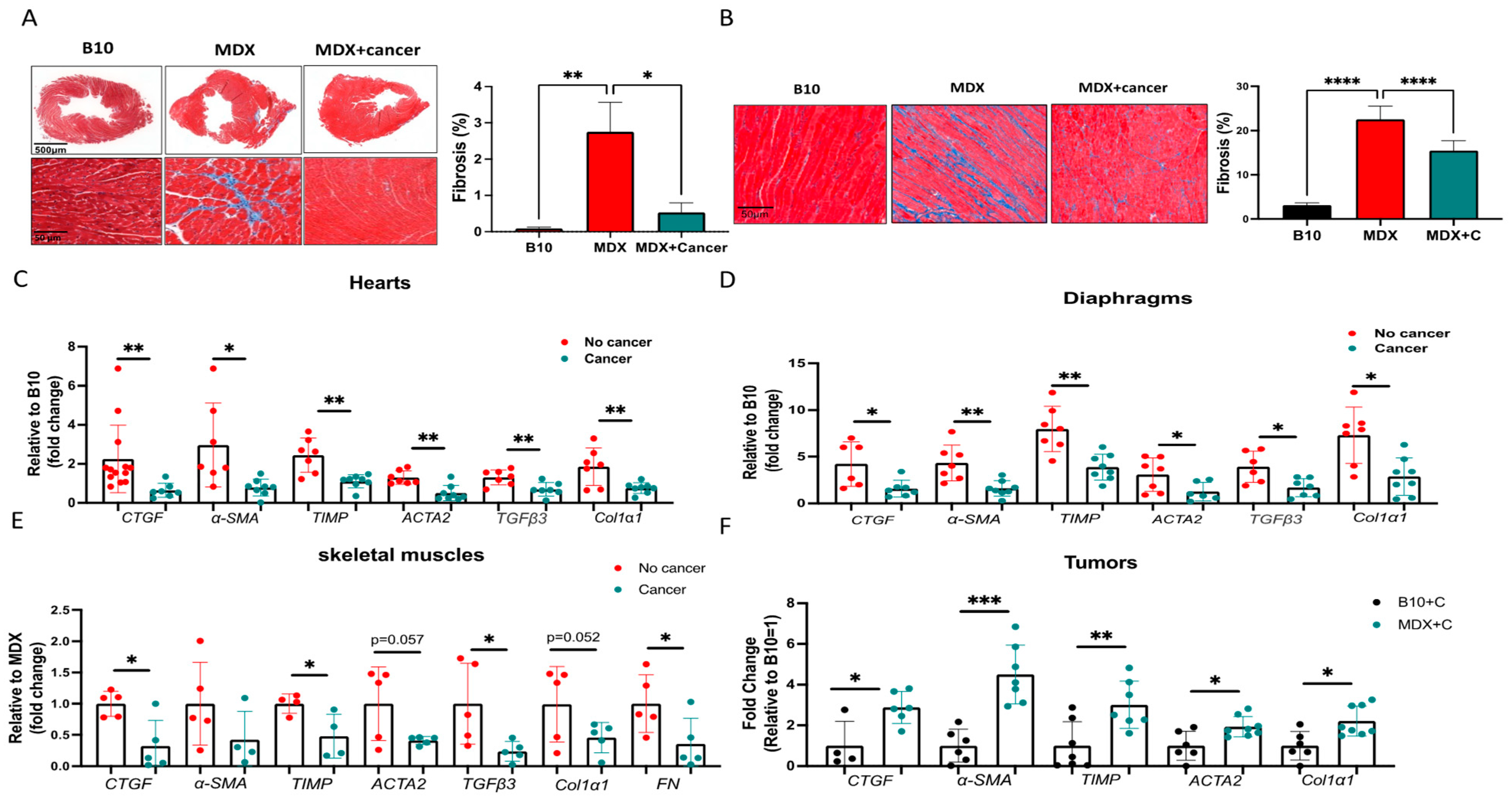
2.1. Tumor Growth Significantly Improved the Contractile Function of MDX Heart and Diaphragm Muscles
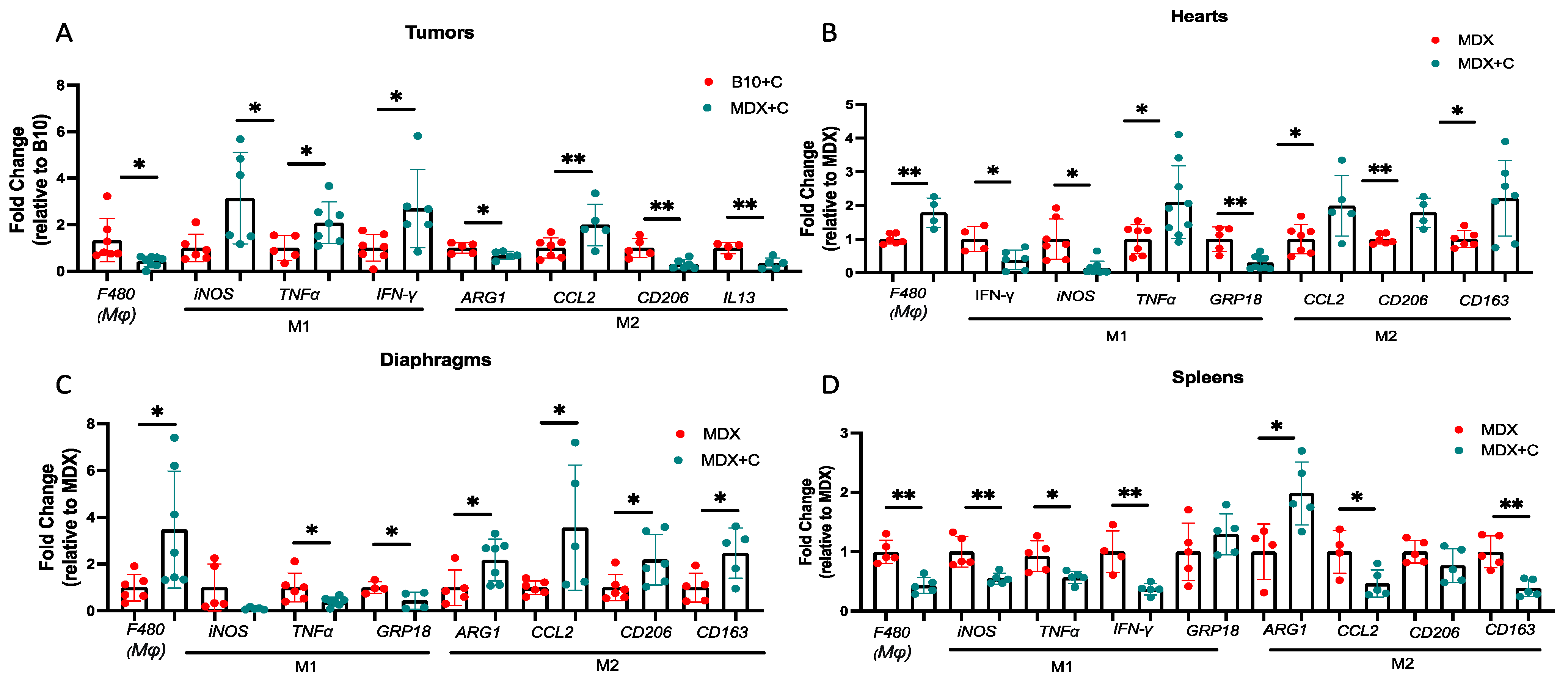
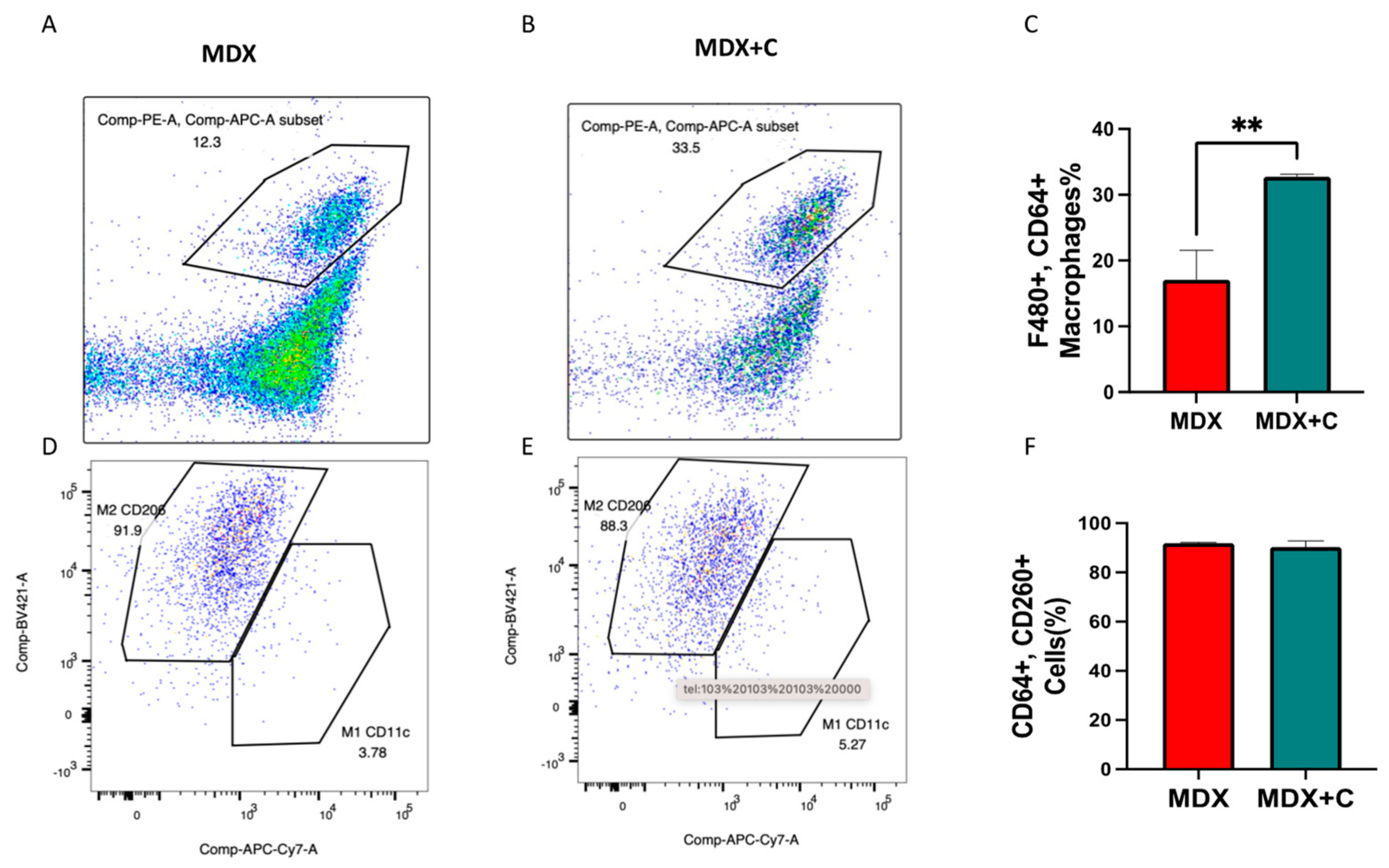
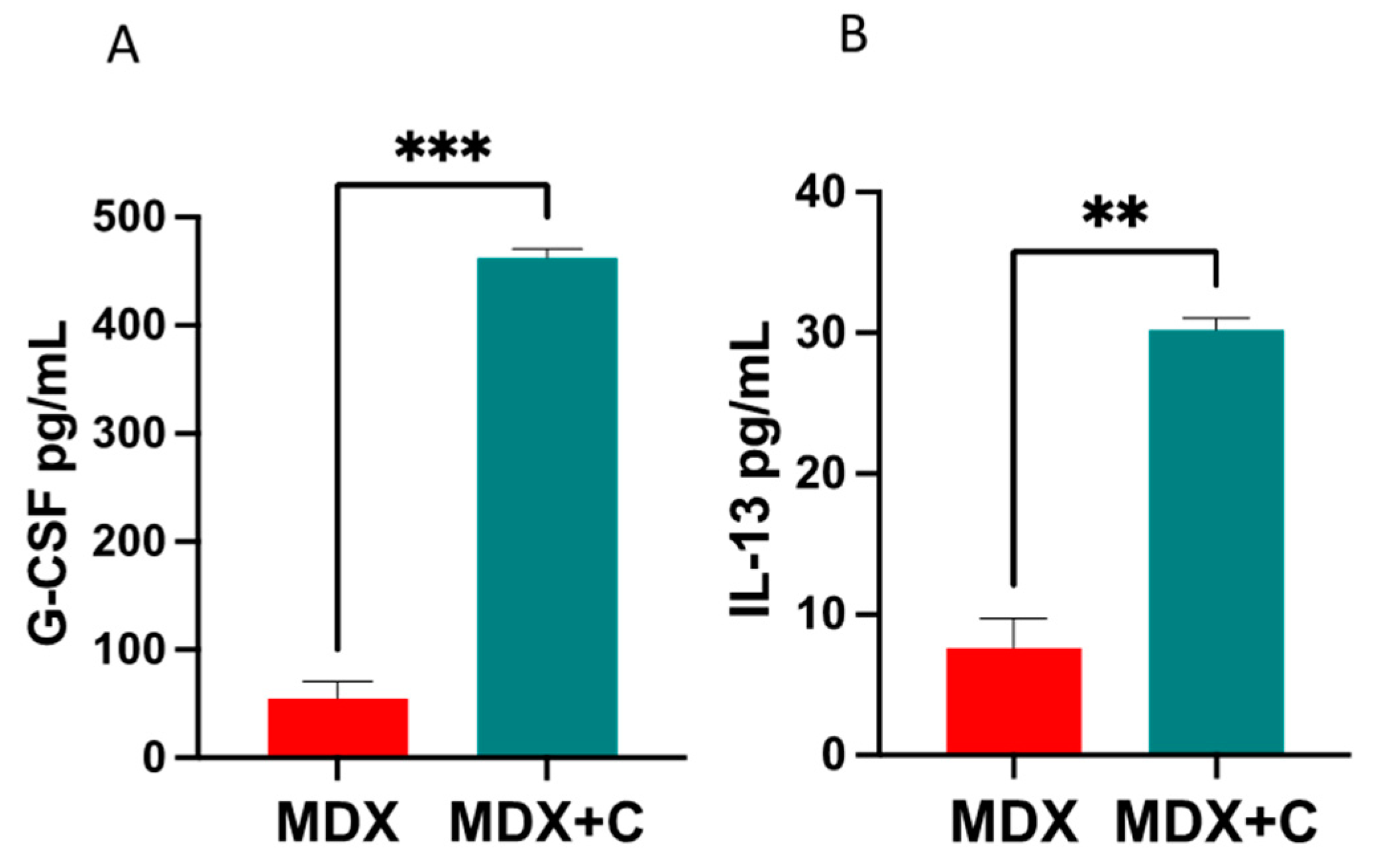
2.2. Improved Contractile Function and Reduction in Fibrosis in Tumor-Bearing Mice Is Mediated by M2 Macrophage Recruitment to the Heart
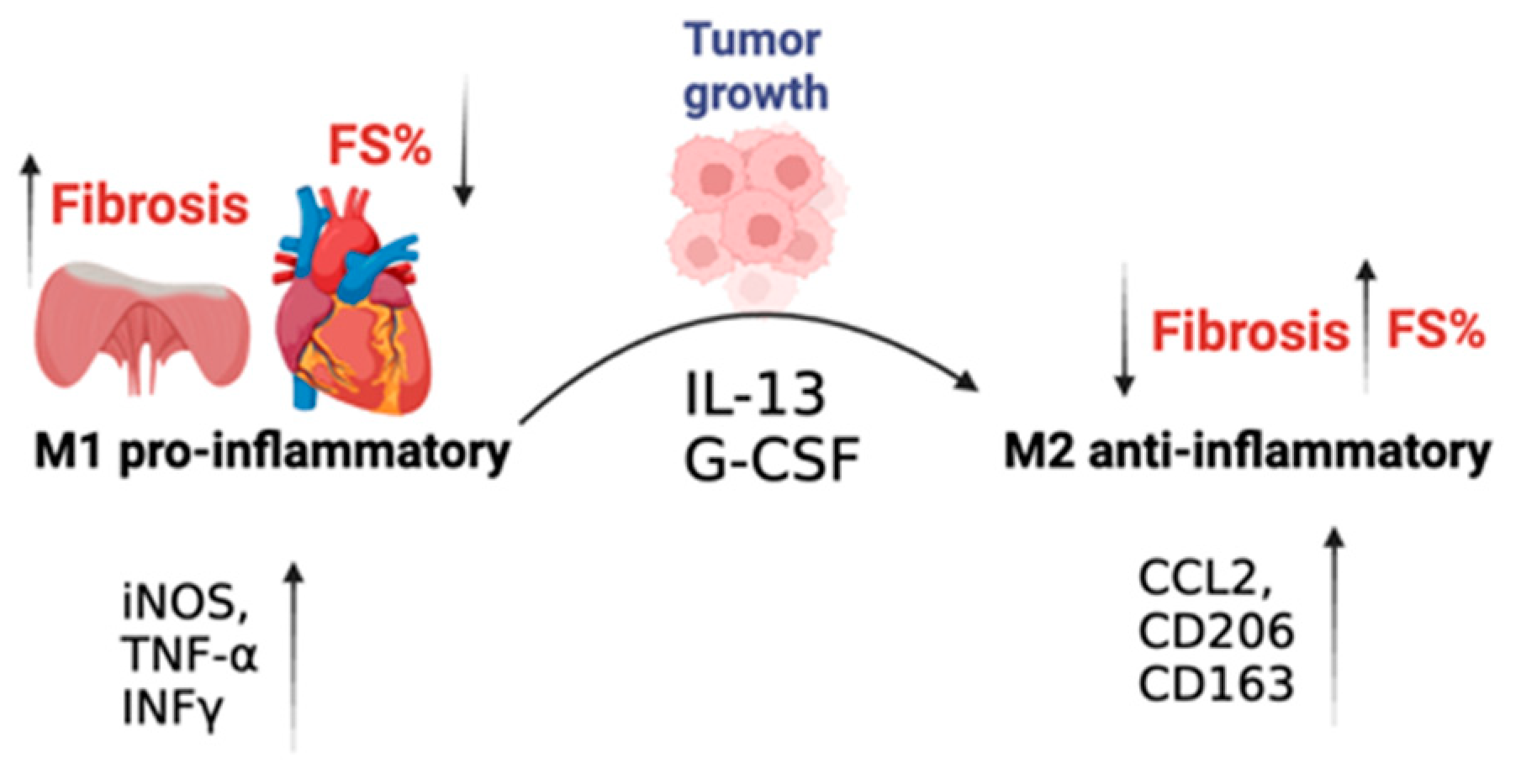
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Culture
4.3. Cancer Cell Implantation
4.4. Echocardiography
4.5. RNA Extraction
4.6. Quantitative Real-Time PCR
4.7. Fibrosis Staining
4.8. Heart Single-Cell Suspension and Flow Cytometry
4.9. ELISA
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Darby, I.A.; Laverdet, B.; Bonte, F.; Desmouliere, A. Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol. 2014, 7, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Diez, J.; de Boer, R.A. Management of cardiac fibrosis is the largest unmet medical need in heart failure. Cardiovasc. Res. 2022, 118, e20–e22. [Google Scholar] [CrossRef]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Wijsenbeek, M.; Cottin, V. Spectrum of Fibrotic Lung Diseases. N. Engl. J. Med. 2020, 383, 958–968. [Google Scholar] [CrossRef]
- Murtha, L.A.; Schuliga, M.J.; Mabotuwana, N.S.; Hardy, S.A.; Waters, D.W.; Burgess, J.K.; Knight, D.A.; Boyle, A.J. The Processes and Mechanisms of Cardiac and Pulmonary Fibrosis. Front. Physiol. 2017, 8, 777. [Google Scholar] [CrossRef] [Green Version]
- Avraham, S.; Abu-Sharki, S.; Shofti, R.; Haas, T.; Korin, B.; Kalfon, R.; Friedman, T.; Shiran, A.; Saliba, W.; Shaked, Y.; et al. Early Cardiac Remodeling Promotes Tumor Growth and Metastasis. Circulation 2020, 142, 670–683. [Google Scholar] [CrossRef]
- Awwad, L.; Aronheim, A. Cardiac Dysfunction Promotes Cancer Progression via Multiple Secreted Factors. Cancer Res. 2022, 82, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Awwad, L.; Goldenberg, T.; Langier-Goncalves, I.; Aronheim, A. Cardiac Remodeling in the Absence of Cardiac Contractile Dysfunction Is Sufficient to Promote Cancer Progression. Cells 2022, 11, 1108. [Google Scholar] [CrossRef]
- Awwad, L.; Shofti, R.; Haas, T.; Aronheim, A. Tumor Growth Ameliorates Cardiac Dysfunction. Cells 2023, 12, 1853. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS−) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Nurakhayev, S.; Nurkesh, A.; Zharkinbekov, Z.; Saparov, A. Macrophage Polarization in Cardiac Tissue Repair Following Myocardial Infarction. Int. J. Mol. Sci. 2021, 22, 2715. [Google Scholar] [CrossRef]
- Simoes, G.F.; Benitez, S.U.; Oliveira, A.L. Granulocyte colony-stimulating factor (G-CSF) positive effects on muscle fiber degeneration and gait recovery after nerve lesion in MDX mice. Brain Behav. 2014, 4, 738–753. [Google Scholar] [CrossRef]
- Yang, H.; Chen, Y.; Gao, C. Interleukin-13 reduces cardiac injury and prevents heart dysfunction in viral myocarditis via enhanced M2 macrophage polarization. Oncotarget 2017, 8, 99495–99503. [Google Scholar] [CrossRef] [Green Version]
- Meijers, W.C.; Maglione, M.; Bakker, S.J.L.; Oberhuber, R.; Kieneker, L.M.; de Jong, S.; Haubner, B.J.; Nagengast, W.B.; Lyon, A.R.; van der Vegt, B.; et al. Heart Failure Stimulates Tumor Growth by Circulating Factors. Circulation 2018, 138, 678–691. [Google Scholar] [CrossRef]
- Koelwyn, G.J.; Newman, A.A.C.; Afonso, M.S.; van Solingen, C.; Corr, E.M.; Brown, E.J.; Albers, K.B.; Yamaguchi, N.; Narke, D.; Schlegel, M.; et al. Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nat. Med. 2020, 26, 1452–1458. [Google Scholar] [CrossRef]
- Koelwyn, G.J.; Aboumsallem, J.P.; Moore, K.J.; de Boer, R.A. Reverse cardio-oncology: Exploring the effects of cardiovascular disease on cancer pathogenesis. J. Mol. Cell Cardiol. 2022, 163, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, H.; Takahashi, M.; Izawa, A.; Ise, H.; Hongo, M.; Kolattukudy, P.E.; Ikeda, U. Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice prevents cardiac dysfunction and remodeling after myocardial infarction. Circ. Res. 2006, 99, 891–899. [Google Scholar] [CrossRef]
- Ley, K. M1 Means Kill; M2 Means Heal. J. Immunol. 2017, 199, 2191–2193. [Google Scholar] [CrossRef] [Green Version]
- Sager, H.B.; Kessler, T.; Schunkert, H. Monocytes and macrophages in cardiac injury and repair. J. Thorac. Dis. 2017, 9, S30–S35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, R.; Banete, A.; Basta, S. Spleen-derived macrophages are readily polarized into classically activated (M1) or alternatively activated (M2) states. Immunobiology 2014, 219, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.; Kong, Y.; Zhao, H.Y.; Zhang, Y.Y.; Han, T.T.; Wang, Y.; Xu, L.P.; Zhang, X.H.; Huang, X.J. G-CSF-induced macrophage polarization and mobilization may prevent acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2019, 54, 1419–1433. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidball, J.G. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 482–496. [Google Scholar] [CrossRef] [Green Version]
- Wolford, C.C.; McConoughey, S.J.; Jalgaonkar, S.P.; Leon, M.; Merchant, A.S.; Dominick, J.L.; Yin, X.; Chang, Y.; Zmuda, E.J.; O’Toole, S.A.; et al. Transcription factor ATF3 links host adaptive response to breast cancer metastasis. J. Clin. Investig. 2013, 123, 2893–2906. [Google Scholar] [CrossRef] [Green Version]
- Aronoff, L.; Epelman, S.; Clemente-Casares, X. Isolation and Identification of Extravascular Immune Cells of the Heart. J. Vis. Exp. 2018, 138, 58114. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Achlaug, L.; Awwad, L.; Langier Goncalves, I.; Goldenberg, T.; Aronheim, A. Tumor Growth Ameliorates Cardiac Dysfunction and Suppresses Fibrosis in a Mouse Model for Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2023, 24, 12595. https://doi.org/10.3390/ijms241612595
Achlaug L, Awwad L, Langier Goncalves I, Goldenberg T, Aronheim A. Tumor Growth Ameliorates Cardiac Dysfunction and Suppresses Fibrosis in a Mouse Model for Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2023; 24(16):12595. https://doi.org/10.3390/ijms241612595
Chicago/Turabian StyleAchlaug, Laris, Lama Awwad, Irina Langier Goncalves, Tomer Goldenberg, and Ami Aronheim. 2023. "Tumor Growth Ameliorates Cardiac Dysfunction and Suppresses Fibrosis in a Mouse Model for Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 24, no. 16: 12595. https://doi.org/10.3390/ijms241612595
APA StyleAchlaug, L., Awwad, L., Langier Goncalves, I., Goldenberg, T., & Aronheim, A. (2023). Tumor Growth Ameliorates Cardiac Dysfunction and Suppresses Fibrosis in a Mouse Model for Duchenne Muscular Dystrophy. International Journal of Molecular Sciences, 24(16), 12595. https://doi.org/10.3390/ijms241612595